
Impact of Matrix Surface Area on Griseofulvin Release from Extrudates Prepared via Nanoextrusion

 ,
, 


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Wet Stirred Media Milling Process
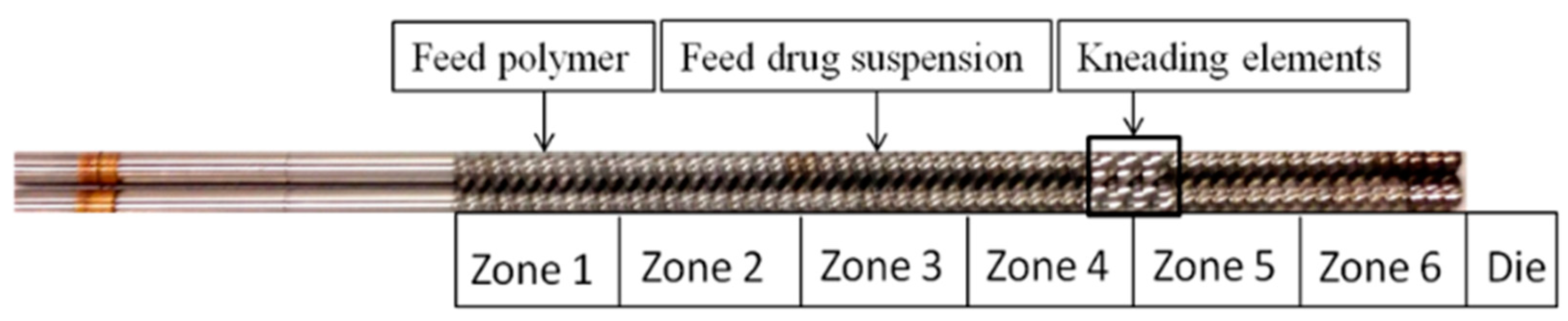
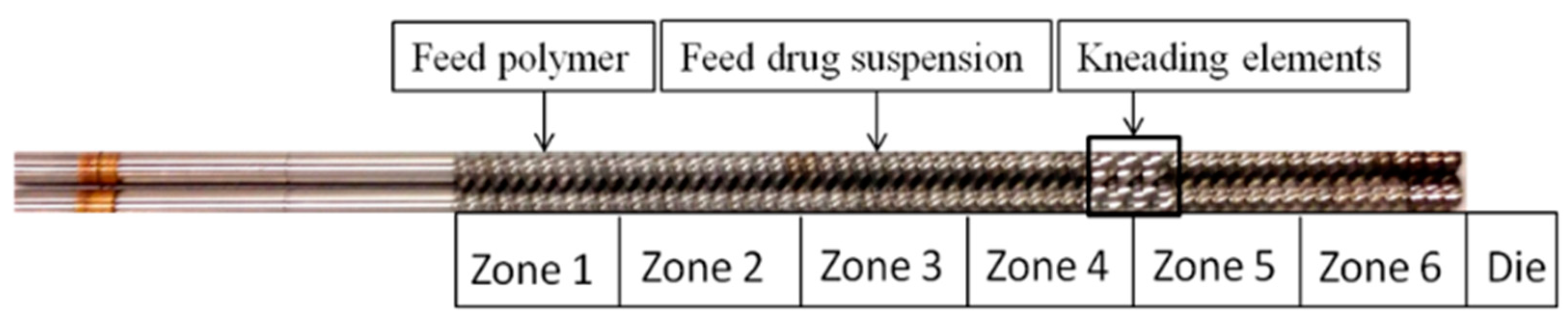
2.2.2. Preparation of Extrudate Powders
2.2.3. Measurement of Particle Size
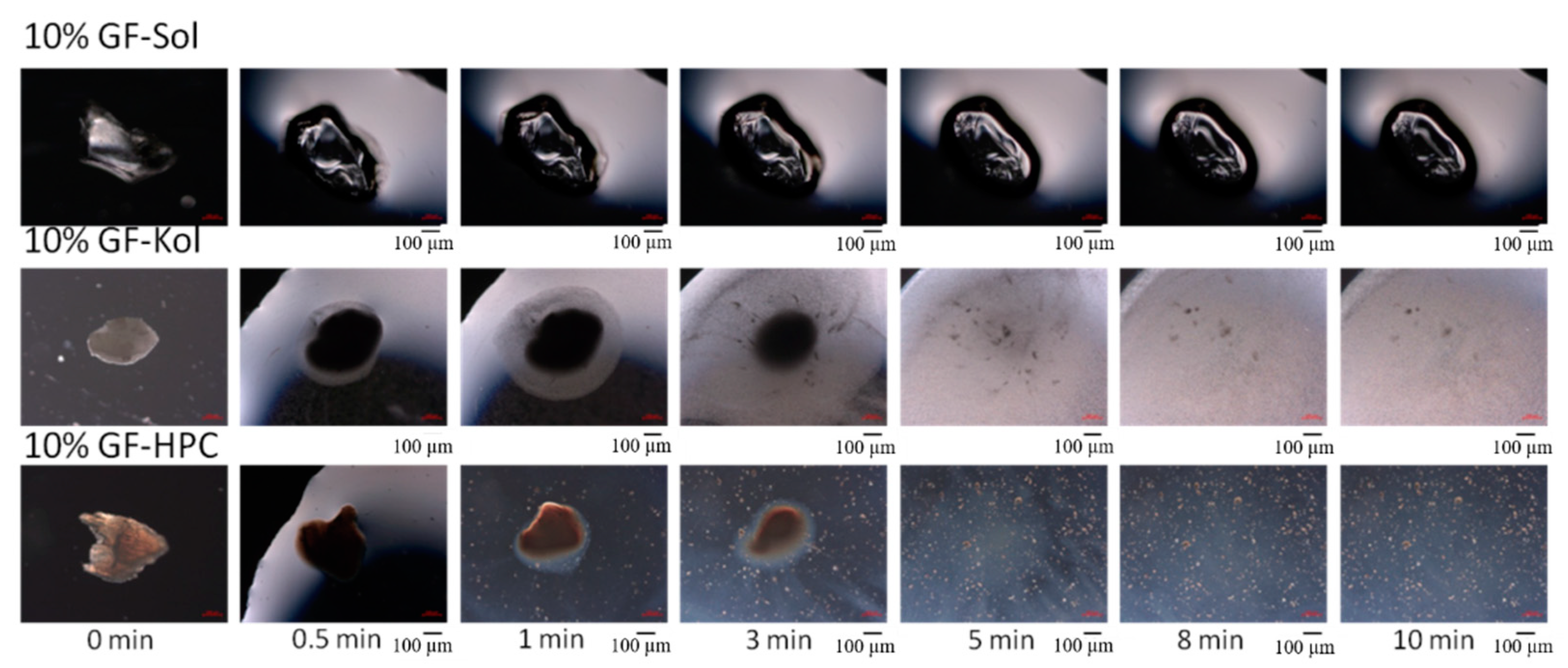
2.2.4. Microscopy
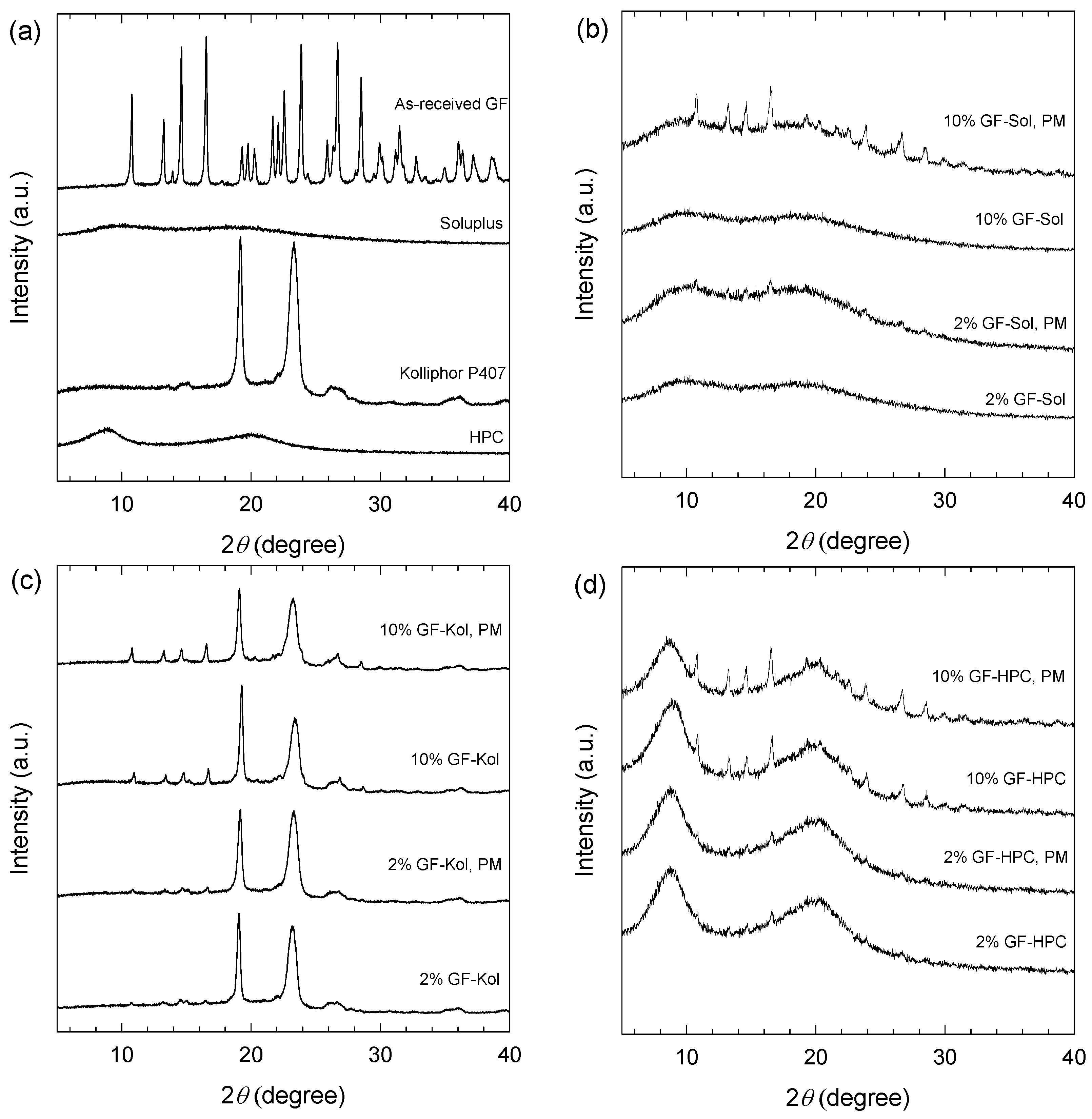
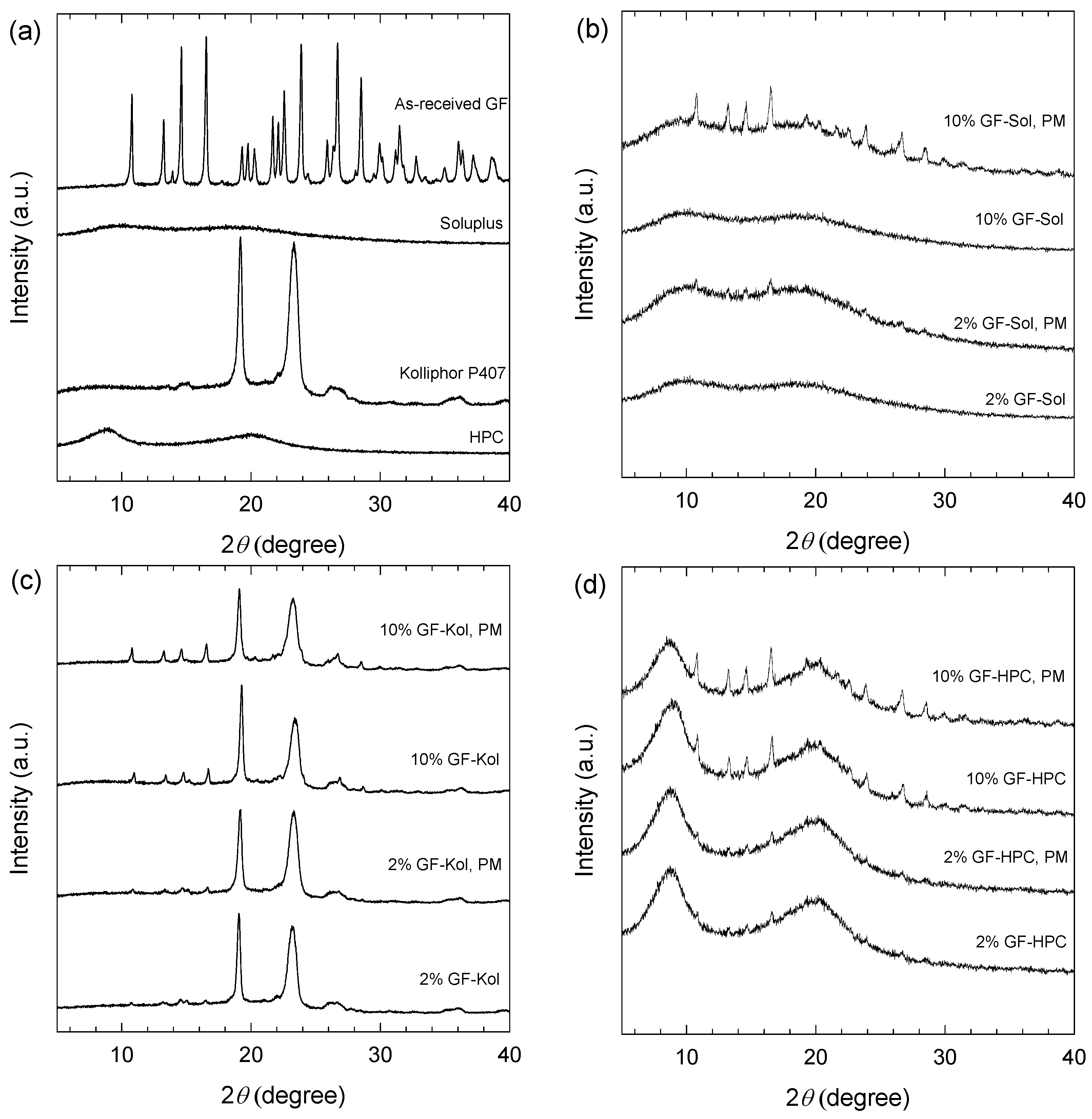
2.2.5. X-ray Powder Diffraction (XRPD)
2.2.6. Thermogravimetric Analysis (TGA)
2.2.7. Wetting of GF Powder by Soluplus®, Kolliphor® P407, and HPC Solutions
2.2.8. GF Content in the Extrudate Powders and Their Dissolution
2.2.9. Solvent-Shift Method for Study of GF Precipitation Inhibition by Polymers
3. Results and Discussion
3.1. Wet Stirred Media Milling of Drug Suspensions
3.2. Preparation and Characterization of the Extrudates
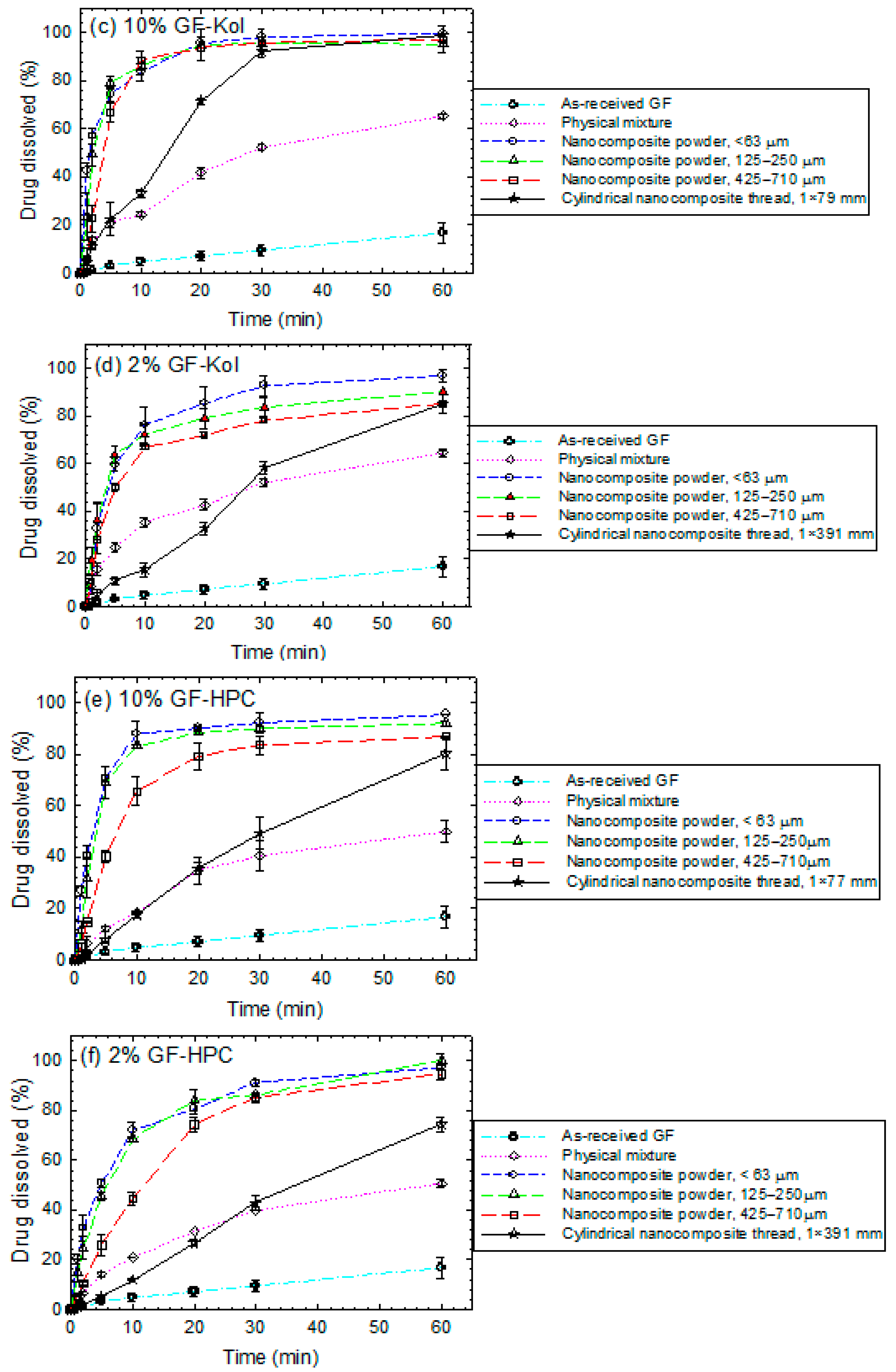
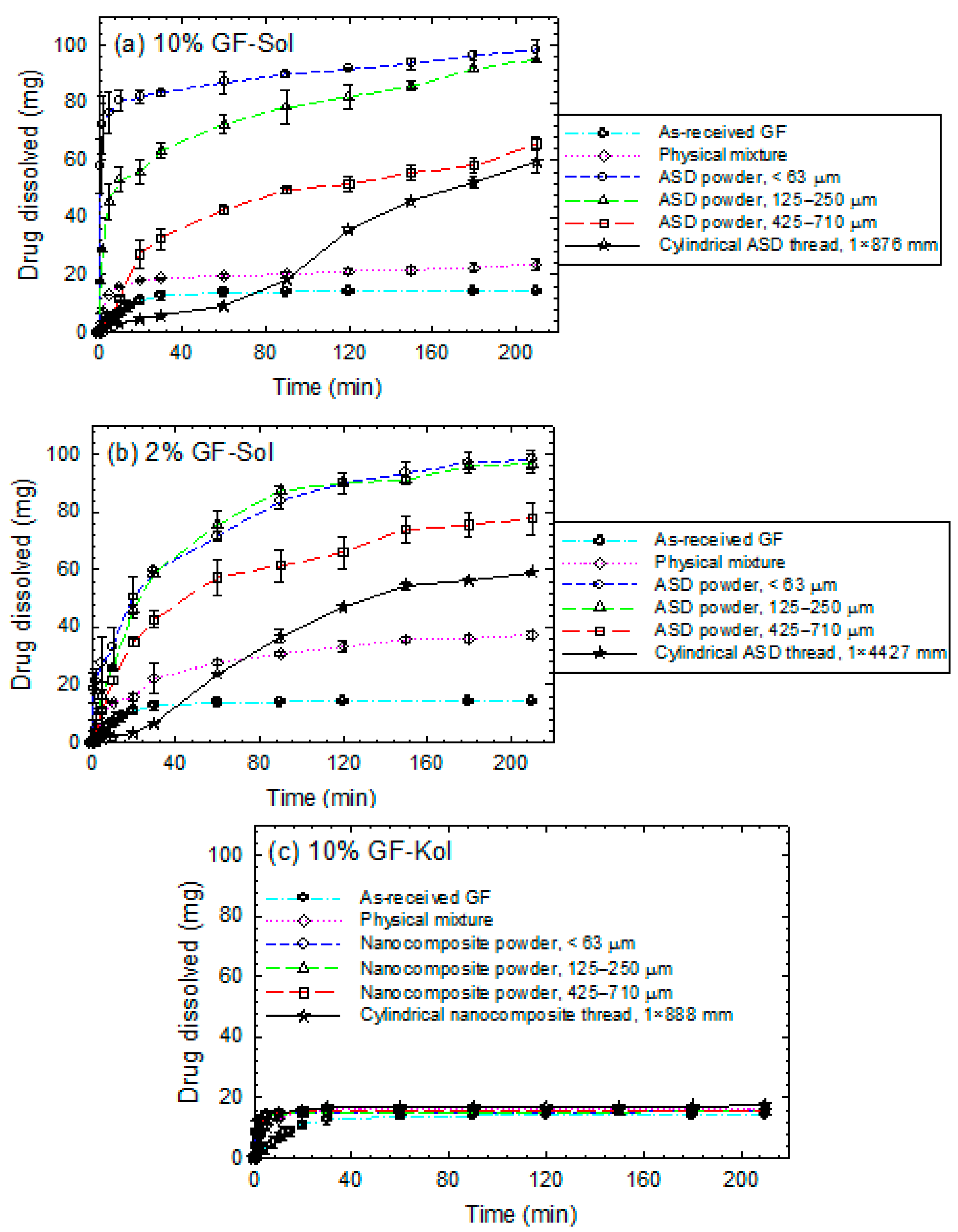
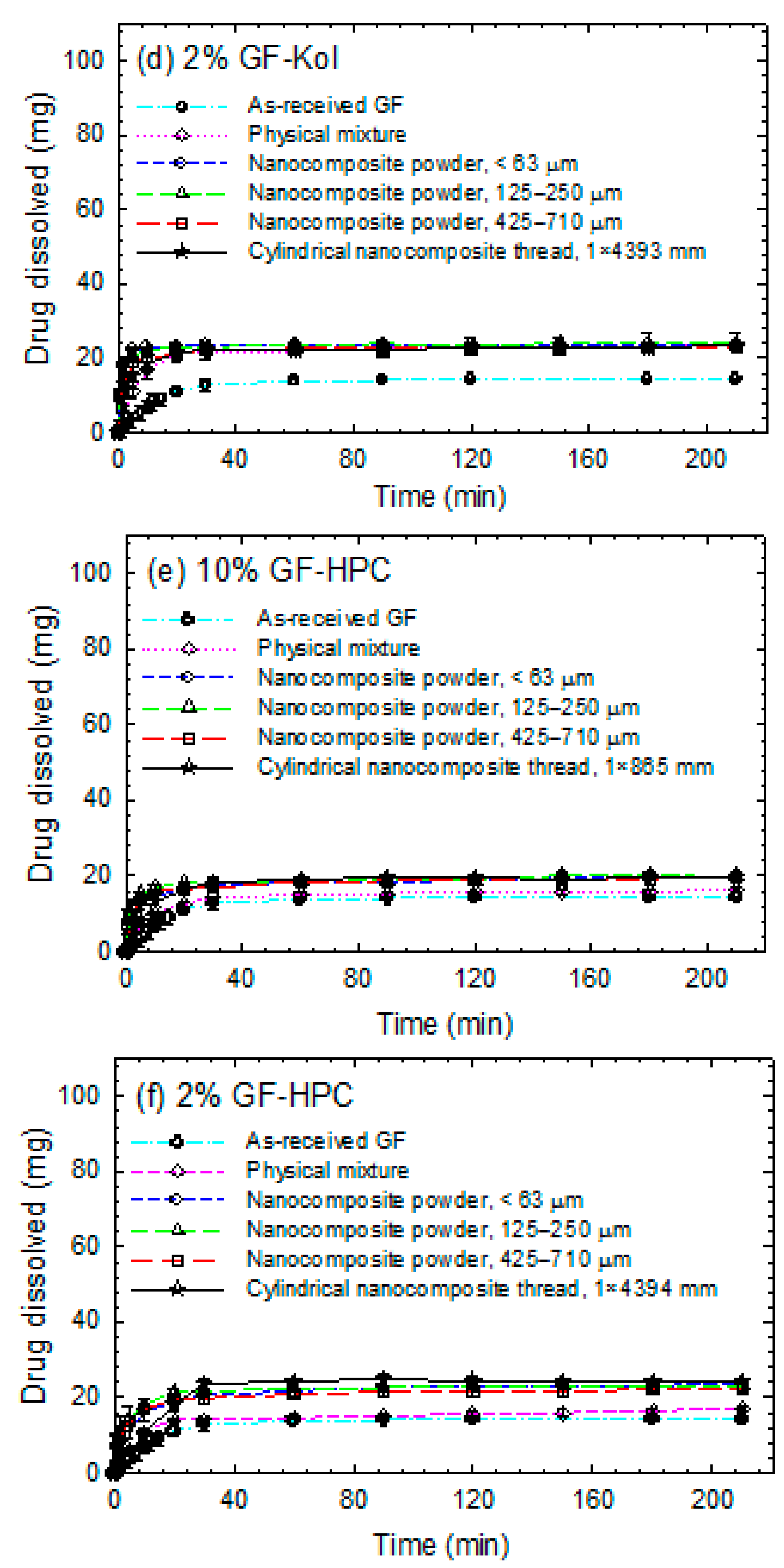
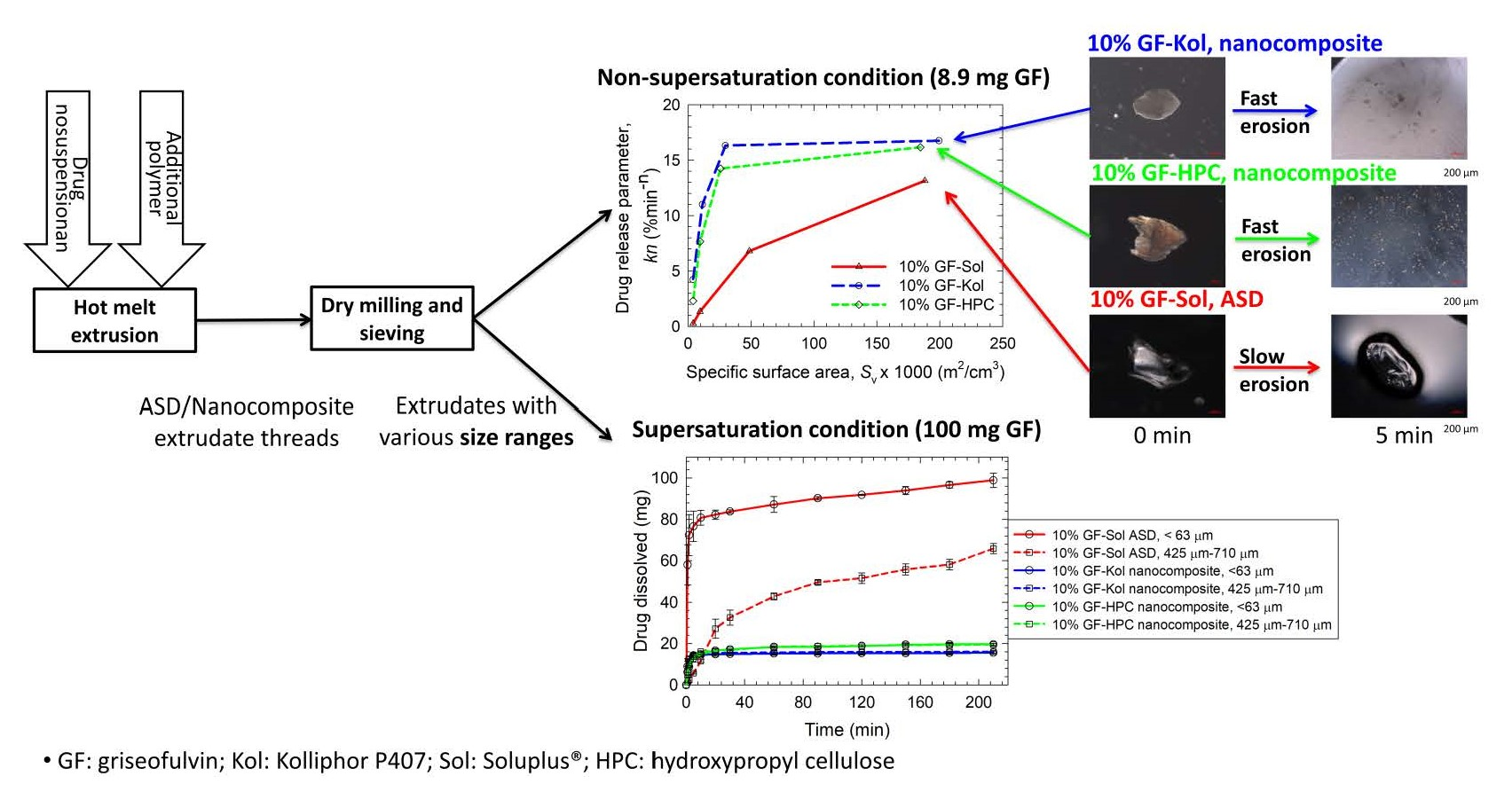
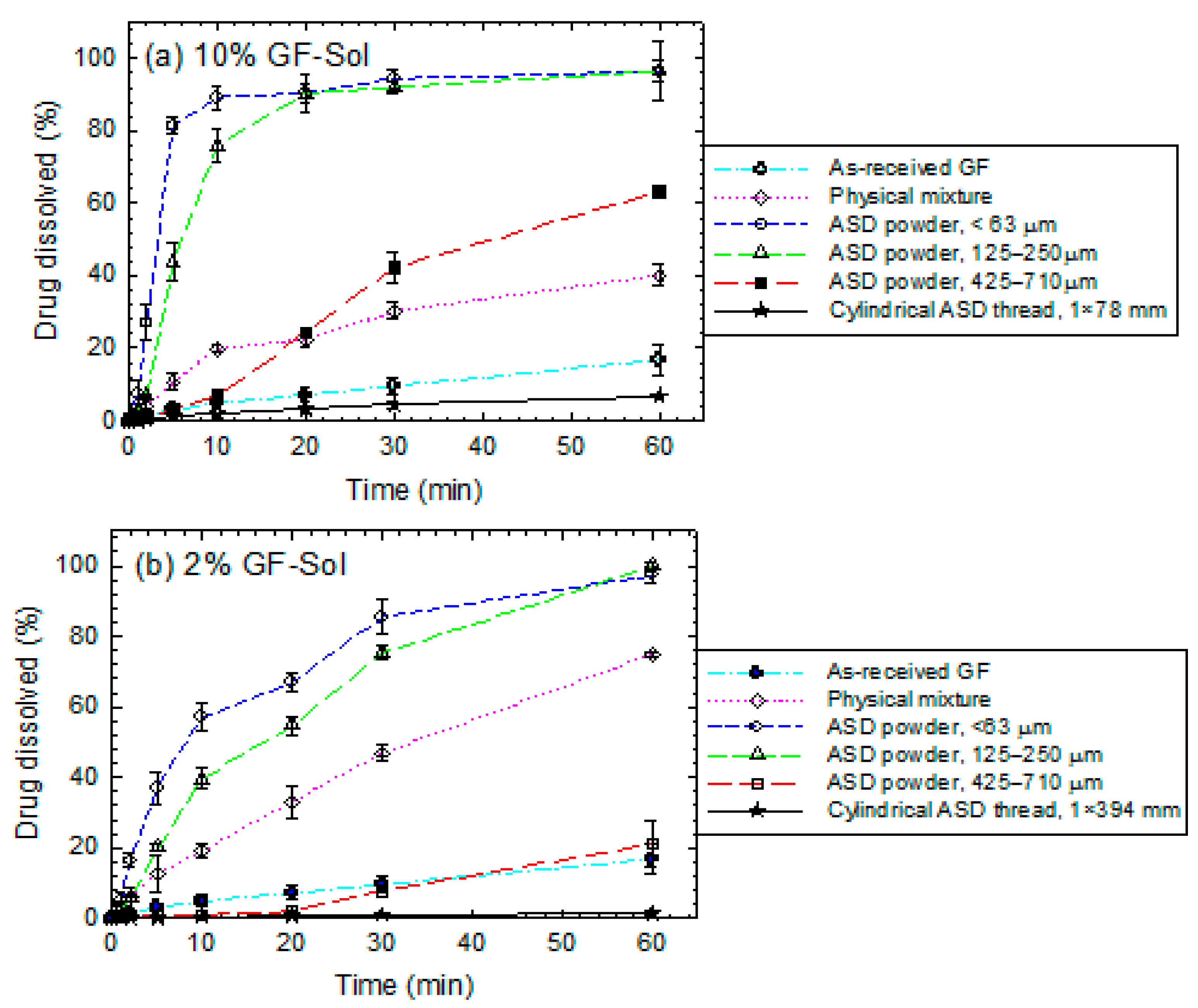
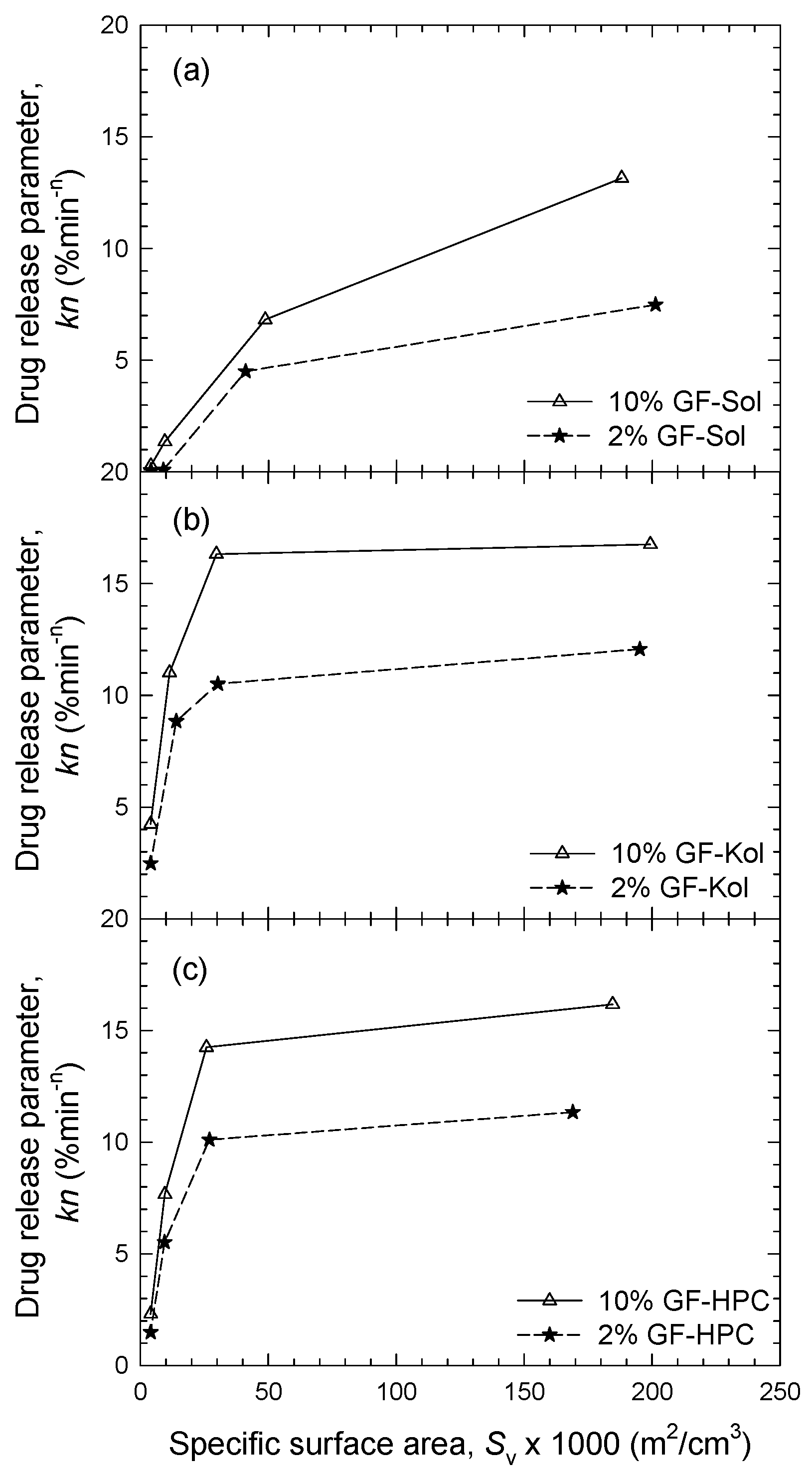
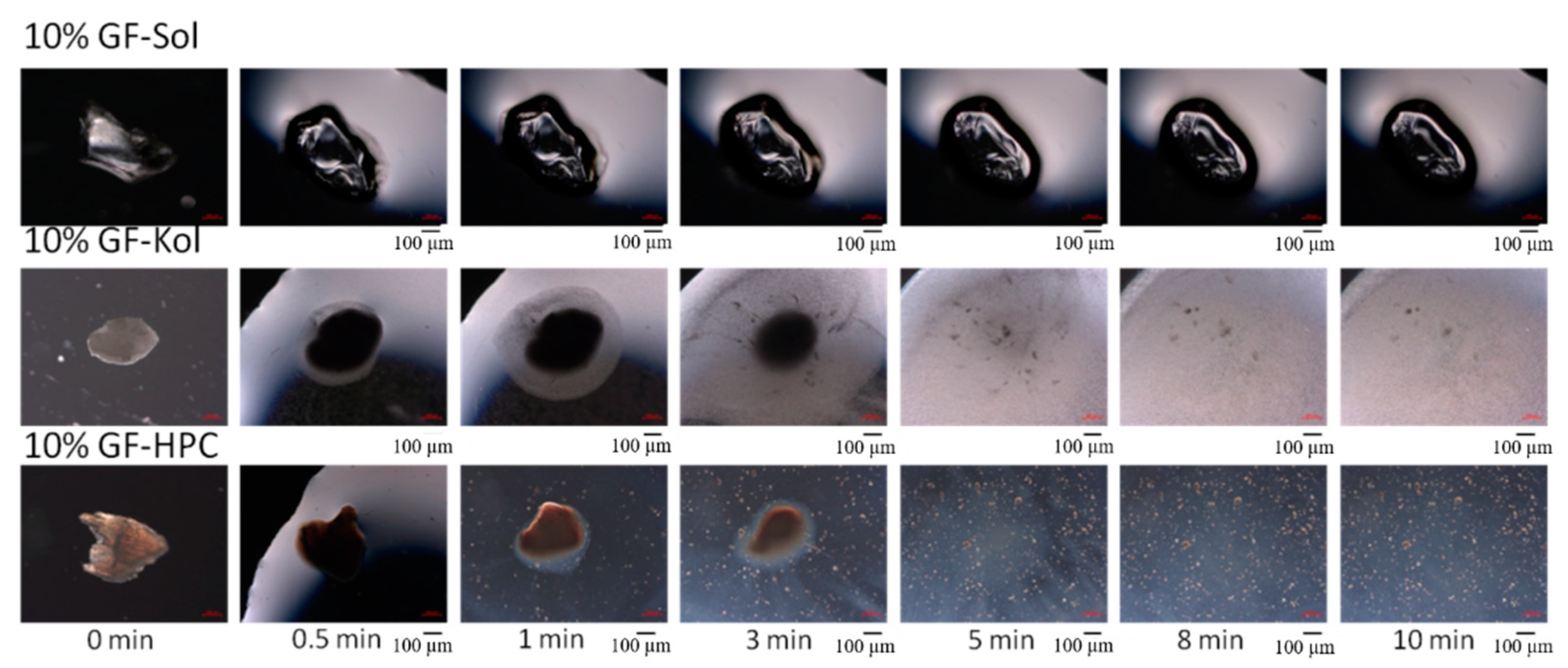
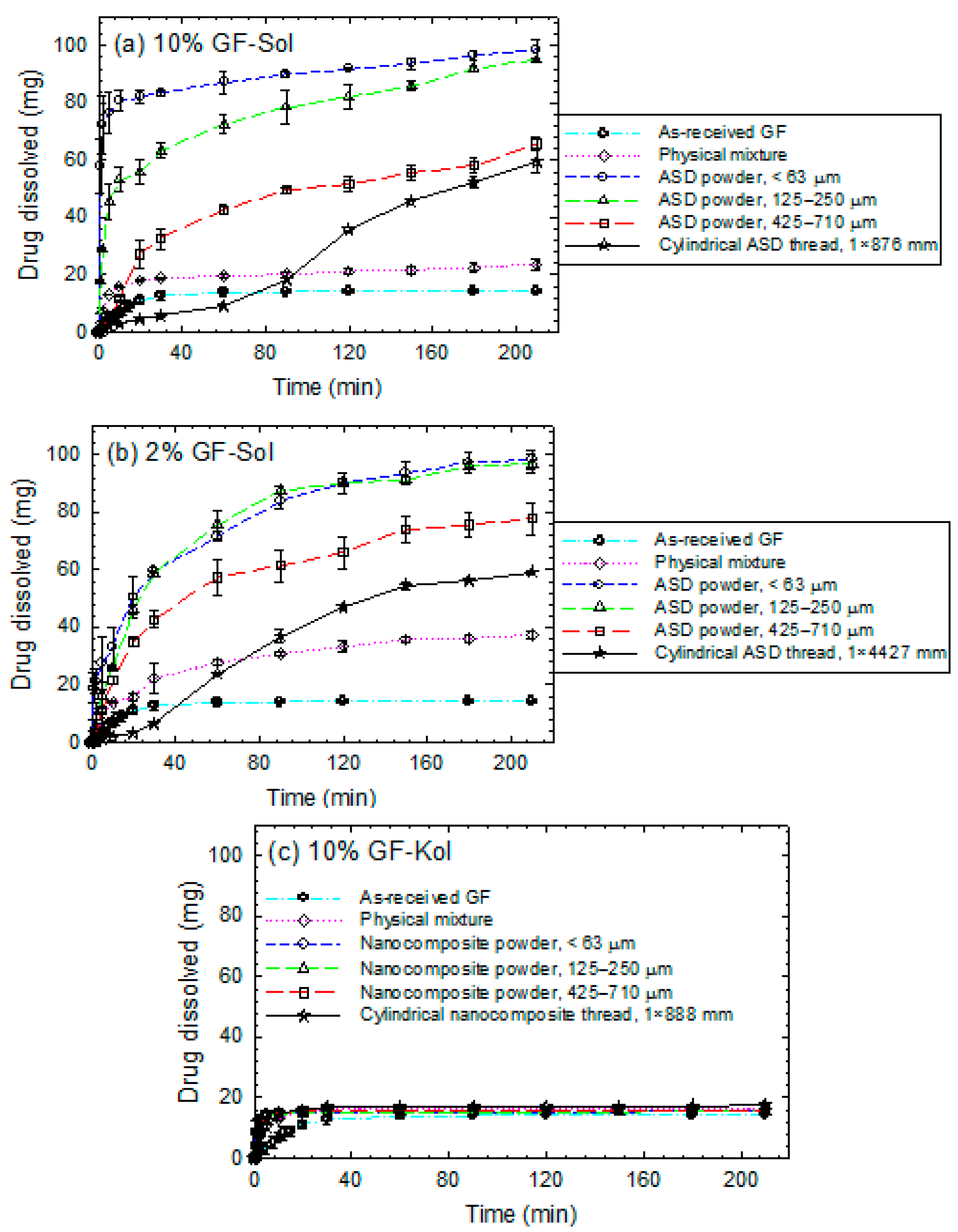
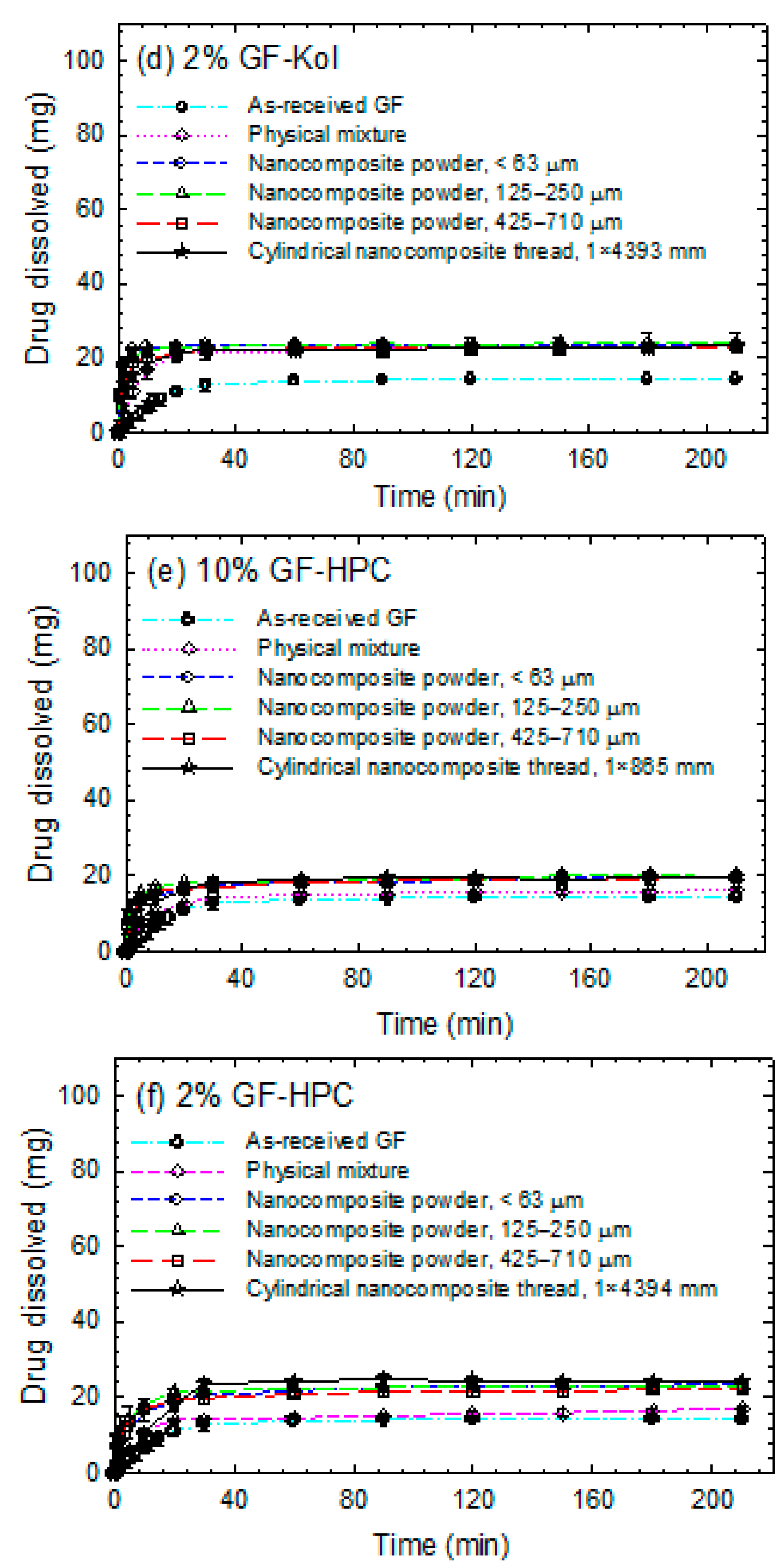
3.3. Impact of Matrix Surface Area at the Non-Supersaturating Dissolution Condition
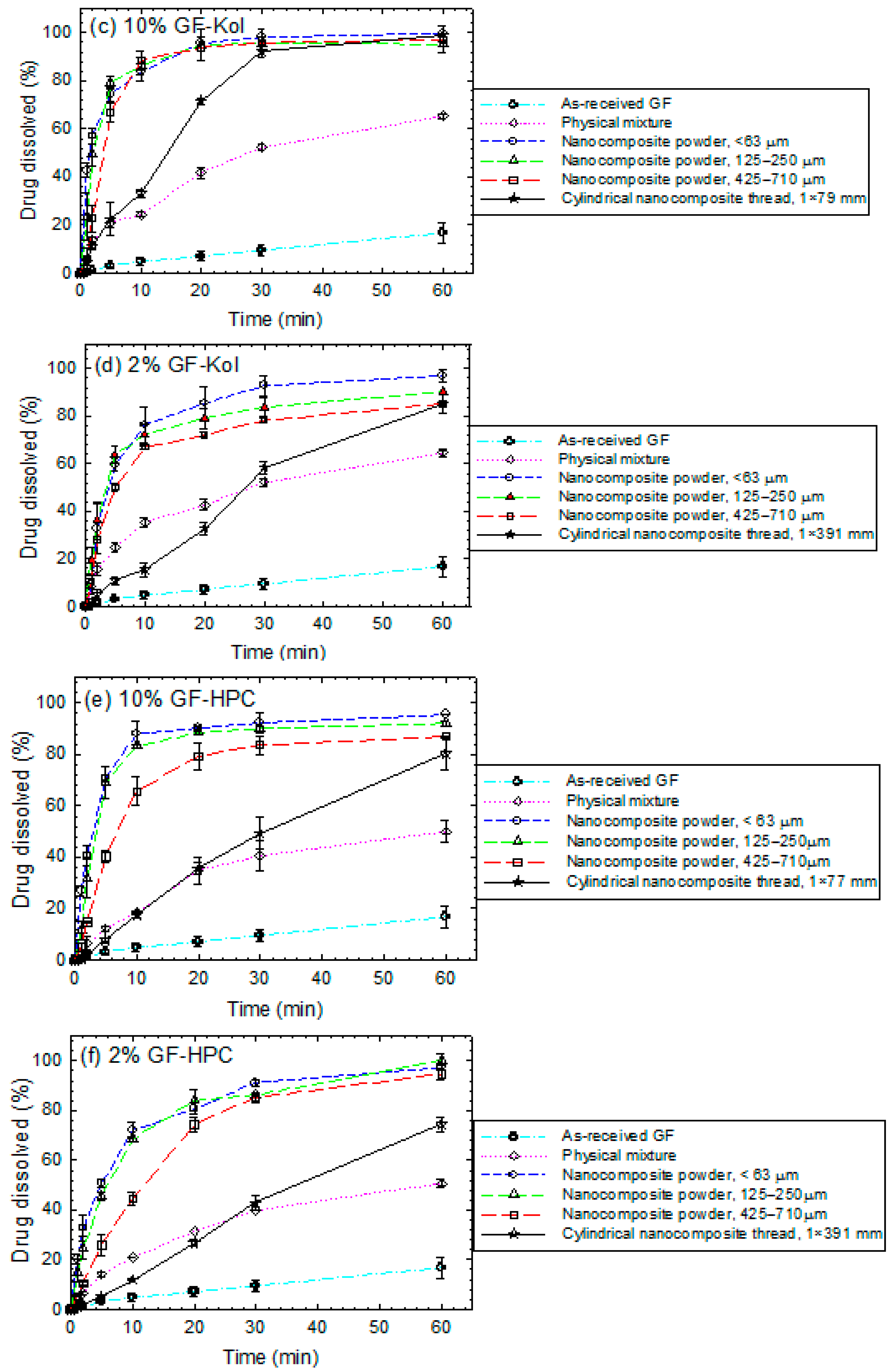
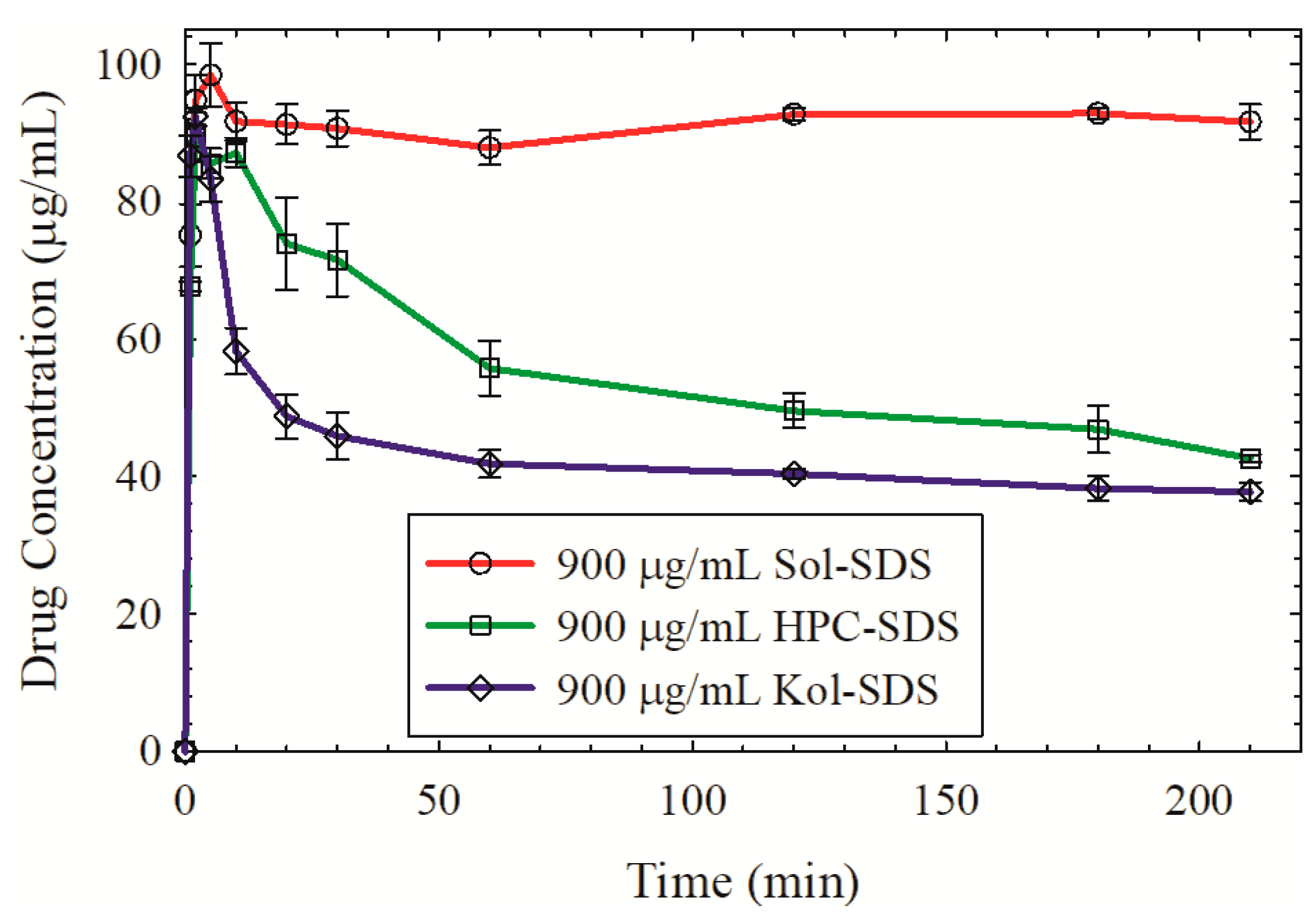
3.4. Impact of Matrix Surface Area at the Supersaturating Dissolution Condition
3.5. The Interplay between Matrix Size and Drug Loading at Supersaturating Dissolution Condition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical Cocrystals: New Solid Phase Modification Approaches for the Formulation of APIs. Pharmaceutics 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Jafari, M.; Padrela, L.; Walker, G.; Croker, D. Creating Cocrystals: A Review of Pharmaceutical Cocrystal Preparation Routes and Applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar] [CrossRef]
- Singh, A.; Mooter, G.V.D. Spray drying formulation of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef]
- Serajuddin, A.T. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and re-cent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, R.J.; Zia, H.; Sandhu, H.K.; Shah, N.H.; Malick, W.A. Improving the Dissolution Rate of Poorly Water Soluble Drug by Solid Dispersion and Solid Solution—Pros and Cons. Drug Deliv. 2007, 14, 33–45. [Google Scholar] [CrossRef]
- Delori, A.; Maclure, P.; Bhardwaj, R.M.; Johnston, A.; Florence, A.J.; Sutcliffe, O.B.; Oswald, I.D.H. Drug solid solutions—A method for tuning phase transformations. Cryst. Eng. Comm. 2014, 16, 5827–5831. [Google Scholar] [CrossRef]
- Gupta, D.; Bhatia, D.; Dave, V.; Sutariya, V.; Gupta, S.V. Salts of Therapeutic Agents: Chemical, Physicochemical, and Biological Considerations. Molecules 2018, 23, 1719. [Google Scholar] [CrossRef] [Green Version]
- Bastin, R.J.; Bowker, M.J.; Slater, B.J. Salt Selection and Optimisation Procedures for Pharmaceutical New Chemical Entities. Org. Process. Res. Dev. 2000, 4, 427–435. [Google Scholar] [CrossRef]
- McInnes, G.T.; Asbury, M.J.; Ramsay, L.E.; Shelton, J.R.; Harrison, I.R. Effect of micronization on the bioavailability and pharmacologic activity of spironolactone. J. Clin. Pharmacol. 1982, 22, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, J.C. Micronization: A method of improving the bioavailability of poorly soluble drugs. Methods Find. Exp. Clin. Pharmacol. 1998, 20, 211–216. [Google Scholar]
- Li, M.; Azad, M.; Davé, R.; Bilgili, E. Nanomilling of Drugs for Bioavailability Enhancement: A Holistic Formulation-Process Perspective. Pharmaceutics 2016, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Malamatari, M.; Taylor, K.M.; Malamataris, S.; Douroumis, D.; Kachrimanis, K. Pharmaceutical nanocrystals: Production by wet milling and applications. Drug Discov. Today 2018, 23, 534–547. [Google Scholar] [CrossRef]
- Brough, C.; Williams, R. Amorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug delivery. Int. J. Pharm. 2013, 453, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Panmai, S.; Wu, Y. Nanosizing—Oral formulation development and biopharmaceutical evaluation. Adv. Drug Deliv. Rev. 2007, 59, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Bhakay, A.; Rahman, M.; Dave, R.N.; Bilgili, E. Bioavailability Enhancement of Poorly Water-Soluble Drugs via Nanocomposites: Formulation–Processing Aspects and Challenges. Pharmaceutics 2018, 10, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, R.H.; Junghanns, J.-U.A.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar] [CrossRef] [Green Version]
- Müller, R.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy: Rationale for development and what we can expect for the future. Adv. Drug Deliv. Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- Forster, A.; Hempenstall, J.; Rades, T. Characterization of glass solutions of poorly water-soluble drugs produced by melt extrusion with hydrophilic amorphous polymers. J. Pharm. Pharmacol. 2010, 53, 303–315. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the True Solubility Advantage for Amorphous Pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Gao, Y.; Zhou, D.; Mo, H.; Zhang, G.G.; Taylor, L.S. Dissolution and Precipitation Behavior of Amorphous Solid Dispersions. J. Pharm. Sci. 2011, 100, 3316–3331. [Google Scholar] [CrossRef]
- Pui, Y.; Chen, Y.; Chen, H.; Wang, S.; Liu, C.; Tonnis, W.; Chen, L.; Serno, P.; Bracht, S.; Qian, F. Maintaining Supersaturation of Nimodipine by PVP with or without the Presence of Sodium Lauryl Sulfate and Sodium Taurocholate. Mol. Pharm. 2018, 15, 2754–2763. [Google Scholar] [CrossRef]
- Mersmann, A.; Heyer, C.; Eble, A. Crystallization Technology Handbook, 2nd ed.; Mersmann, A., Ed.; Marcel Dekker: New York, NY, USA, 2001; pp. 45–144. [Google Scholar]
- Prasad, D.; Chauhan, H.; Atef, E. Role of Molecular Interactions for Synergistic Precipitation Inhibition of Poorly Soluble Drug in Supersaturated Drug–Polymer–Polymer Ternary Solution. Mol. Pharm. 2016, 13, 756–765. [Google Scholar] [CrossRef]
- Rahman, M.; Coelho, A.; Tarabokija, J.; Ahmad, S.; Radgman, K.; Bilgili, E. Synergistic and antagonistic effects of various amphiphilic polymer combinations in enhancing griseofulvin release from ternary amorphous solid dispersions. Eur. J. Pharm. Sci. 2020, 150, 105354. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.L.-N.; Park, C.; Lee, B.-J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef]
- LeLeux, J.; Iii, R.W. Recent advancements in mechanical reduction methods: Particulate systems. Drug Dev. Ind. Pharm. 2013, 40, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals–Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef]
- Basa, S.; Muniyappan, T.; Karatgi, P.; Prabhu, R.; Pillai, R. Production and in vitro characterization of solid dos-age form incorporating drug nanoparticles. Drug Dev. Ind. Pharm. 2008, 34, 1209–1218. [Google Scholar]
- Bhakay, A.; Azad, M.; Bilgili, E.; Dave, R. Redispersible fast dissolving nanocomposite microparticles of poorly water-soluble drugs. Int. J. Pharm. 2014, 461, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Van Eerdenbrugh, B.; Froyen, L.; Van Humbeeck, J.; Martens, J.A.; Augustijns, P.; Mooter, G.V.D. Drying of crystalline drug nanosuspensions—The importance of surface hydrophobicity on dissolution behavior upon redispersion. Eur. J. Pharm. Sci. 2008, 35, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.; Arteaga, C.; Abdelmalek, B.; Dave, R.; Bilgili, E. Spray drying of drug-swellable dispersant suspensions for preparation of fast-dissolving, high drug-loaded, surfactant-free nanocomposites. Drug Dev. Ind. Pharm. 2014, 41, 1–15. [Google Scholar] [CrossRef]
- Lee, J. Drug Nano- and Microparticles Processed into Solid Dosage Forms: Physical Properties. J. Pharm. Sci. 2003, 92, 2057–2068. [Google Scholar] [CrossRef]
- Wang, Y.; Kho, K.; Cheow, W.S.; Hadinoto, K. A comparison between spray drying and spray freeze drying for dry powder inhaler formulation of drug-loaded lipid–polymer hybrid nanoparticles. Int. J. Pharm. 2012, 424, 98–106. [Google Scholar] [CrossRef]
- Cheow, W.S.; Ng, M.L.L.; Kho, K.; Hadinoto, K. Spray-freeze-drying production of thermally sensitive polymeric nanoparticle aggregates for inhaled drug delivery: Effect of freeze-drying adjuvants. Int. J. Pharm. 2011, 404, 289–300. [Google Scholar] [CrossRef]
- De Waard, H.; Hinrichs, W.; Frijlink, H.W. A novel bottom–up process to produce drug nanocrystals: Controlled crystallization during freeze-drying. J. Control. Release 2008, 128, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Layre, A.-M.; Couvreur, P.; Richard, J.; Requier, D.; Ghermani, N.E.; Gref, R. Freeze-Drying of Composite Core-Shell Nanoparticles. Drug Dev. Ind. Pharm. 2006, 32, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Park, C.H.; Lee, J. Effect of Polymer Molecular Weight on Nanocomminution of Poorly Soluble Drug. Drug Deliv. 2008, 15, 347–353. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J. Effective polymeric dispersants for vacuum, convection and freeze drying of drug nanosuspensions. Int. J. Pharm. 2010, 397, 218–224. [Google Scholar] [CrossRef]
- Li, M.; Lopez, N.; Bilgili, E. A study of the impact of polymer–surfactant in drug nanoparticle coated pharmatose composites on dissolution performance. Adv. Powder Technol. 2016, 27, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, R.; Eitzlmayr, A.; Matsko, N.; Tetyczka, C.; Khinast, J.; Roblegg, E. Nano-extrusion: A promising tool for continuous manufacturing of solid nano-formulations. Int. J. Pharm. 2014, 477, 1–11. [Google Scholar] [CrossRef]
- Li, M.; Ioannidis, N.; Gogos, C.; Bilgili, E. A comparative assessment of nanocomposites vs. amorphous solid dispersions prepared via nanoextrusion for drug dissolution enhancement. Eur. J. Pharm. Biopharm. 2017, 119, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Repka, M.A.; Bandari, S.; Kallakunta, V.R.; Vo, A.Q.; McFall, H.; Pimparade, M.B.; Bhagurkar, A.M. Melt extrusion with poorly soluble drugs—An integrated review. Int. J. Pharm. 2018, 535, 68–85. [Google Scholar] [CrossRef]
- Langham, Z.A.; Booth, J.; Hughes, L.P.; Reynolds, G.K.; Wren, S.A.C. Mechanistic Insights into the Dissolution of Spray-Dried Amorphous Solid Dispersions. J. Pharm. Sci. 2012, 101, 2798–2810. [Google Scholar] [CrossRef]
- Fule, R.; Paithankar, V.; Amin, P. Hot melt extrusion based solid solution approach: Exploring polymer comparison, physicochemical characterization and in-vivo evaluation. Int. J. Pharm. 2016, 499, 280–294. [Google Scholar] [CrossRef]
- Baumgartner, R.; Matić, J.; Schrank, S.; Laske, S.; Khinast, J.; Roblegg, E. NANEX: Process design and optimization. Int. J. Pharm. 2016, 506, 35–45. [Google Scholar] [CrossRef]
- Khinast, J.; Baumgartner, R.; Roblegg, E. Nano-extrusion: A One-Step Process for Manufacturing of Solid Nanoparticle Formulations Directly from the Liquid Phase. AAPS Pharm. Sci. Tech. 2013, 14, 601–604. [Google Scholar] [CrossRef]
- Ye, X.; Patil, H.; Feng, X.; Tiwari, R.V.; Lu, J.; Gryczke, A.; Kolter, K.; Langley, N.; Majumdar, S.; Neupane, D.; et al. Conjugation of Hot-Melt Extrusion with High-Pressure Homogenization: A Novel Method of Continuously Preparing Nanocrystal Solid Dispersions. AAPS Pharm. Sci. Tech. 2015, 17, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Feng, X.; Patil, H.; Tiwari, R.V.; Repka, M.A. Coupling 3D printing with hot-melt extrusion to produce controlled-release tablets. Int. J. Pharm. 2017, 519, 186–197. [Google Scholar] [CrossRef]
- Juluri, A.; Popescu, C.; Zhou, L.; Murthy, R.N.; Gowda, V.K.; Kumar, P.C.; Pimparade, M.B.; Repka, M.A.; Murthy, S.N. Taste Masking of Griseofulvin and Caffeine Anhydrous Using Kleptose Linecaps DE17 by Hot Melt Extrusion. AAPS Pharm. Sci. Tech. 2015, 17, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Pudlas, M.; Kyeremateng, S.O.; Williams, L.A.; Kimber, J.A.; van Lishaut, H.; Kazarian, S.G.; Woehrle, G.H. Analyzing the impact of different excipients on drug release behavior in hot-melt extrusion formulations using FTIR spectroscopic imaging. Eur. J. Pharm. Sci. 2015, 67, 21–31. [Google Scholar] [CrossRef]
- Javeer, S.; Amin, P. Solubility and dissolution enhancement of HPMC-based solid dispersions of carbamazepine by hot-melt extrusion technique. Asian J. Pharm. 2014, 8, 119. [Google Scholar] [CrossRef]
- Nagy, Z.K.; Balogh, A.; Vajna, B.; Farkas, A.; Patyi, G.; Kramarics, Á.; Marosi, G. Comparison of Electrospun and Extruded Soluplus®-Based Solid Dosage Forms of Improved Dissolution. J. Pharm. Sci. 2012, 101, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.; Andrews, G.; Jones, D.; Walker, G. Analysis of in vitro drug dissolution from PCL melt extrusion. Chem. Eng. J. 2010, 164, 359–370. [Google Scholar] [CrossRef]
- Ghebremeskel, A.N.; Vemavarapu, C.; Lodaya, M. Use of Surfactants as Plasticizers in Preparing Solid Dispersions of Poorly Soluble API: Stability Testing of Selected Solid Dispersions. Pharm. Res. 2006, 23, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Marcos, L.; Lamprou, D.A.; McBurney, R.T.; Halbert, G.W. A novel hot-melt extrusion formulation of albendazole for increasing dissolution properties. Int. J. Pharm. 2016, 499, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Lee, Y.-C.; Shabani, Z.; Lamm, C.F.; Zhu, W.; Li, Y.; Templeton, A. Processing Impact on Performance of Solid Dispersions. Pharmaceutics 2018, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, K.; Lin, Z.; Capece, M.; Kunnath, K.; Chen, L.; Davé, R.N. Effect of Particle Size and Polymer Loading on Dissolution Behavior of Amorphous Griseofulvin Powder. J. Pharm. Sci. 2019, 108, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Zhang, G.G.Z.; Law, D.; Grant, D.J.W.; Schmitt, E.A. Thermodynamics, Molecular Mobility and Crystallization Kinetics of Amorphous Griseofulvin. Mol. Pharm. 2008, 5, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Nisso. Hydroxypropyl Cellulose. Technical Data Sheet. 2011. Available online: https://pdf4pro.com/view/technical-data-sheet-nisso-hpc-3326f6.html (accessed on 5 July 2021).
- Altamimi, M.; Elzayat, E.M.; Qamar, W.; AlShehri, S.M.; Sherif, A.Y.; Haq, N.; Shakeel, F. Evaluation of the bioavailability of hydrocortisone when prepared as solid dispersion. Saudi Pharm. J. 2019, 27, 629–636. [Google Scholar] [CrossRef]
- Terife, G.; Wang, P.; Faridi, N.; Gogos, C.G. Hot melt mixing and foaming of soluplus® and indomethacin. Polym. Eng. Sci. 2012, 52, 1629–1639. [Google Scholar] [CrossRef]
- Hammouda, B. Temperature effect on the nanostructure of SDS micelles in water. J. Res. Natl. Inst. Stand. Technol. 2013, 118, 151. [Google Scholar] [CrossRef]
- Bilgili, E.; Afolabi, A. A combined microhydrodynamics–polymer adsorption analysis for elucidation of the roles of stabilizers in wet stirred media milling. Int. J. Pharm. 2012, 439, 193–206. [Google Scholar] [CrossRef]
- Bilgili, E.; Li, M.; Afolabi, A. Is the combination of cellulosic polymers and anionic surfactants a good strategy for ensuring physical stability of BCS Class II drug nanosuspensions? Pharm. Dev. Technol. 2015, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Justine, T.; Pieere, L.; Chole, V.; Martine, A.; Laureanne, N.; Eric, Z.; Philippe, H.; Krier, F.; Evrard, B. Continuous production of itraconazole-based solid dispersions by hot melt extrusion: Preformulation, optimization and design space determination. Int. J. Pharm. 2016, 151, 114–124. [Google Scholar] [CrossRef]
- Knoblauch, J.; Zimmermann, I. Thermochemical analysis of the dissolution process of Griseofulvin. Eur. J. Pharm. Biopharm. 2007, 67, 743–751. [Google Scholar] [CrossRef]
- Albadarin, A.; Potter, C.; Davis, M.T.; Iqbal, J.; Korde, S.; Pagire, S.; Paradkar, A.; Walker, G. Development of stability-enhanced ternary solid dispersions via combinations of HPMCP and Soluplus® processed by hot melt extrusion. Int. J. Pharm. 2017, 532, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Goyanes, A.; Allahham, N.; Trenfield, S.J.; Stoyanov, E.; Gaisford, S.; Basit, A.W. Direct powder extrusion 3D printing: Fabrication of drug products using a novel single-step process. Int. J. Pharm. 2019, 567, 118471. [Google Scholar] [CrossRef] [PubMed]
- Kolter, K.; Karl, M.; Gryczke, A.; Ludwigshafen am Rhein, B. Hot-melt Extrusion with BASF Pharma Polymers: Ex-trusion Compendium; BASF: Ludwigshafen, Germany, 2012. [Google Scholar]
- Hołownia, D.; Kwiatkowska, I.; Hupka, J. An investigation on wetting of porous materials. Physicochem. Probl. Miner. Process. 2008, 42, 251–262. [Google Scholar]
- Washburn, E.W. The Dynamics of Capillary Flow. Phys. Rev. 1921, 17, 273–283. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Peppas, N. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar]
- Li, M.; Suriel, I.; Vekaria, J.; Proske, J.; Orbe, P.; Armani, M.; Dave, R.; Bilgili, E. Impact of dispersants on dissolution of itraconazole from drug-loaded, surfactant-free, spray-dried nanocomposites. Powder Technol. 2018, 339, 281–295. [Google Scholar] [CrossRef]
- Rahman, M.; Arevalo, F.; Coelho, A.; Bilgili, E. Hybrid nanocrystal–amorphous solid dispersions (HyNASDs) as alternative to ASDs for enhanced release of BCS Class II drugs. Eur. J. Pharm. Biopharm. 2019, 145, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Keck, C.M.; Müller, R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur. J. Pharm. Biopharm. 2006, 62, 3–16. [Google Scholar] [CrossRef]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef]
- Onoue, S.; Takahashi, H.; Kawabata, Y.; Seto, Y.; Hatanaka, J.; Timmermann, B.; Yamada, S. Formulation Design and Photochemical Studies on Nanocrystal Solid Dispersion of Curcumin with Improved Oral Bioavailability. J. Pharm. Sci. 2010, 99, 1871–1881. [Google Scholar] [CrossRef]
- Cerdeira, A.M.; Mazzotti, M.; Gander, B. Miconazole nanosuspensions: Influence of formulation variables on particle size reduction and physical stability. Int. J. Pharm. 2010, 396, 210–218. [Google Scholar] [CrossRef]
- Cerdeira, A.M.; Mazzotti, M.; Gander, B. Formulation and drying of miconazole and itraconazole nanosuspensions. Int. J. Pharm. 2013, 443, 209–220. [Google Scholar] [CrossRef]
- Rahman, M.; Ahmad, S.; Tarabokija, J.; Bilgili, E. Roles of surfactant and polymer in drug release from spray-dried hybrid nanocrystal-amorphous solid dispersions (HyNASDs). Powder Technol. 2020, 361, 663–678. [Google Scholar] [CrossRef]
- Knieke, C.; Azad, M.; Davé, R.; Bilgili, E. A study of the physical stability of wet media-milled fenofibrate suspensions using dynamic equilibrium curves. Chem. Eng. Res. Des. 2013, 91, 1245–1258. [Google Scholar] [CrossRef]
- Li, M.; Alvarez, P.; Orbe, P.; Bilgili, E. Multi-faceted Characterization of Wet-milled Griseofulvin Nanosuspensions for Elucidation of Aggregation State and Stabilization Mechanisms. AAPS Pharm. Sci. Tech. 2018, 19, 1789–1801. [Google Scholar] [CrossRef]
- Winnik, F.M.; Winnik, M.A. The Interaction of Sodium Dodecylsulfate with (Hydroxypropyl)Cellulose. Polym. J. 1990, 22, 482–488. [Google Scholar] [CrossRef] [Green Version]
- França, M.T.; Pereira, R.N.; Riekes, M.K.; Pinto, J.M.O.; Stulzer, H.K. Investigation of novel supersaturating drug delivery systems of chlorthalidone: The use of polymer-surfactant complex as an effective carrier in solid dispersions. Eur. J. Pharm. Sci. 2018, 111, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Yu, H.; Tao, J.; Zeng, J.; Zhu, Q.; Zhu, C.; Gan, Y. Supersaturated polymeric micelles for oral cyclosporine A delivery: The role of Soluplus–sodium dodecyl sulfate complex. Colloids Surf. B Biointerfaces 2016, 141, 301–310. [Google Scholar] [CrossRef]
- Berglund, K.D.; Przybycien, T.M.; Tilton, R.D. Coadsorption of Sodium Dodecyl Sulfate with Hydrophobically Modified Nonionic Cellulose Polymers. 1. Role of Polymer Hydrophobic Modification. Langmuir 2003, 19, 2705–2713. [Google Scholar] [CrossRef]
- Berglund, K.D.; Przybycien, T.M.; Tilton, R.D. Coadsorption of Sodium Dodecyl Sulfate with Hydrophobically Modified Nonionic Cellulose Polymers. 2. Role of Surface Selectivity in Adsorption Hysteresis. Langmuir 2003, 19, 2714–2721. [Google Scholar] [CrossRef]
- Kissa, E. Dispersions: Characterization, Testing, and Measurement; Routledge: England, UK, 1999. [Google Scholar]
- Deng, Z.; Xu, S.; Li, S. Understanding a relaxation behavior in a nanoparticle suspension for drug delivery applications. Int. J. Pharm. 2008, 351, 236–243. [Google Scholar] [CrossRef]
- Leuner, C. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Skorda, D.; Kontoyannis, C.G. Identification and quantitative determination of atorvastatin calcium polymorph in tablets using FT-Raman spectroscopy. Talanta 2008, 74, 1066–1070. [Google Scholar] [CrossRef]
- Craig, D.Q. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [CrossRef]
- Marsac, P.J.; Li, T.; Taylor, L.S. Estimation of Drug–Polymer Miscibility and Solubility in Amorphous Solid Dispersions Using Experimentally Determined Interaction Parameters. Pharm. Res. 2009, 26, 139–151. [Google Scholar] [CrossRef]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef]
- Thakral, S.; Thakral, N.K. Prediction of Drug–Polymer Miscibility through the use of Solubility Parameter based Flory–Huggins Interaction Parameter and the Experimental Validation: PEG as Model Polymer. J. Pharm. Sci. 2013, 102, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.; Kavassalis, T.A.; Rudin, A. Estimation of Hansen Solubility Parameters for (Hydroxyethyl) and (Hydroxypropyl) cellulose through Molecular Simulation. Ind. Eng. Chem. Res. 1994, 33, 3154–3159. [Google Scholar] [CrossRef]
- Hülsmann, S.; Backensfeld, T.; Keitel, S.; Bodmeier, R. Melt extrusion—An alternative method for enhancing the dissolution rate of 17β-estradiol hemihydrate. Eur. J. Pharm. Biopharm. 2000, 49, 237–242. [Google Scholar] [CrossRef]
- Xie, T.; Taylor, L.S. Improved Release of Celecoxib from High Drug Loading Amorphous Solid Dispersions Formulated with Polyacrylic Acid and Cellulose Derivatives. Mol. Pharm. 2016, 13, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, Y.; Zhang, L.; Wang, Q.; Zhang, D. Stability of nanosuspensions in drug delivery. J. Control. Release 2013, 172, 1126–1141. [Google Scholar] [CrossRef]
- Zecevic, D.E.; Meier, R.; Daniels, R.; Wagner, K.-G. Site specific solubility improvement using solid dispersions of HPMC-AS/HPC SSL–Mixtures. Eur. J. Pharm. Biopharm. 2014, 87, 264–270. [Google Scholar] [CrossRef]
- Zhang, K.; Yu, H.; Luo, Q.; Yang, S.; Lin, X.; Zhang, Y.; Tian, B.; Tang, X. Increased dissolution and oral absorption of itraconazole/Soluplus extrudate compared with itraconazole nanosuspension. Eur. J. Pharm. Biopharm. 2013, 85, 1285–1292. [Google Scholar] [CrossRef]
- Reintjes, T. Solubility Enhancement with BASF Pharma Polymers. Solubilizer Compendium. 2011. Available online: https://pharmaceutical.basf.com/global/images/b_03_110921e_solubility_enhance_compendium.pdf (accessed on 5 July 2021).
- Alopaeus, J.F.; Hagesæther, E.; Tho, I. Micellisation mechanism and behaviour of Soluplus®–furosemide mi-celles: Preformulation studies of an oral nanocarrier-based system. Pharmaceuticals 2019, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Maintaining Supersaturation in Aqueous Drug Solutions: Impact of Different Polymers on Induction Times. Cryst. Growth Des. 2012, 13, 740–751. [Google Scholar] [CrossRef]
- Warren, D.B.; Benameur, H.; Porter, C.; Pouton, C. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: A mechanistic basis for utility. J. Drug Target. 2010, 18, 704–731. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Rades, T.; Boyd, B. The Precipitation Behavior of Poorly Water-Soluble Drugs with an Emphasis on the Digestion of Lipid Based Formulations. Pharm. Res. 2016, 33, 548–562. [Google Scholar] [CrossRef]
- BASF. Soluplus®-BASF Technical Information Document. Available online: https://pharma.basf.com/technicalinformation/30446233/soluplus (accessed on 23 June 2021).
- BASF. Kolliphor® P 407-BASF Technical Information Document. Available online: https://documents.basf.com/4c09c1d5b700752ef8dc94d24eaffc2322dc9107?response-content-disposition=in-line (accessed on 23 June 2021).
- Fedors, R.F. A method for estimating both the solubility parameters and molar volumes of liquids. Polym. Eng. Sci. 1974, 14, 147–154. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Understanding Polymer Properties Important for Crystal Growth Inhibition—Impact of Chemically Diverse Polymers on Solution Crystal Growth of Ritonavir. Cryst. Growth Des. 2012, 12, 3133–3143. [Google Scholar] [CrossRef]
- Guan, J.; Huan, X.; Liu, Q.; Jin, L.; Wu, H.; Zhang, X.; Mao, S. Synergetic effect of nucleation and crystal growth inhibitor on in vitro-in vivo performance of supersaturable lacidipine solid dispersion. Int. J. Pharm. 2019, 566, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Taylor, L.S. Tailoring supersaturation from amorphous solid dispersions. J. Control. Release 2018, 279, 114–125. [Google Scholar] [CrossRef]
- Xie, T.; Taylor, L.S. Dissolution Performance of High Drug Loading Celecoxib Amorphous Solid Dispersions Formulated with Polymer Combinations. Pharm. Res. 2016, 33, 739–750. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation ID | Polymer a | Drug Particle Size after Milling (nm) | ||
|---|---|---|---|---|
| D10 ± SD | D50 ± SD | D90 ± SD | ||
| 10% GF-Sol | Soluplus® | 113 ± 0 | 156 ± 1 | 228 ± 1 |
| 2% GF-Sol | Soluplus® | 113 ± 0 | 156 ± 1 | 228 ± 1 |
| 10% GF-Kol | Kolliphor® P407 | 100 ± 8 | 185 ± 4 | 358 ± 49 |
| 2% GF-Kol | Kolliphor® P407 | 100 ± 8 | 185 ± 4 | 358 ± 49 |
| 10% GF-HPC | HPC | 121 ± 1 | 159 ± 0 | 215 ± 1 |
| 2% GF-HPC | HPC | 121 ± 1 | 159 ± 0 | 215 ± 1 |
| Formulation ID | Polymer | Feeding Rate (g/min) | Processing Temperature (°C) | Residence Time (s) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Polymer a | Suspension b | Zone 1 | Zone 2 | Zone 3 | Zone 4 | Zone 5 | Zone 6 | Die | |||
| 10% GF-Sol | Soluplus® | 2.9 | 1.4 | 70 | 70 | 100 | 100 | 165 | 165 | 165 | 120 |
| 2% GF-Sol | Soluplus® | 2.7 | 0.25 | 70 | 70 | 90 | 100 | 165 | 165 | 165 | 115 |
| 10% GF-Kol | Kolliphor® P407 | 2.9 | 1.4 | 10 | 25 | 100 | 100 | 110 | 110 | 90 | 120 |
| 2% GF-Kol | Kolliphor® P407 | 2.8 | 0.28 | 10 | 25 | 95 | 100 | 110 | 110 | 85 | 110 |
| 10% GF-HPC | HPC | 2.8 | 1.4 | 70 | 120 | 120 | 140 | 140 | 140 | 140 | 116 |
| 2% GF-HPC | HPC | 2.8 | 0.27 | 70 | 120 | 120 | 140 | 140 | 140 | 140 | 120 |
| Formulation ID | Drug Content (RSD) (% w/w) a | Moisture Content ± SD (% w/w) a,b | <63 µm | 125–250 µm | 425–710 µm | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D10 (µm), SD (µm) | D50 (µm), SD (µm) | D90 (µm), SD (µm) | D10 (µm), SD (µm) | D50 (µm), SD (µm) | D90 (µm), SD (µm) | D10 (µm), SD (µm) | D50 (µm), SD (µm) | D90 (µm), SD (µm) | |||
| 10% GF-Sol | 9.8 (1.5) | 3.6 ± 0.0 | 12.9, 0.1 | 51.7, 0.1 | 104, 0.2 | 104, 1.6 | 146, 0.8 | 195, 3.7 | 469, 17 | 716, 20 | 845, 3.5 |
| 2% GF-Sol | 1.9 (1.9) | 3.2 ± 0.5 | 13.7, 0.3 | 42.8, 0.6 | 90.7, 0.5 | 104, 1.1 | 158, 2.5 | 230, 8.2 | 480, 20 | 726, 32 | 847, 4.3 |
| 10% GF-Kol | 9.7 (2.2) | 0.8 ± 0.2 c | 12.7, 3.4 | 40.0, 2.3 | 81.0, 9.9 | 123, 17 | 195, 9.9 | 278, 6.8 | 371, 7.7 | 497, 29 | 581, 14 |
| 2% GF-Kol | 2.1 (1.9) | 0.7 ± 0.1 c | 17.1, 0.9 | 39.6, 1.2 | 67.5, 6.7 | 138, 2.3 | 210, 8.7 | 280, 3.4 | 380, 11 | 492, 24 | 604, 37 |
| 10% GF-HPC | 10.3 (1.0) | 2.9 ± 0.2 | 16.9, 1.7 | 43.9, 9.1 | 115, 7.6 | 126, 3.3 | 195, 5.1 | 294, 16 | 464, 21 | 634, 17 | 812, 8.1 |
| 2% GF-HPC | 2.1 (1.4) | 2.8 ± 0.2 | 21.3, 1.4 | 57.1, 0.6 | 123, 19 | 127, 2.4 | 185, 8.8 | 266, 16 | 446, 36 | 616, 33 | 799, 21 |
| Formulation ID | Specification of Size (µm) a | Korsmeyer-Peppas Model a | kn (%min−n) | ||
|---|---|---|---|---|---|
| n (-) | k (%min−n) | R2 (-) | |||
| As-received GF | – | 0.746 | 0.785 | 0.992 | 0.58 |
| 10% GF-Sol | PM | 0.530 | 4.72 | 0.965 | 2.50 |
| <63 | 1.30 | 10.1 | 0.991 | 13.2 | |
| 125–250 | 1.10 | 6.19 | 0.953 | 6.81 | |
| 425–710 | 0.932 | 1.46 | 0.963 | 1.36 | |
| Thread | 0.697 | 0.40 | 0.988 | 0.28 | |
| 2% GF-Sol | PM | 0.737 | 3.69 | 0.999 | 2.72 |
| <63 | 0.568 | 13.2 | 0.950 | 7.86 | |
| 125–250 | 0.745 | 6.04 | 0.979 | 4.50 | |
| 425–710 | 1.43 | 0.06 | 0.989 | 0.09 | |
| Thread | 0.628 | 0.11 | 0.948 | 0.07 | |
| 10% GF-Kol | PM | 0.492 | 9.06 | 0.976 | 4.46 |
| <63 | 0.389 | 43.1 | 0.981 | 16.8 | |
| 125–250 | 0.527 | 31.0 | 0.939 | 16.3 | |
| 425–710 | 1.27 | 8.66 | 0.990 | 11.0 | |
| Thread | 0.903 | 4.70 | 0.984 | 4.24 | |
| 2% GF-Kol | PM | 0.406 | 12.7 | 0.981 | 5.14 |
| <63 | 0.616 | 19.6 | 0.900 | 12.1 | |
| 125–250 | 0.355 | 29.6 | 0.939 | 10.5 | |
| 425–710 | 0.376 | 23.5 | 0.934 | 8.84 | |
| Thread | 0.824 | 3.01 | 0.981 | 2.48 | |
| 10% GF-HPC | PM | 0.546 | 5.76 | 0.953 | 3.14 |
| <63 | 0.603 | 26.8 | 1.000 | 16.2 | |
| 125–250 | 0.963 | 14.8 | 0.978 | 14.2 | |
| 425–710 | 0.894 | 8.59 | 0.977 | 7.67 | |
| Thread | 0.826 | 2.80 | 0.991 | 2.31 | |
| 2% GF-HPC | PM | 0.528 | 6.11 | 0.981 | 3.23 |
| <63 | 0.521 | 21.8 | 0.995 | 11.3 | |
| 125–250 | 0.641 | 15.8 | 0.997 | 10.1 | |
| 425–710 | 0.795 | 6.95 | 0.997 | 5.52 | |
| Thread | 0.945 | 1.58 | 0.994 | 1.49 | |
| Liquid a | Slope, (g2/s) | R2 (-) | η (cP) | ρ (g/mL) | γ (mN/m) | cosθss/cosθw (-) |
|---|---|---|---|---|---|---|
| Water | 7.00 × 10−3 | 0.990 | 0.890 | 1.00 | 66.5 | 1 |
| Aq. Soluplus® | 6.00 × 10−4 | 0.993 | 12.0 | 1.01 | 40.6 | 1.86 |
| Aq. Kolliphor | 1.70 × 10−3 | 0.999 | 29.5 | 1.01 | 35.9 | 14.6 |
| Aq. HPC | 2.00 × 10−4 | 0.999 | 403 | 1.02 | 37.9 | 22.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Furey, C.; Skros, J.; Xu, O.; Rahman, M.; Azad, M.; Dave, R.; Bilgili, E. Impact of Matrix Surface Area on Griseofulvin Release from Extrudates Prepared via Nanoextrusion. Pharmaceutics 2021, 13, 1036. https://doi.org/10.3390/pharmaceutics13071036
Li M, Furey C, Skros J, Xu O, Rahman M, Azad M, Dave R, Bilgili E. Impact of Matrix Surface Area on Griseofulvin Release from Extrudates Prepared via Nanoextrusion. Pharmaceutics. 2021; 13(7):1036. https://doi.org/10.3390/pharmaceutics13071036
Chicago/Turabian StyleLi, Meng, Casey Furey, Jeffrey Skros, Olivia Xu, Mahbubur Rahman, Mohammad Azad, Rajesh Dave, and Ecevit Bilgili. 2021. "Impact of Matrix Surface Area on Griseofulvin Release from Extrudates Prepared via Nanoextrusion" Pharmaceutics 13, no. 7: 1036. https://doi.org/10.3390/pharmaceutics13071036
APA StyleLi, M., Furey, C., Skros, J., Xu, O., Rahman, M., Azad, M., Dave, R., & Bilgili, E. (2021). Impact of Matrix Surface Area on Griseofulvin Release from Extrudates Prepared via Nanoextrusion. Pharmaceutics, 13(7), 1036. https://doi.org/10.3390/pharmaceutics13071036