
Insight into the Web of Stress Responses Triggered at Gene Expression Level by Porphyrin-PDT in HT29 Human Colon Carcinoma Cells

 , and
, and 
Abstract
:1. Introduction
2. Results
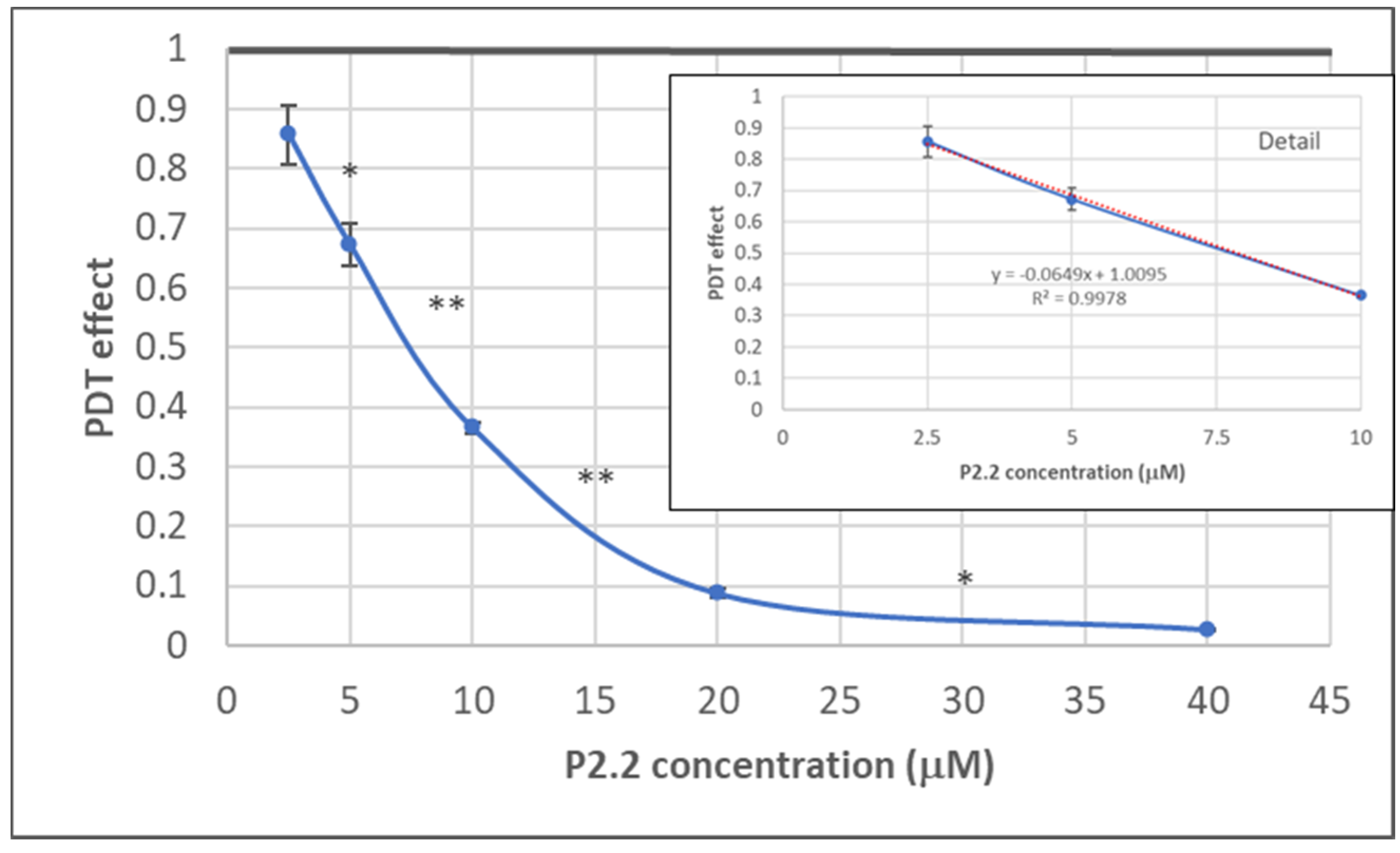
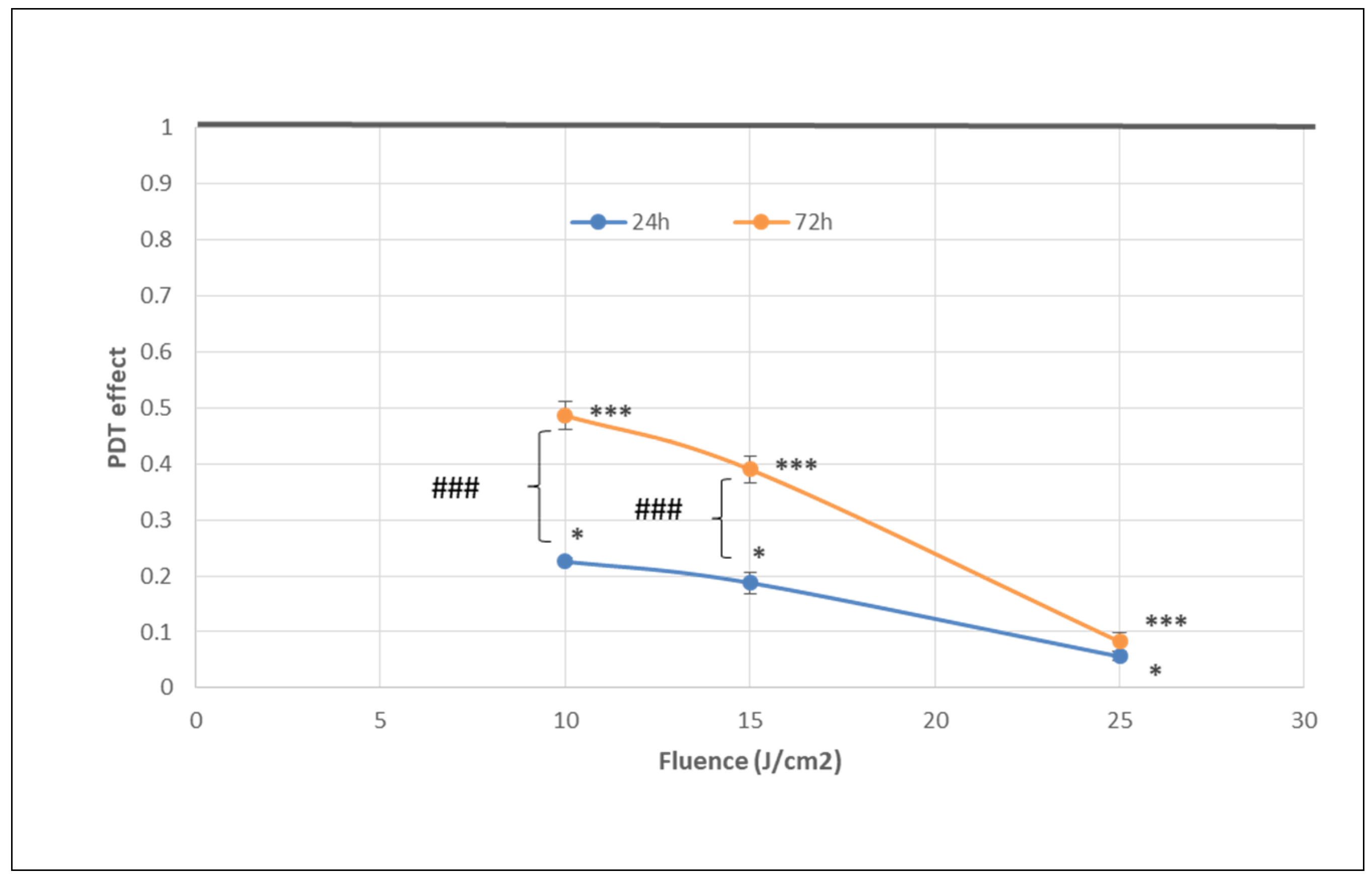
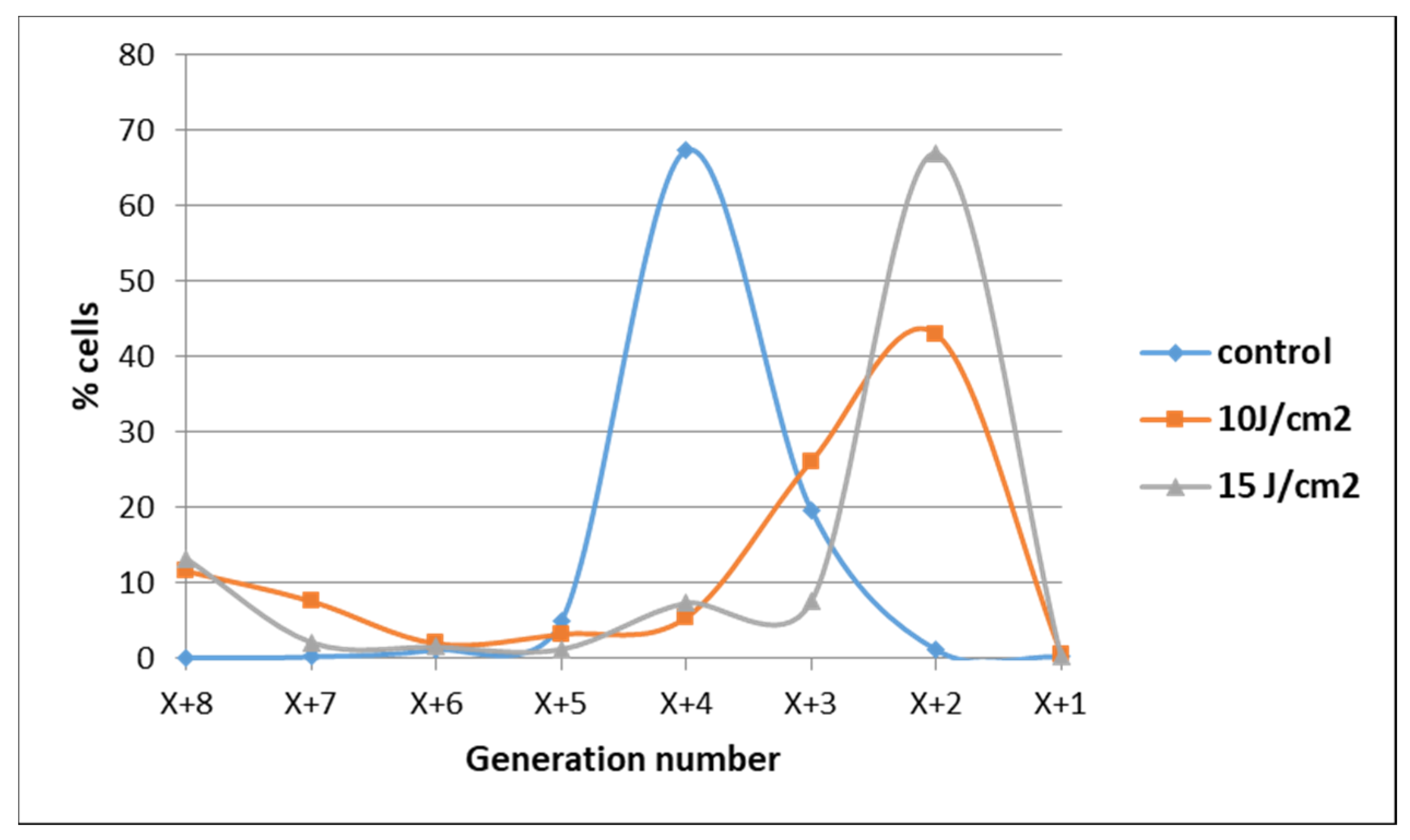
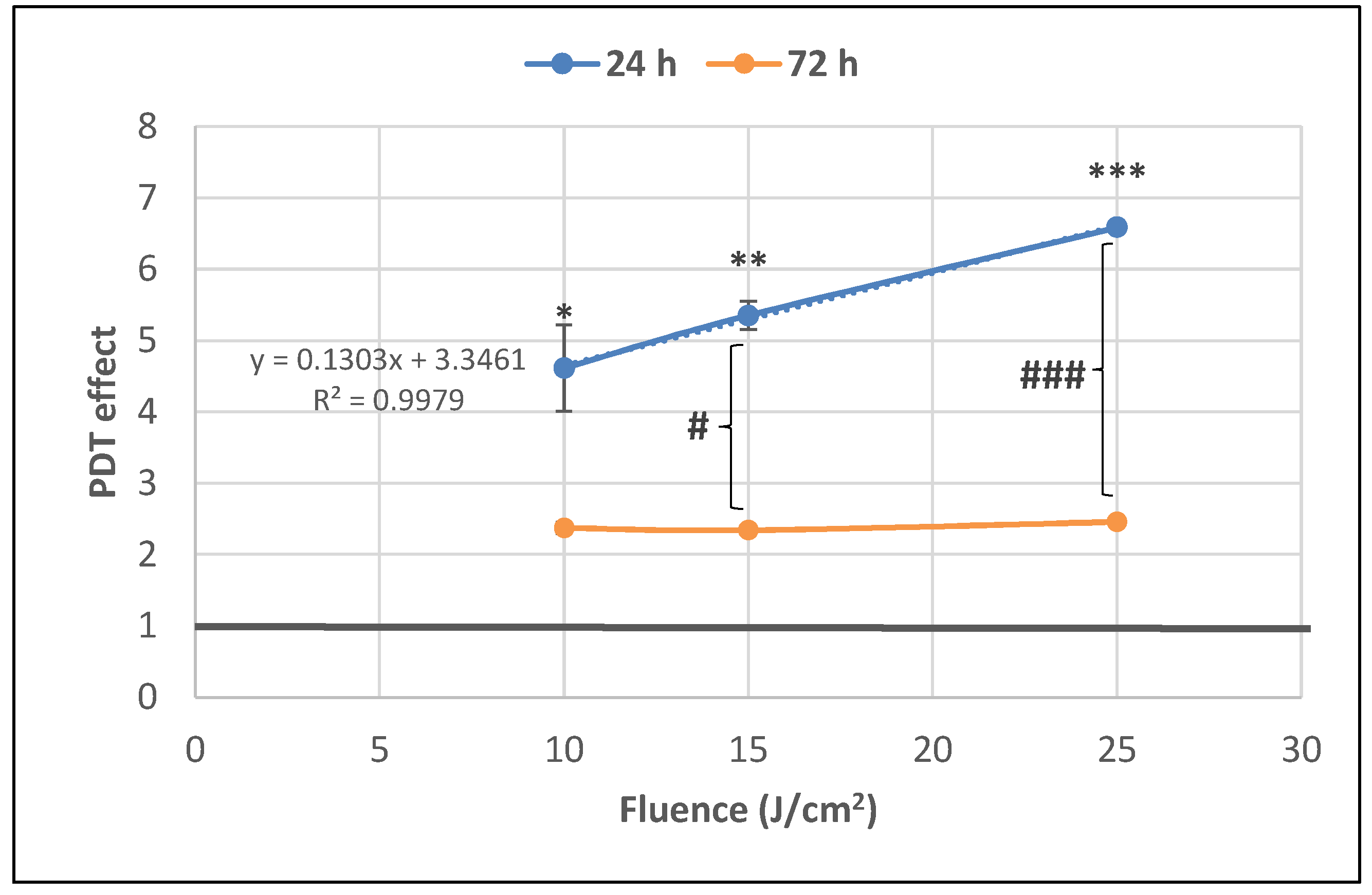
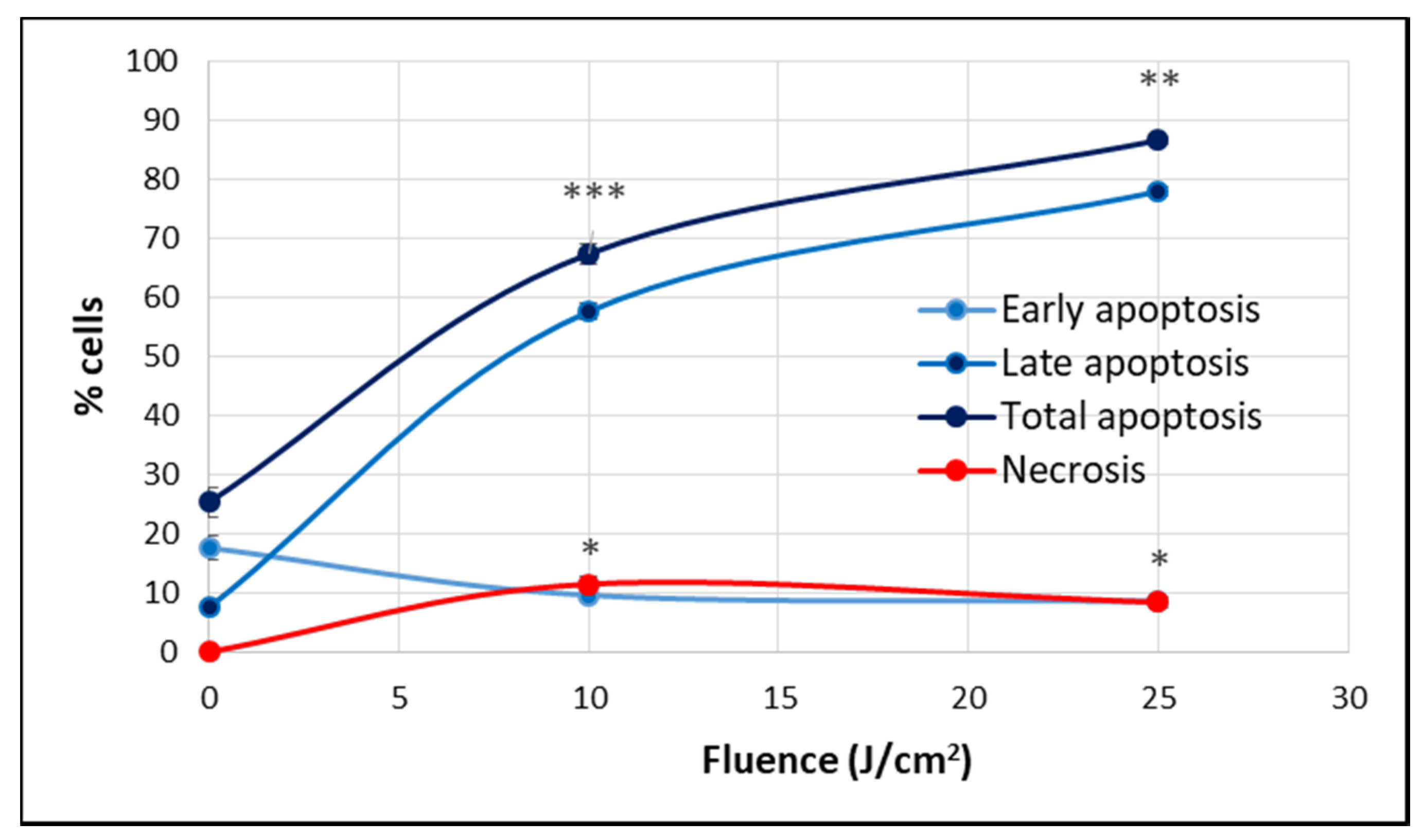
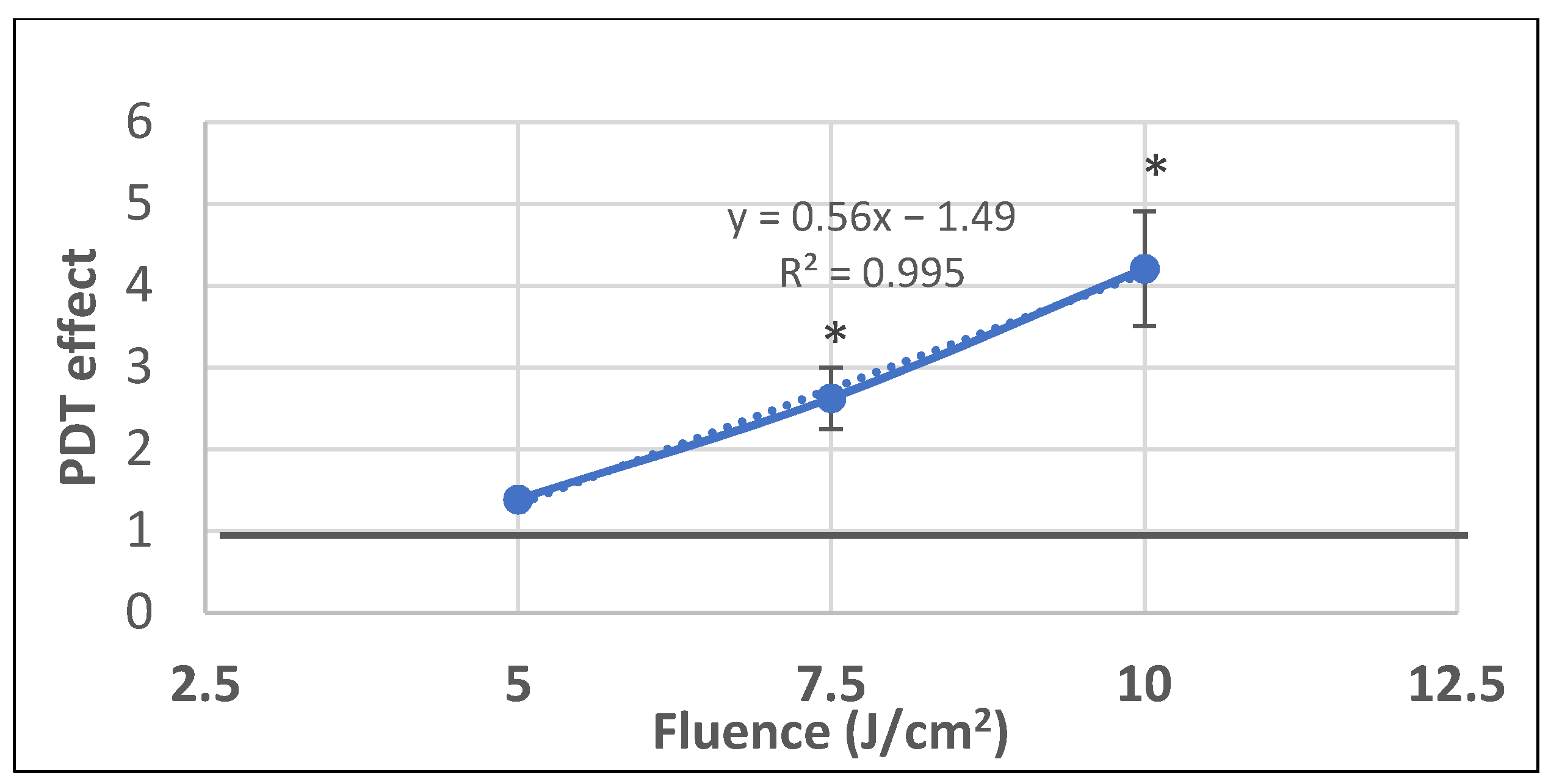
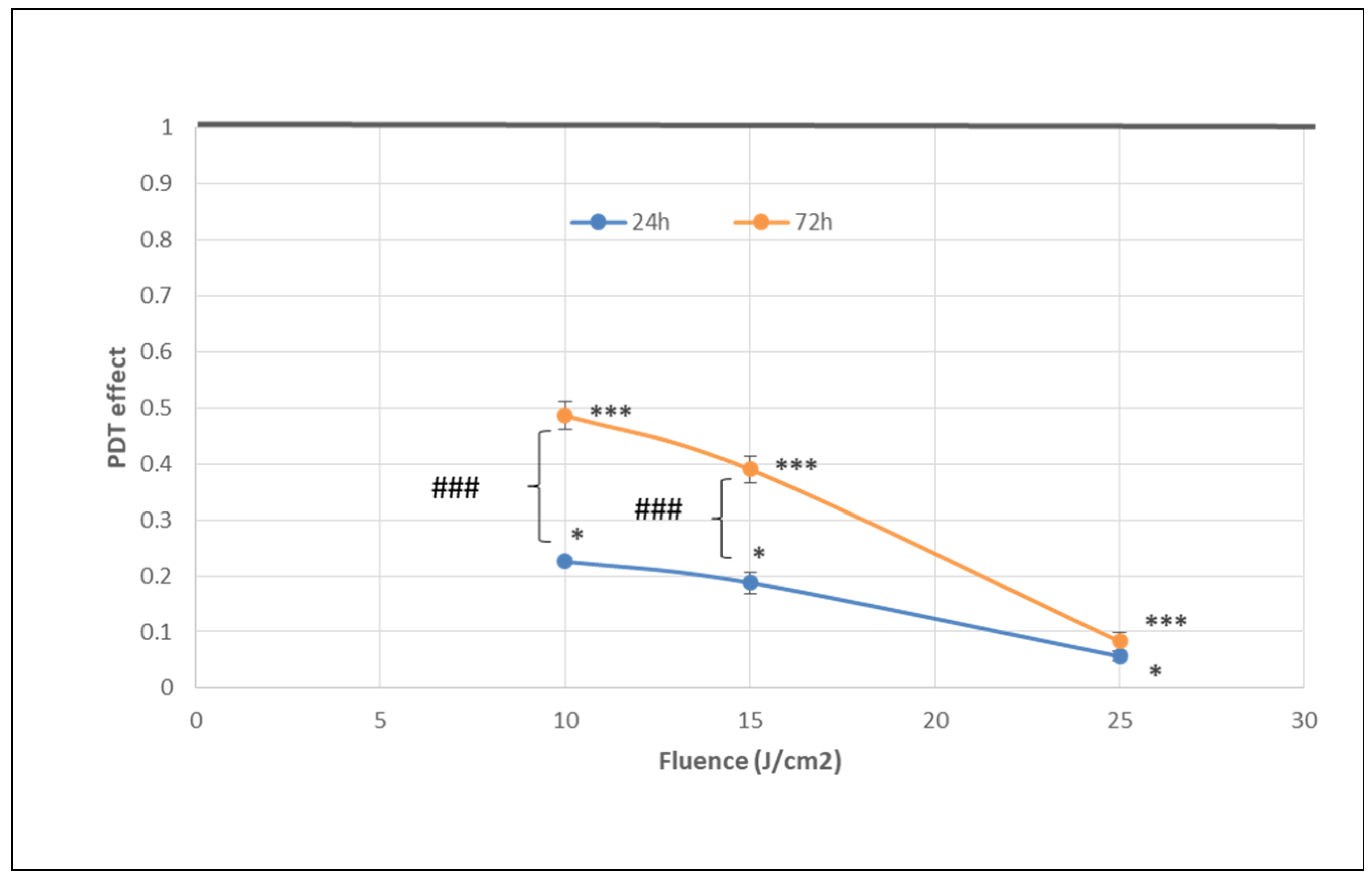
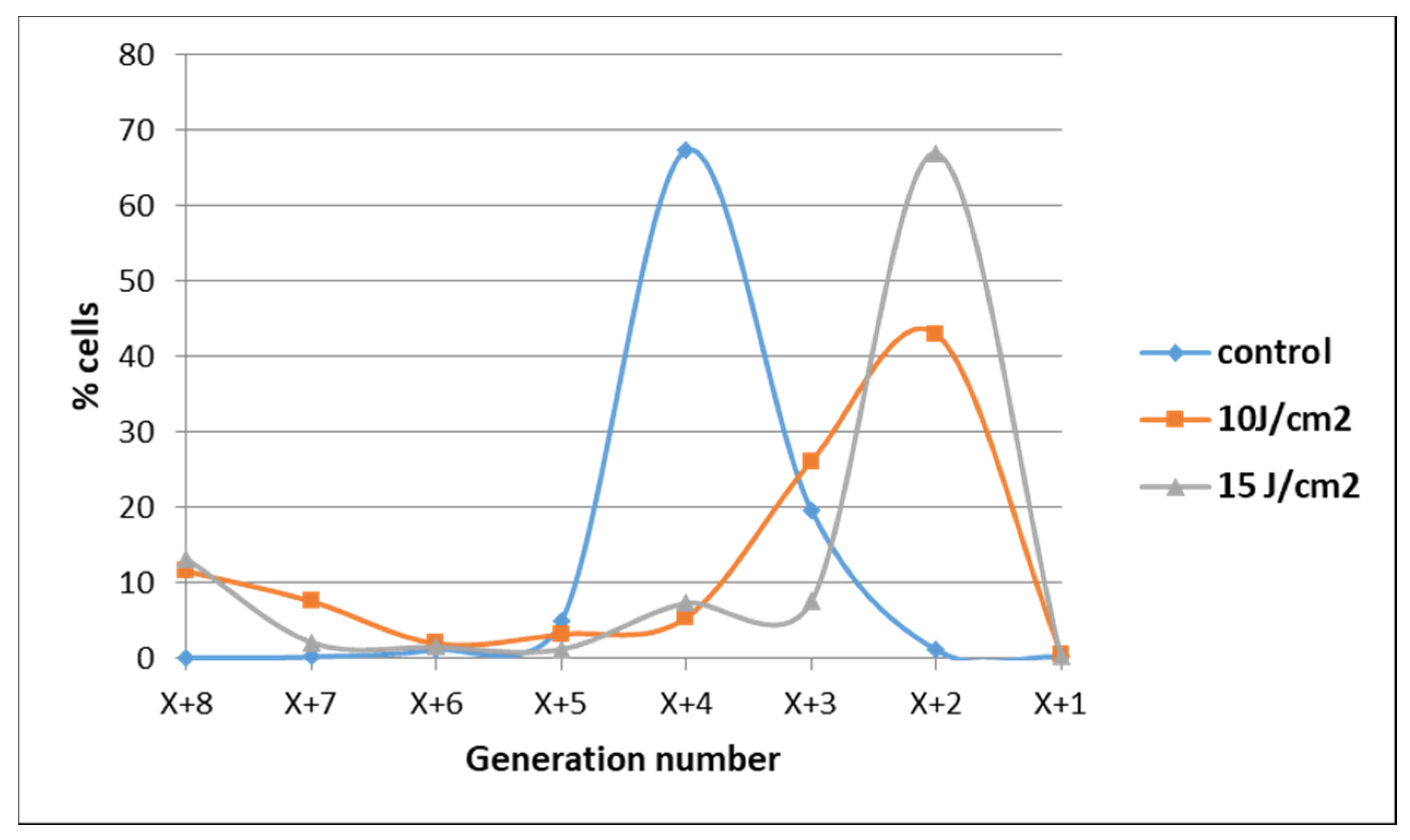
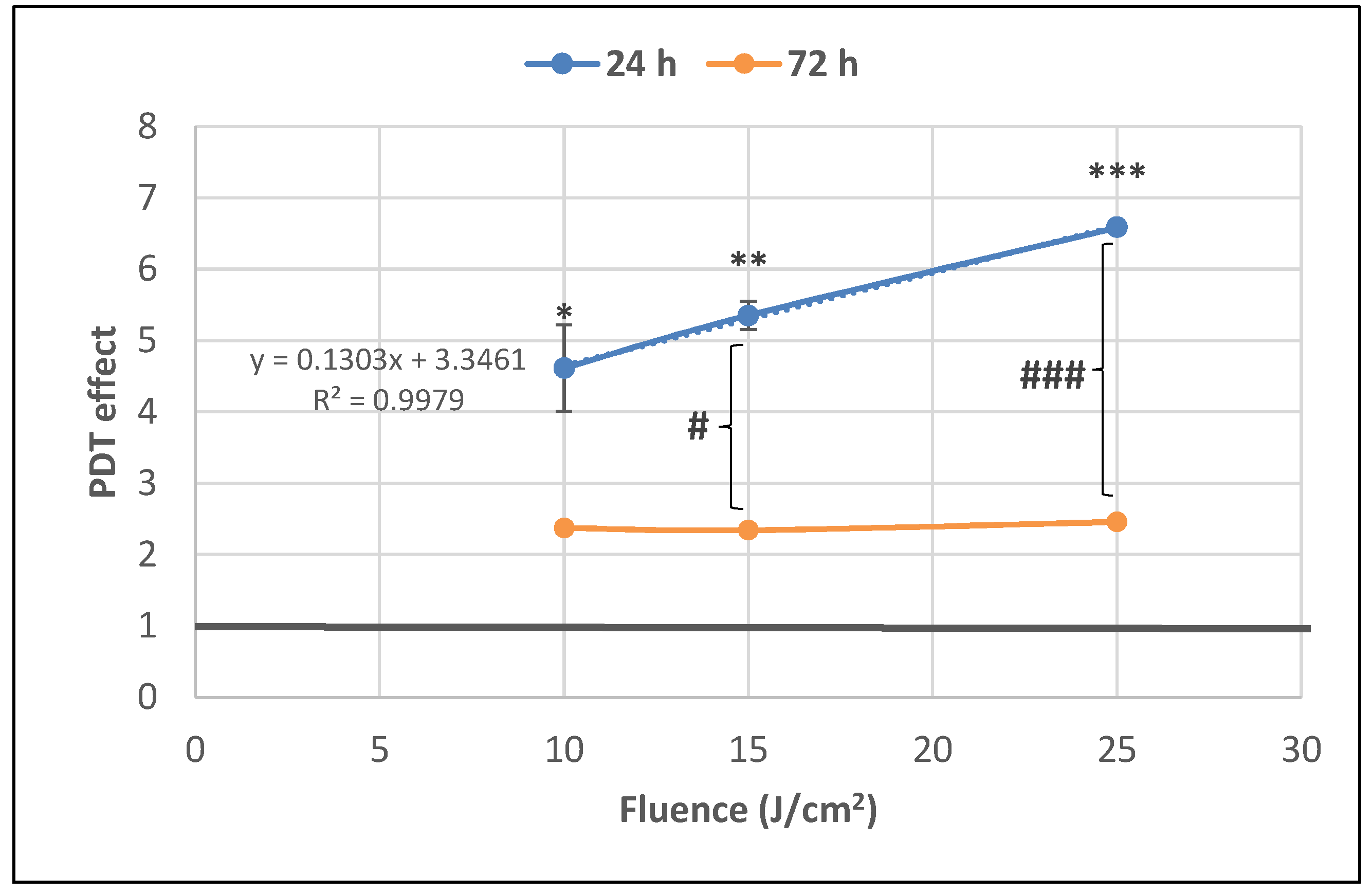
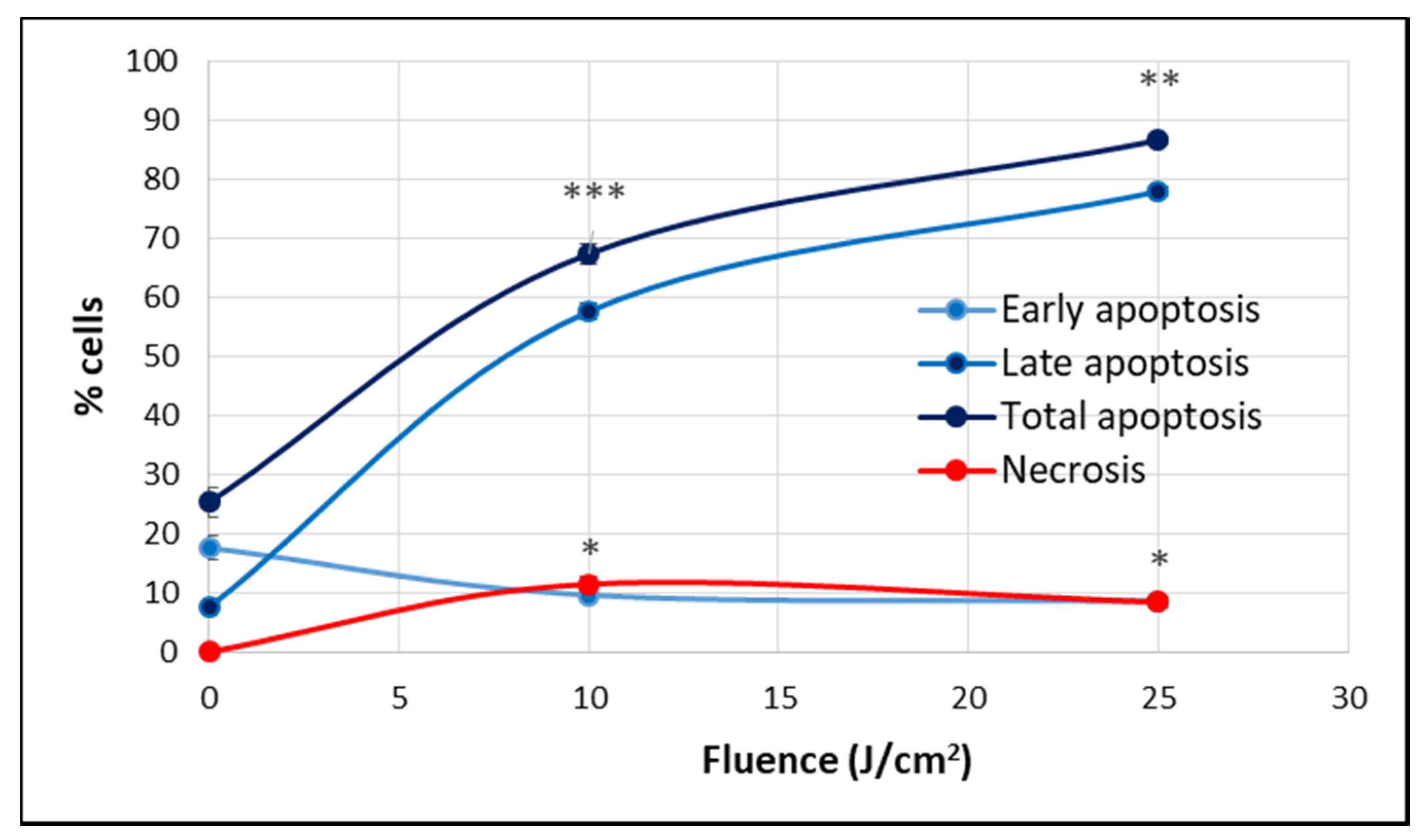
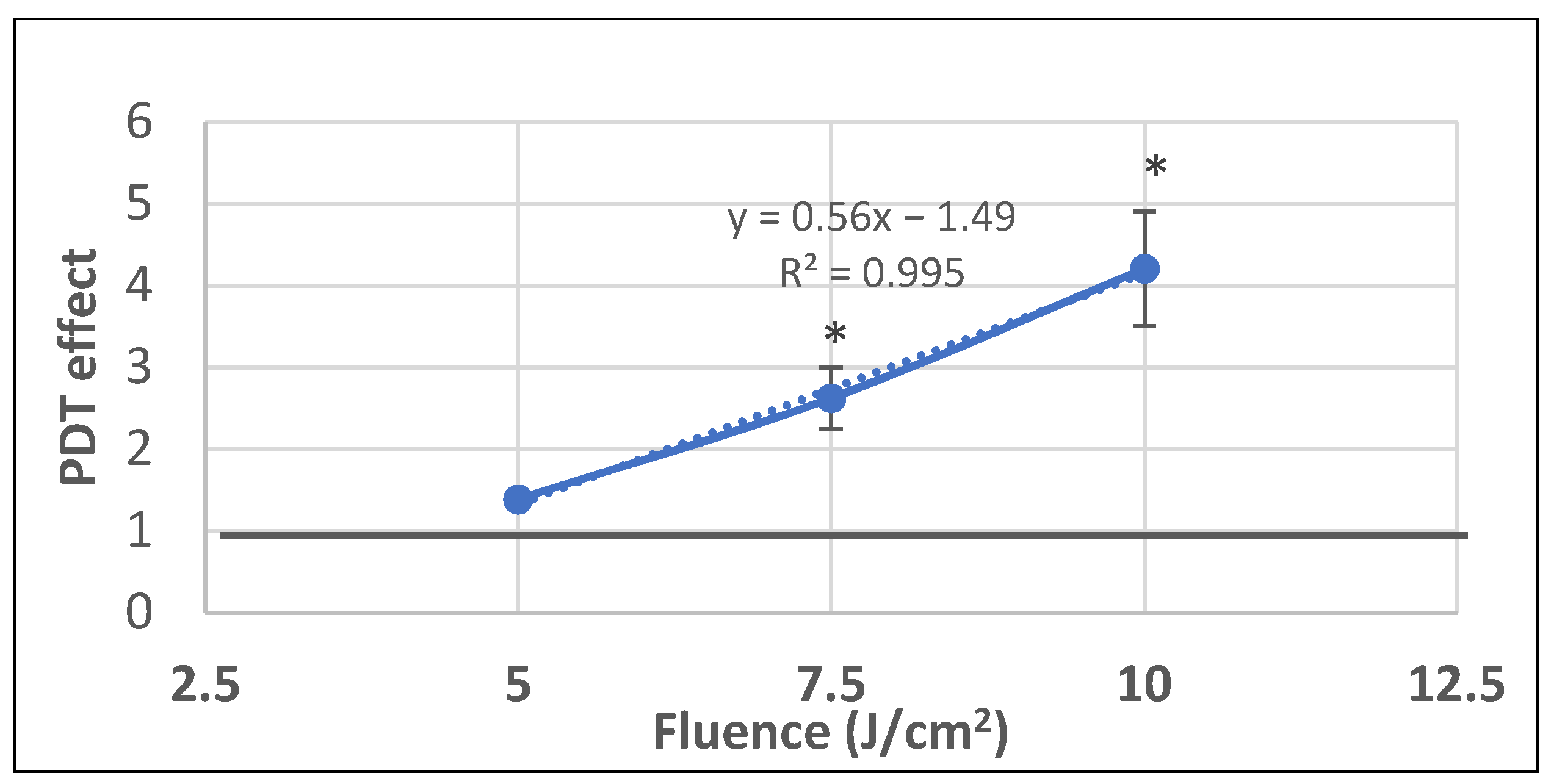
2.1. PDT-Induced Changes of Cell Viability
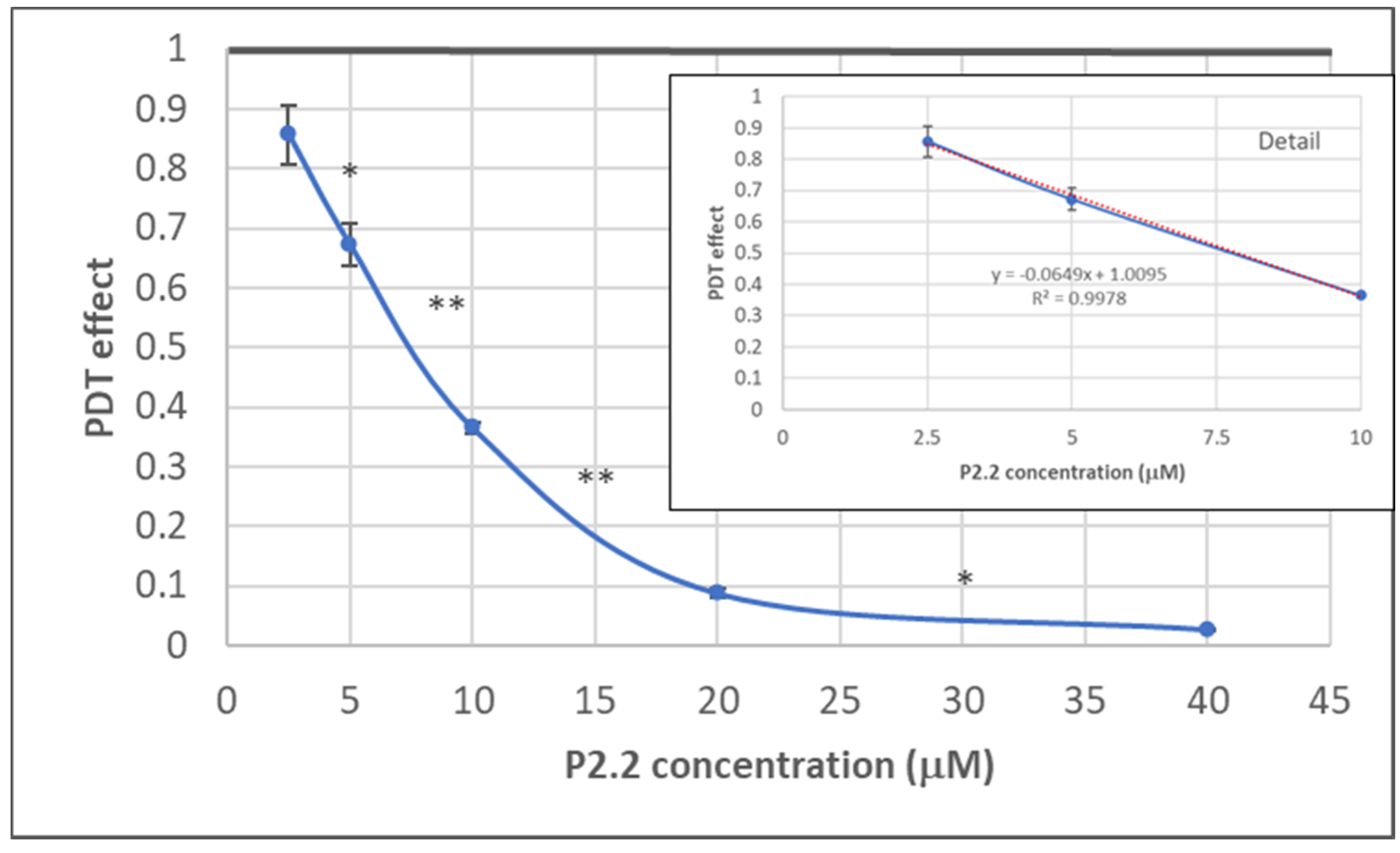
2.1.1. PDT-Induced Decrease of Viable Tumor Cells
2.1.2. PDT-Induced Alteration of Membrane Integrity
2.2. PDT-Induced Gene Expression Changes
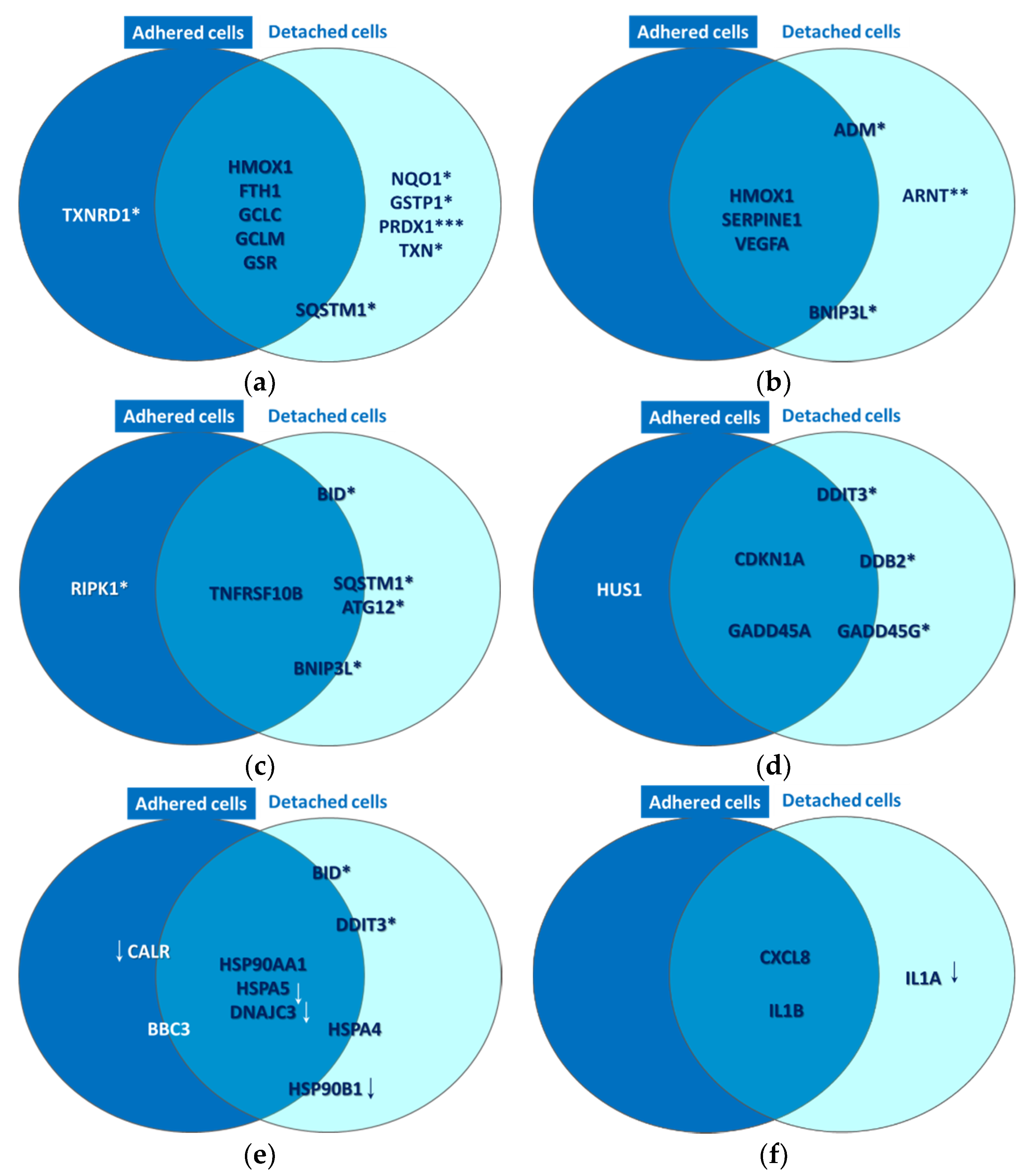
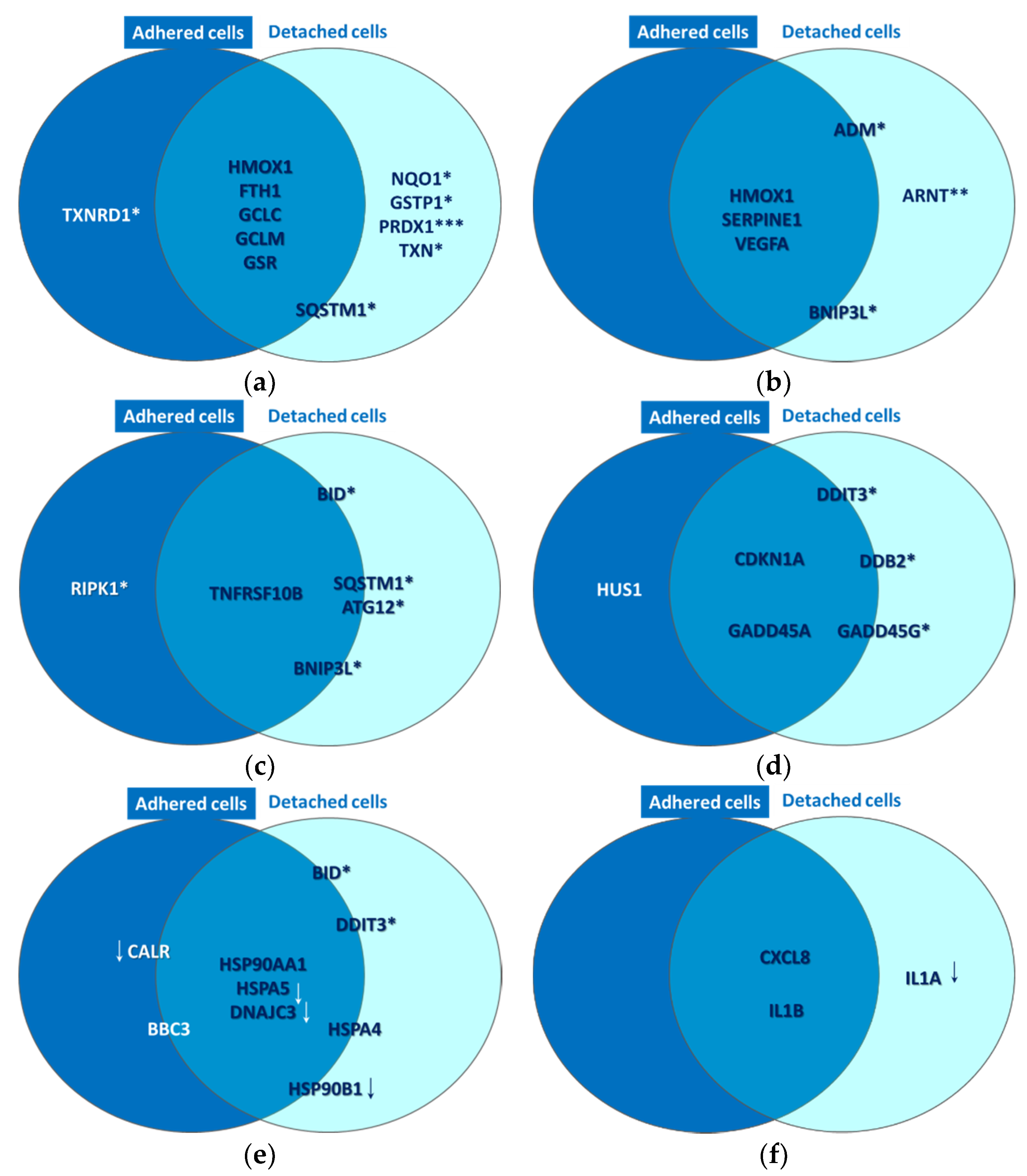
2.2.1. Oxidative Stress
2.2.2. Hypoxia Signaling
2.2.3. Cell Death
2.2.4. DNA Damage
2.2.5. Unfolded Protein Response
2.2.6. Inflammation
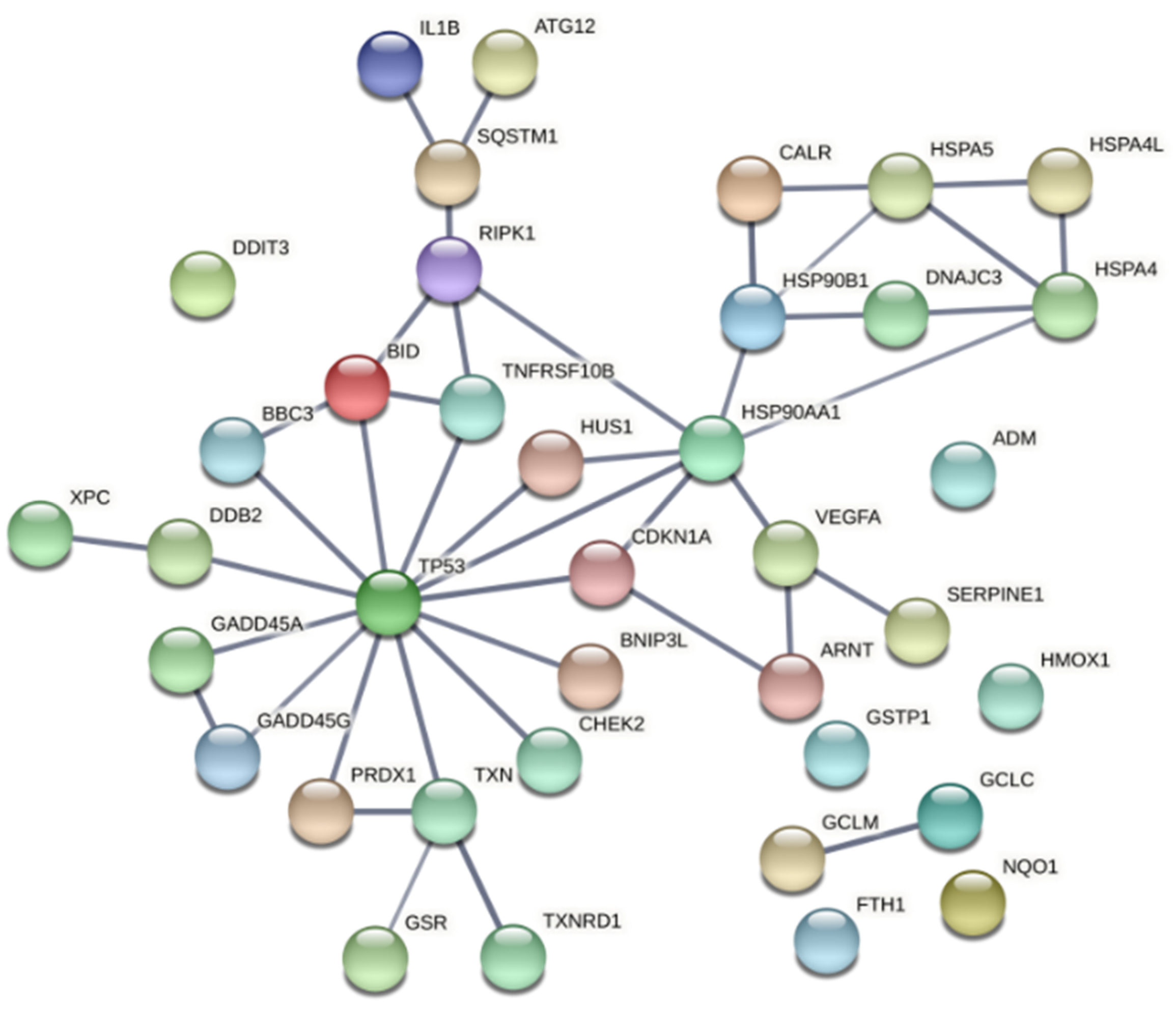
3. Discussion
4. Materials and Methods
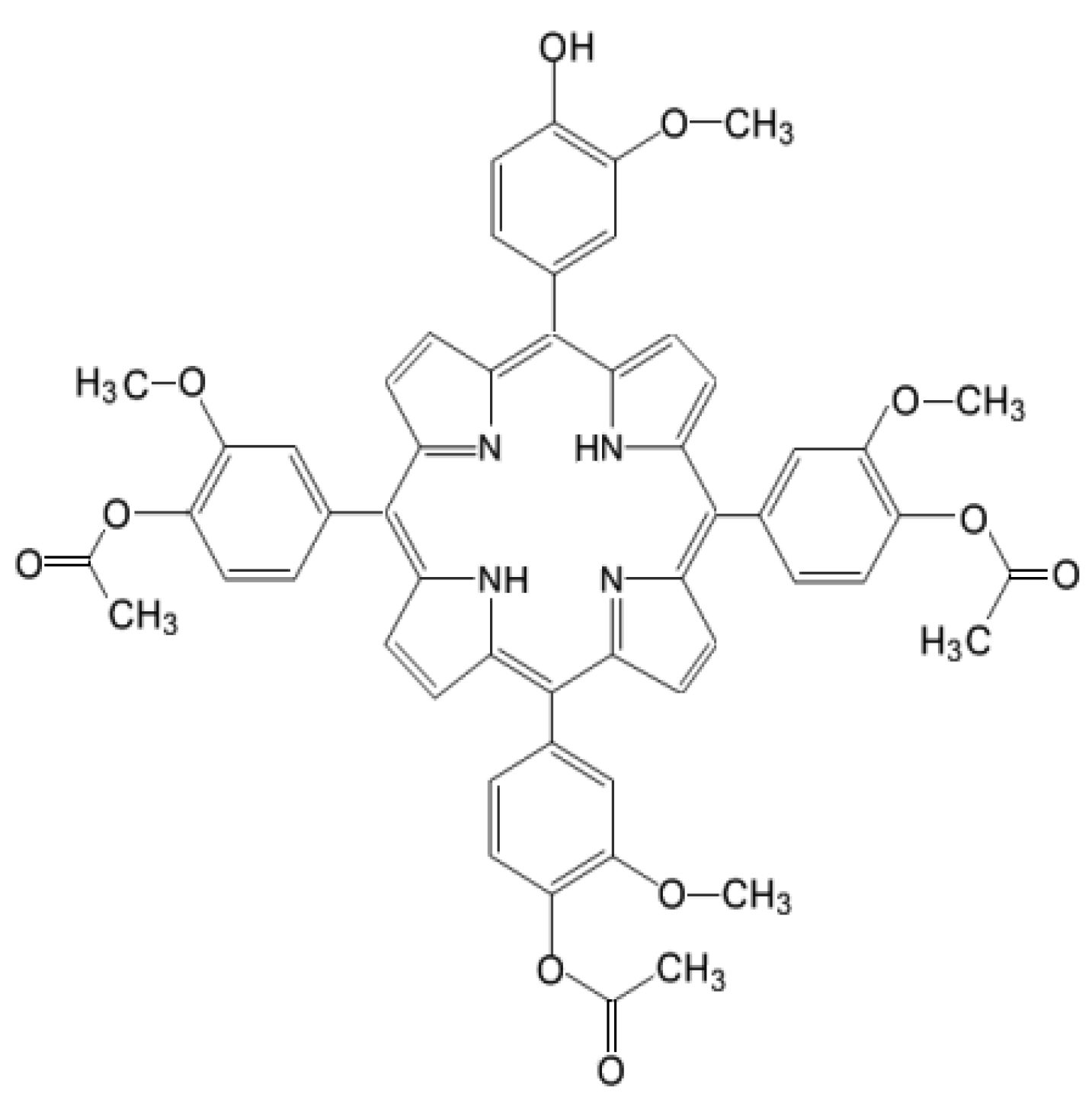
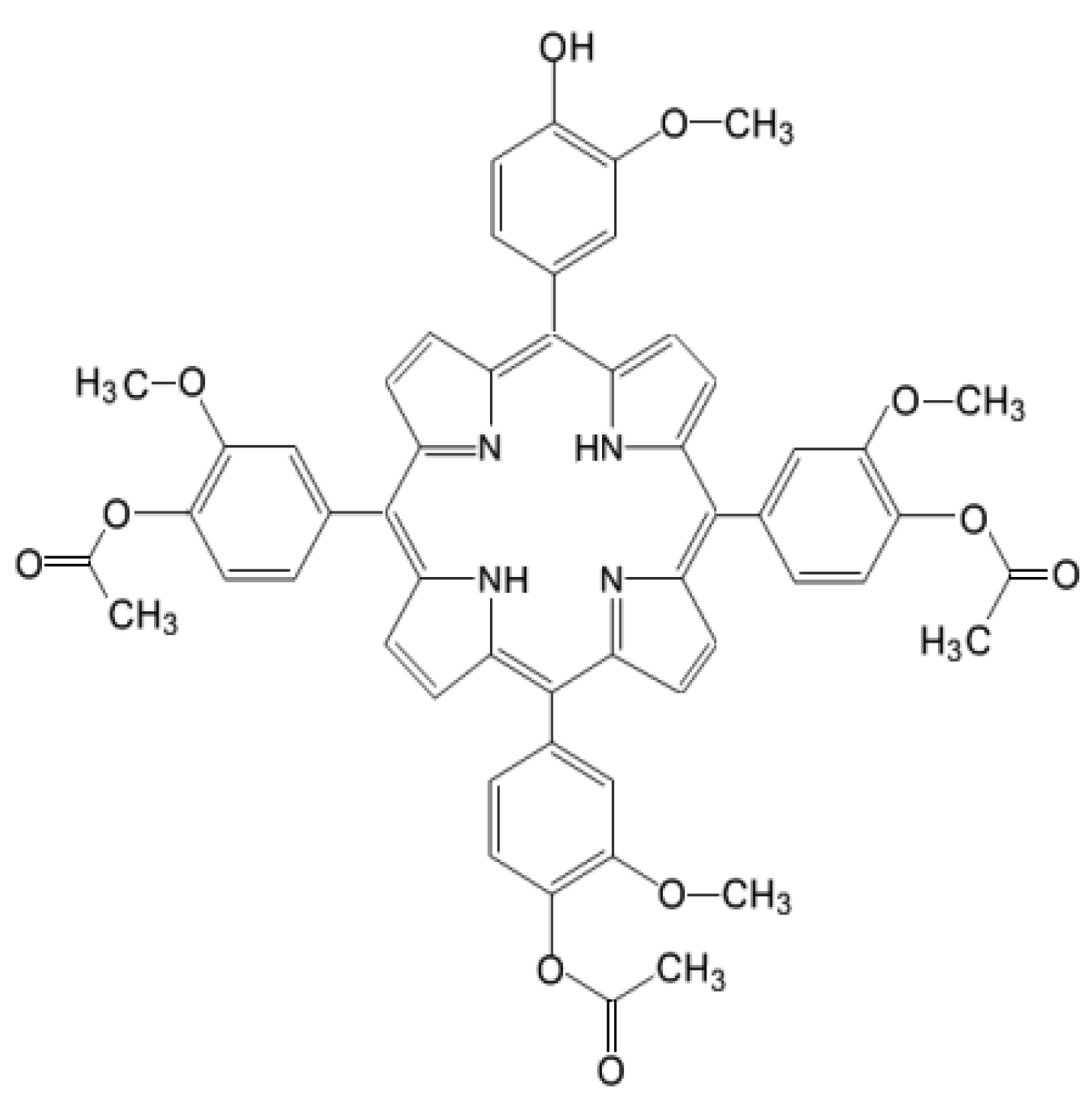
4.1. Photosensitizer
4.2. Cells
4.3. Loading of Cells with P2.2
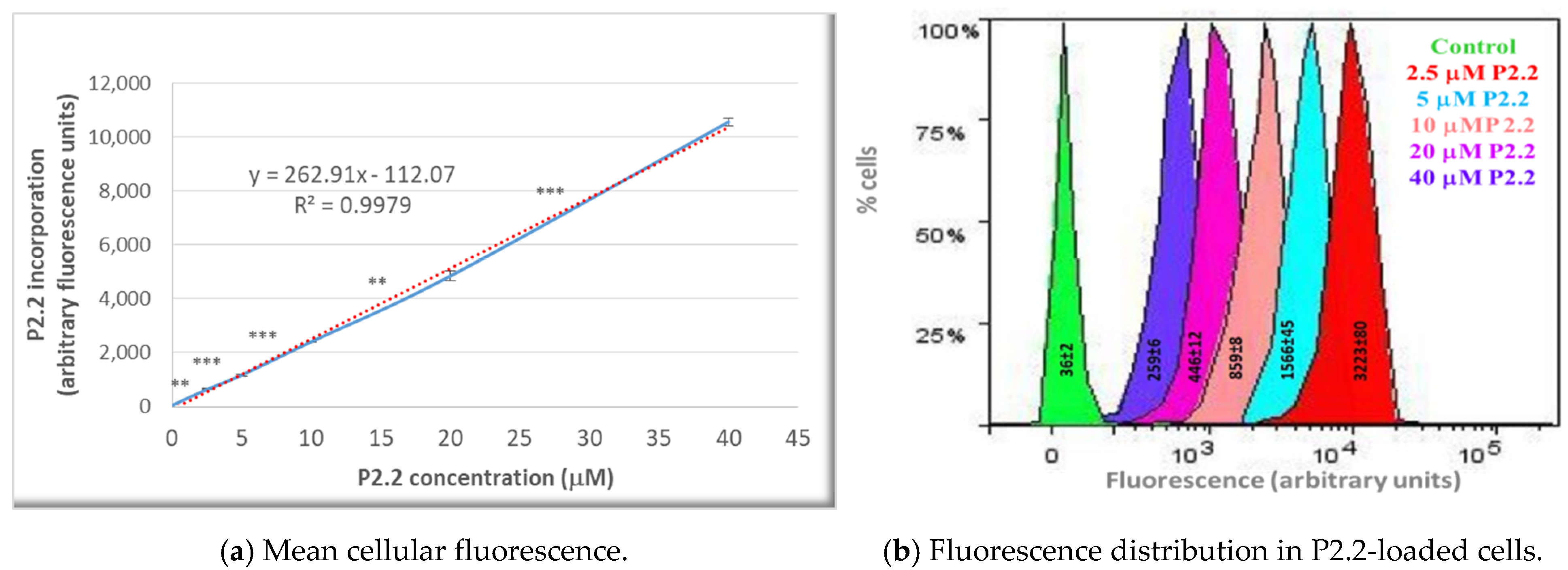
4.4. P2.2 Uptake in HT29 Cells
4.5. In Vitro PDT
4.6. Post-PDT Investigations
4.6.1. Preparation of Samples for Post-PDT Investigations
- Cell culture supernatants were harvested from samples cultivated for 24 h post-PDT and were centrifuged for eliminating detached cells. These cell-free culture supernatants were used for the LDH release assay at 24 h post-PDT. Cellular sediments resulting following centrifugation were suspended in a small volume of complete culture medium. Parts of these cells were used for post-PDT investigations at 24 h and parts for cell cultures were analyzed at 72 h post-PDT, as will be described below.
- Adhered cells were detached at 24 h post-PDT with Trypsin-EDTA (see Section 4.2). The resulting cell suspension was centrifuged and the sediment was suspended in complete culture medium. Parts of these cells were used for investigations at 24 h post-PDT and parts were plated for cell cultures to be analyzed at 72 h post-PDT, as will be described below.
- Detached and adhered cells harvested at 24 h post-PDT were mixed. Cells in non-treated samples were counted and the volume containing 10,000 control cells was calculated. For MTS reduction and LDH release (see below Section 4.6.2 and Section 4.6.3, respectively), the previously calculated cell suspension volume was collected from all samples, both PDT-treated and non-treated, was placed in 96 well plates and the culture volume was adjusted to 100 µL complete culture medium in each well. Triplicate samples containing only culture medium constituted the background control for these colorimetric tests. Cell cultures were incubated for another 48 h at 37 °C in 5% CO2 atmosphere and were analyzed at 72 h post-PDT.
4.6.2. MTS Reduction
4.6.3. LDH Release
4.6.4. Apoptosis and Necrosis Evaluation by Flow Cytometry
4.6.5. Cell Proliferation Evaluation
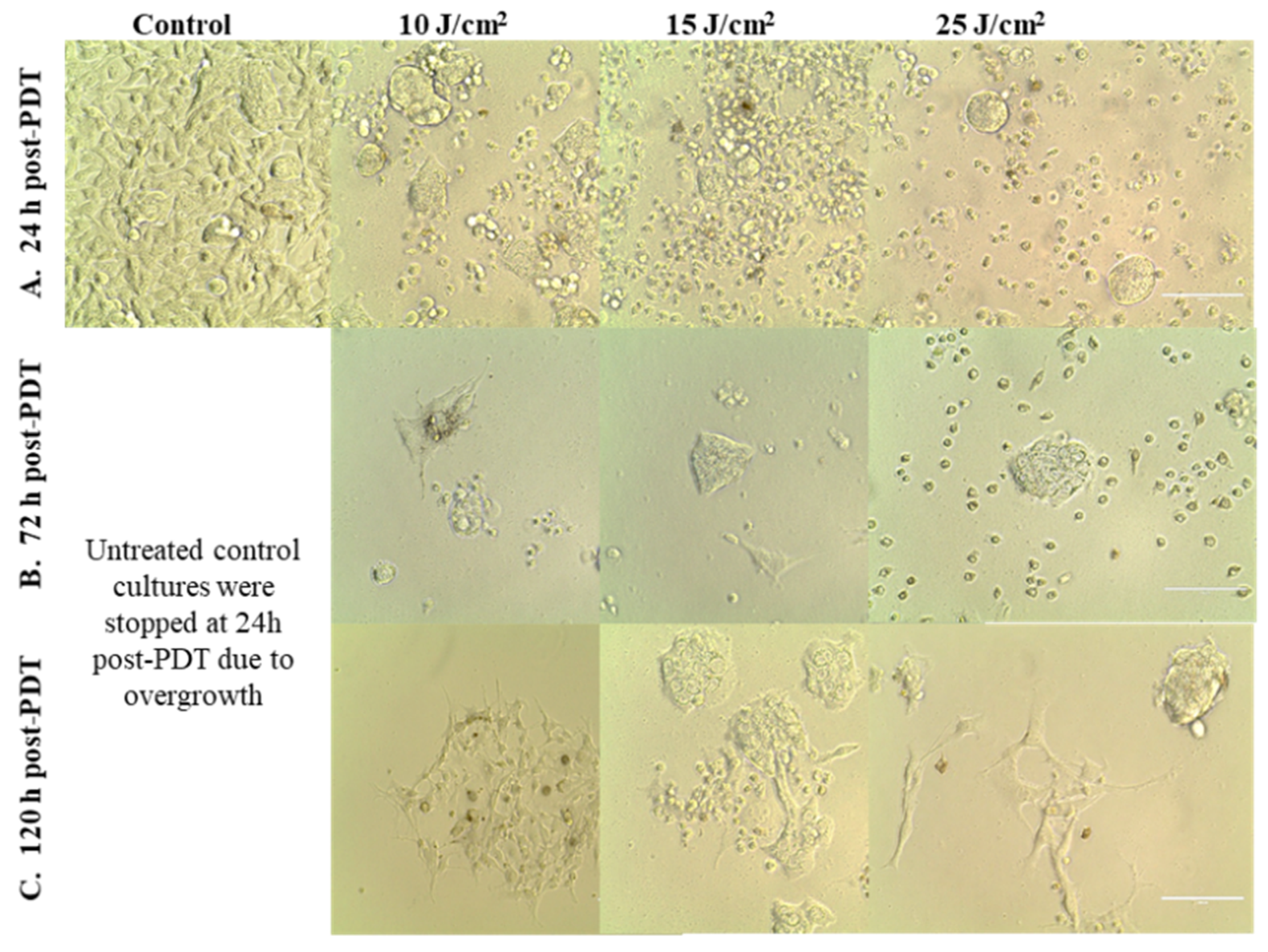
4.6.6. Microscopic Monitoring of Cell Cultures
4.6.7. Gene Expression
4.7. Data Processing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA. Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Falk-Mahapatra, R.; Gollnick, S.O. Photodynamic Therapy and Immunity: An Update. Photochem. Photobiol. 2020, 96, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manda, G.; Hinescu, M.E.; Neagoe, I.V.; Ferreira, L.F.V.; Boscencu, R.; Vasos, P.; Basaga, S.H.; Cuadrado, A. Emerging Therapeutic Targets in Oncologic Photodynamic Therapy. Curr. Pharm. Des. 2018, 24, 5268–5295. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Tan, L.-C.; Dong, L.-W.; Zhang, W.-Q.; Shen, X.-X.; Lu, X.; Zheng, H.; Lu, Y.-G. Susceptibility and Resistance Mechanisms During Photodynamic Therapy of Melanoma. Front. Oncol. 2020, 10, 597. [Google Scholar] [CrossRef]
- Donohoe, C.; Senge, M.O.; Arnaut, L.G.; Gomes-da-Silva, L.C. Cell death in photodynamic therapy: From oxidative stress to anti-tumor immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188308. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. DAMPs and PDT-mediated photo-oxidative stress: Exploring the unknown. Photochem. Photobiol. Sci. 2011, 10, 670–680. [Google Scholar] [CrossRef] [Green Version]
- Bigot, E.; Bataille, R.; Patrice, T. Increased singlet oxygen-induced secondary ROS production in the serum of cancer patients. J. Photochem. Photobiol. B 2012, 107, 14–19. [Google Scholar] [CrossRef]
- Boscencu, R.; Manda, G.; Radulea, N.; Socoteanu, R.P.; Ceafalan, L.C.; Neagoe, I.V.; Ferreira Machado, I.; Basaga, S.H.; Vieira Ferreira, L.F. Studies on the Synthesis, Photophysical and Biological Evaluation of Some Unsymmetrical Meso-Tetrasubstituted Phenyl Porphyrins. Molecules 2017, 22, 1815. [Google Scholar] [CrossRef] [Green Version]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Ros, U.; Peña-Blanco, A.; Hänggi, K.; Kunzendorf, U.; Krautwald, S.; Wong, W.W.-L.; García-Sáez, A.J. Necroptosis Execution Is Mediated by Plasma Membrane Nanopores Independent of Calcium. Cell Rep. 2017, 19, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one-photosensitizers, photochemistry and cellular localization. Photodiagn. Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Looft, A.; Pfitzner, M.; Preuß, A.; Röder, B. In vivo singlet molecular oxygen measurements: Sensitive to changes in oxygen saturation during PDT. Photodiagn. Photodyn. Ther. 2018, 23, 325–330. [Google Scholar] [CrossRef]
- Shen, Y.; Li, X.; Zhao, B.; Xue, Y.; Wang, S.; Chen, X.; Yang, J.; Lv, H.; Shang, P. Iron metabolism gene expression and prognostic features of hepatocellular carcinoma. J. Cell. Biochem. 2018, 119, 9178–9204. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Yadav, S. The glutathione cycle: Glutathione metabolism beyond the γ-glutamyl cycle. IUBMB Life 2018, 70, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, F.; Soltani, A.; Ghahremanloo, A.; Javid, H.; Hashemy, S.I. The thioredoxin system and cancer therapy: A review. Cancer Chemother. Pharmacol. 2019, 84, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-X.; Sun, X.-Y.; Yang, S.-M.; Wang, Q.; Wang, Z.-Y. Peroxiredoxin 1 promoted tumor metastasis and angiogenesis in colorectal cancer. Pathol. Res. Pract. 2018, 214, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Wei, Y.; Jiang, T.; Wang, S. Correlation of Nrf2, NQO1, MRP1, cmyc and p53 in colorectal cancer and their relationships to clinicopathologic features and survival. Int. J. Clin. Exp. Pathol. 2014, 7, 1124–1131. [Google Scholar] [PubMed]
- Beaver, S.K.; Mesa-Torres, N.; Pey, A.L.; Timson, D.J. NQO1: A target for the treatment of cancer and neurological diseases, and a model to understand loss of function disease mechanisms. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Kosumi, K.; Masugi, Y.; Yang, J.; Qian, Z.R.; Kim, S.A.; Li, W.; Shi, Y.; da Silva, A.; Hamada, T.; Liu, L.; et al. Tumor SQSTM1 (p62) expression and T cells in colorectal cancer. Oncoimmunology 2017, 6, e1284720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yang, S.; Xu, B.; Wang, T.; Zheng, Y.; Liu, F.; Ren, F.; Jiang, J.; Shi, H.; Zou, B.; et al. p62 functions as an oncogene in colorectal cancer through inhibiting apoptosis and promoting cell proliferation by interacting with the vitamin D receptor. Cell Prolif. 2019, 52, e12585. [Google Scholar] [CrossRef]
- Li, S.; Wei, X.; He, J.; Tian, X.; Yuan, S.; Sun, L. Plasminogen activator inhibitor-1 in cancer research. Biomed. Pharmacother. 2018, 105, 83–94. [Google Scholar] [CrossRef]
- Wang, L.; Gala, M.; Yamamoto, M.; Pino, M.S.; Kikuchi, H.; Shue, D.S.; Shirasawa, S.; Austin, T.R.; Lynch, M.P.; Rueda, B.R.; et al. Adrenomedullin is a therapeutic target in colorectal cancer. Int. J. Cancer 2014, 134, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.-P.; Wu, K.-J. Epigenetic regulation of hypoxia-responsive gene expression: Focusing on chromatin and DNA modifications. Int. J. Cancer 2014, 134, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, S.; Kikuchi, H.; Iino, I.; Uehara, T.; Setoguchi, T.; Fujita, T.; Hiramatsu, Y.; Ohta, M.; Kamiya, K.; Kitagawa, K.; et al. Anti-VEGF antibody therapy induces tumor hypoxia and stanniocalcin 2 expression and potentiates growth of human colon cancer xenografts. Int. J. Cancer 2014, 135, 295–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, P.; Wang, W.; Kim, S.; Wang, S.; Burns, T.F.; Sax, J.K.; Buzzai, M.; Dicker, D.T.; McKenna, W.G.; Bernhard, E.J.; et al. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell 2004, 6, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Chau, L.-Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [Green Version]
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef]
- Babinčák, M.; Jendželovský, R.; Košuth, J.; Majerník, M.; Vargová, J.; Mikulášek, K.; Zdráhal, Z.; Fedoročko, P. Death Receptor 5 (TNFRSF10B) Is Upregulated and TRAIL Resistance Is Reversed in Hypoxia and Normoxia in Colorectal Cancer Cell Lines after Treatment with Skyrin, the Active Metabolite of Hypericum spp. Cancers 2021, 13, 1646. [Google Scholar] [CrossRef] [PubMed]
- Gahl, R.F.; Dwivedi, P.; Tjandra, N. Bcl-2 proteins bid and bax form a network to permeabilize the mitochondria at the onset of apoptosis. Cell Death Dis. 2016, 7, e2424. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Wang, L.; Chang, X.; Feng, J.; Yu, J.; Chen, G. TRADD Mediates RIPK1-Independent Necroptosis Induced by Tumor Necrosis Factor. Front. Cell Dev. Biol. 2019, 7, 393. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.L.; He, G.Y.; Lan, X.L.; Zeng, Z.C.; Guan, J.; Ding, Y.; Qian, X.L.; Liao, W.T.; Ding, Y.Q.; Liang, L. Inhibition of ATG12-mediated autophagy by miR-214 enhances radiosensitivity in colorectal cancer. Oncogenesis 2018, 7, 16. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 2013, 793, 1–19. [Google Scholar] [CrossRef]
- Xiang, H.; Geng, X.; Ge, W.; Li, H. Meta-analysis of CHEK2 1100delC variant and colorectal cancer susceptibility. Eur. J. Cancer 2011, 47, 2546–2551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-Q.; Zhao, J.-J.; Chen, C.-L.; Liu, Y.; Zeng, J.-X.; Wu, Z.-R.; Tang, Y.; Zhu, Q.; Weng, D.-S.; Xia, J.-C. HUS1 checkpoint clamp component (HUS1) is a potential tumor suppressor in primary hepatocellular carcinoma. Mol. Carcinog. 2019, 58, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Jauhiainen, A.; Thomsen, C.; Strömbom, L.; Grundevik, P.; Andersson, C.; Danielsson, A.; Andersson, M.K.; Nerman, O.; Rörkvist, L.; Ståhlberg, A.; et al. Distinct cytoplasmic and nuclear functions of the stress induced protein DDIT3/CHOP/GADD153. PLoS ONE 2012, 7, e33208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, S.; Raychaudhuri, P. Damaged-DNA Binding Protein-2 Drives Apoptosis Following DNA Damage. Cell Div. 2010, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemzow, L.; Lubin, A.; Zhang, L.; Gong, F. XPC: Going where no DNA damage sensor has gone before. DNA Repair 2015, 36, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Rashid, H.-O.; Yadav, R.K.; Kim, H.-R.; Chae, H.-J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Reimertz, C.; Kögel, D.; Rami, A.; Chittenden, T.; Prehn, J.H.M. Gene expression during ER stress-induced apoptosis in neurons: Induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 2003, 162, 587–597. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 2004, 28, 51–65. [Google Scholar] [CrossRef]
- Theodoraki, M.A.; Caplan, A.J. Quality control and fate determination of Hsp90 client proteins. Biochim. Biophys. Acta 2012, 1823, 683–688. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef]
- Van Huizen, R.; Martindale, J.L.; Gorospe, M.; Holbrook, N.J. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. J. Biol. Chem. 2003, 278, 15558–15564. [Google Scholar] [CrossRef] [Green Version]
- Feng, R.; Ye, J.; Zhou, C.; Qi, L.; Fu, Z.; Yan, B.; Liang, Z.; Li, R.; Zhai, W. Calreticulin down-regulation inhibits the cell growth, invasion and cell cycle progression of human hepatocellular carcinoma cells. Diagn. Pathol. 2015, 10, 149. [Google Scholar] [CrossRef] [Green Version]
- Cohen, I.; Idan, C.; Rider, P.; Peleg, R.; Vornov, E.; Elena, V.; Tomas, M.; Martin, T.; Tudor, C.; Cicerone, T.; et al. IL-1α is a DNA damage sensor linking genotoxic stress signaling to sterile inflammation and innate immunity. Sci. Rep. 2015, 5, 14756. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Lu, M.; Petersen, S.; Ashkenazi, A. Apoptosis Initiation Through the Cell-Extrinsic Pathway. Methods Enzymol. 2014, 544, 99–128. [Google Scholar] [CrossRef]
- Oliver, P.G.; LoBuglio, A.F.; Zinn, K.R.; Kim, H.; Nan, L.; Zhou, T.; Wang, W.; Buchsbaum, D.J. Treatment of human colon cancer xenografts with TRA-8 anti-death receptor 5 antibody alone or in combination with CPT-11. Clin. Cancer Res. 2008, 14, 2180–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, T.; Shi, Q.; Li, Z.; Feng, Y.; Yang, H.; Tao, Z.; Li, H.; Chen, J.; Wang, S.; Liang, Y.; et al. Combination of long-acting TRAIL and tumor cell-targeted photodynamic therapy as a novel strategy to overcome chemotherapeutic multidrug resistance and TRAIL resistance of colorectal cancer. Theranostics 2021, 11, 4281–4297. [Google Scholar] [CrossRef] [PubMed]
- Billen, L.P.; Shamas-Din, A.; Andrews, D.W. Bid: A Bax-like BH3 protein. Oncogene 2008, 27 (Suppl. S1), S93–S104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 2001, 98, 11318–11323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhao, D.; Wang, G.; Wang, Y.; Cao, L.; Sun, J.; Jiang, Q.; He, Z. Recent progress of hypoxia-modulated multifunctional nanomedicines to enhance photodynamic therapy: Opportunities, challenges, and future development. Acta Pharm. Sin. B 2020, 10, 1382–1396. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, K. RIPK1 and RIPK3: Critical regulators of inflammation and cell death. Trends Cell Biol. 2015, 25, 347–353. [Google Scholar] [CrossRef]
- Martins, W.K.; Belotto, R.; Silva, M.N.; Grasso, D.; Suriani, M.D.; Lavor, T.S.; Itri, R.; Baptista, M.S.; Tsubone, T.M. Autophagy Regulation and Photodynamic Therapy: Insights to Improve Outcomes of Cancer Treatment. Front. Oncol. 2020, 10, 610472. [Google Scholar] [CrossRef]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Wang, J.Y.J. Cell Death Response to DNA Damage. Yale J. Biol. Med. 2019, 92, 771–779. [Google Scholar]
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res. 2020, 48, 11227–11243. [Google Scholar] [CrossRef]
- Stoyanova, T.; Roy, N.; Kopanja, D.; Raychaudhuri, P.; Bagchi, S. DDB2 (damaged-DNA binding protein 2) in nucleotide excision repair and DNA damage response. Cell Cycle 2009, 8, 4067–4071. [Google Scholar] [CrossRef]
- Barakat, B.M.; Wang, Q.-E.; Han, C.; Milum, K.; Yin, D.-T.; Zhao, Q.; Wani, G.; Arafa, E.-S.A.; El-Mahdy, M.A.; Wani, A.A. Overexpression of DDB2 enhances the sensitivity of human ovarian cancer cells to cisplatin by augmenting cellular apoptosis. Int. J. Cancer 2010, 127, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Bagchi, S.; Raychaudhuri, P. Damaged DNA binding protein 2 in reactive oxygen species (ROS) regulation and premature senescence. Int. J. Mol. Sci. 2012, 13, 11012–11026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minig, V.; Kattan, Z.; van Beeumen, J.; Brunner, E.; Becuwe, P. Identification of DDB2 protein as a transcriptional regulator of constitutive SOD2 gene expression in human breast cancer cells. J. Biol. Chem. 2009, 284, 14165–14176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomioka, Y.; Kushibiki, T.; Awazu, K. Evaluation of oxygen consumption of culture medium and in vitro photodynamic effect of talaporfin sodium in lung tumor cells. Photomed. Laser Surg. 2010, 28, 385–390. [Google Scholar] [CrossRef]
- Hernansanz-Agustín, P.; Izquierdo-Álvarez, A.; Sánchez-Gómez, F.J.; Ramos, E.; Villa-Piña, T.; Lamas, S.; Bogdanova, A.; Martínez-Ruiz, A. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 2014, 71, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Moserova, I.; Kralova, J. Role of ER stress response in photodynamic therapy: ROS generated in different subcellular compartments trigger diverse cell death pathways. PLoS ONE 2012, 7, e32972. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. S2), 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggs, S.; Alario, A.J.; McHorney, C. Health risk behaviors and attempted suicide in adolescents who report prior maltreatment. J. Pediatr. 1990, 116, 815–821. [Google Scholar] [CrossRef]
- Pereira, E.J.; Burns, J.S.; Lee, C.Y.; Marohl, T.; Calderon, D.; Wang, L.; Atkins, K.A.; Wang, C.-C.; Janes, K.A. Sporadic activation of an oxidative stress-dependent NRF2-p53 signaling network in breast epithelial spheroids and premalignancies. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.; Ryoo, I.; Kang, H.C.; Kwak, M.-K. The sensitivity of cancer cells to pheophorbide a-based photodynamic therapy is enhanced by Nrf2 silencing. PLoS ONE 2014, 9, e107158. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Zawacka-Pankau, J.; Krachulec, J.; Grulkowski, I.; Bielawski, K.P.; Selivanova, G. The p53-mediated cytotoxicity of photodynamic therapy of cancer: Recent advances. Toxicol. Appl. Pharmacol. 2008, 232, 487–497. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.E.; Ceder, S.; Bykov, V.J.N.; Wiman, K.G. p53 as a hub in cellular redox regulation and therapeutic target in cancer. J. Mol. Cell Biol. 2019, 11, 330–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, M.J.; Pansa, M.F.; Vera, R.E.; Fernández-Zapico, M.E.; Rumie Vittar, N.B.; Rivarola, V.A. Transcriptional activation of HIF-1 by a ROS-ERK axis underlies the resistance to photodynamic therapy. PLoS ONE 2017, 12, e0177801. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Su, L.; Lei, Y.; Liu, X.; Zhang, Y.; Liu, X. DDIT3 and KAT2A Proteins Regulate TNFRSF10A and TNFRSF10B Expression in Endoplasmic Reticulum Stress-mediated Apoptosis in Human Lung Cancer Cells. J. Biol. Chem. 2015, 290, 11108–11118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Wang, H.-G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, G.P.; O’Connor, H.; Henry, C.M.; Davidovich, P.; Clancy, D.M.; Albert, M.L.; Cullen, S.P.; Martin, S.J. TRAIL Receptors Serve as Stress-Associated Molecular Patterns to Promote ER-Stress-Induced Inflammation. Dev. Cell 2020, 52, 714–730.e5. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Jundi, K.; Greene, C.M. Transcription of Interleukin-8: How Altered Regulation Can Affect Cystic Fibrosis Lung Disease. Biomolecules 2015, 5, 1386–1398. [Google Scholar] [CrossRef]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [Green Version]
- Galdiero, M.R.; Bianchi, P.; Grizzi, F.; Di Caro, G.; Basso, G.; Ponzetta, A.; Bonavita, E.; Barbagallo, M.; Tartari, S.; Polentarutti, N.; et al. Occurrence and significance of tumor-associated neutrophils in patients with colorectal cancer. Int. J. Cancer 2016, 139, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Berry, R.S.; Xiong, M.-J.; Greenbaum, A.; Mortaji, P.; Nofchissey, R.A.; Schultz, F.; Martinez, C.; Luo, L.; Morris, K.T.; Hanson, J.A. High levels of tumor-associated neutrophils are associated with improved overall survival in patients with stage II colorectal cancer. PLoS ONE 2017, 12, e0188799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Papagoras, C.; Miltiades, P.; Angelidou, I.; Mitsios, A.; Kotsianidis, I.; Skendros, P.; Sivridis, E.; et al. Gradient Infiltration of Neutrophil Extracellular Traps in Colon Cancer and Evidence for Their Involvement in Tumour Growth. PLoS ONE 2016, 11, e0154484. [Google Scholar] [CrossRef] [PubMed]
- Bie, Y.; Ge, W.; Yang, Z.; Cheng, X.; Zhao, Z.; Li, S.; Wang, W.; Wang, Y.; Zhao, X.; Yin, Z.; et al. The Crucial Role of CXCL8 and Its Receptors in Colorectal Liver Metastasis. Dis. Markers 2019, 2019, 8023460. [Google Scholar] [CrossRef] [PubMed]
- Samuel, T.; Fadlalla, K.; Gales, D.N.; Putcha, B.D.K.; Manne, U. Variable NF-κB pathway responses in colon cancer cells treated with chemotherapeutic drugs. BMC Cancer 2014, 14, 599. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, L.F.V.; Machado, I.F.; Gama, A.; Socoteanu, R.P.; Boscencu, R.; Manda, G.; Calhelha, R.C.; Ferreira, I.C.F.R. Photochemical /Photocytotoxicity Studies of New Tetrapyrrolic Structures as Potential Candidates for Cancer Theranostics. Curr. Drug Discov. Technol. 2020, 17, 661–669. [Google Scholar] [CrossRef]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidative Stress FTH1, GCLC, GCLM, GSR, GSTP1, HMOX1, NQO1, PRDX1, SQSTM1, TXN, TXNRD1 |
| Hypoxia Signaling ADM, ARNT, BNIP3L, CA9, EPO, HMOX1, LDHA, MMP9, SERPINE1 (PAI-1), SLC2A1, VEGFA |
| Osmotic Stress AKR1B1, AQP1, AQP2, AQP4, CFTR, EDN1, HSPA4L (OSP94), NFAT5, SLC5A3 |
| Cell Death Apoptosis: CASP1 (ICE), FAS, MCL1, TNFRSF10A (TRAIL-R), TNFRSF10B (DR5), TNFRSF1A (TNFR1). AutophagyATG12, ATG5, ATG7, BECN1, FAS, ULK1 Necrosis: FAS, GRB2, PARP1 (ADPRT1), PVR, RIPK1, TNFRSF10A (TRAIL-R), TNFRSF1A (TNFR1), TXNL4B |
| DNA Damage and Repair Cell Cycle Arrest and Checkpoints: CDKN1A (p21CIP1, WAF1), CHEK1, CHEK2 (RAD53), DDIT3 (GADD153, CHOP), HUS1, MRE11, NBN, RAD17, RAD9A Other DNA Damage Responses: ATM, ATR, DDB2, GADD45A, GADD45G, RAD51, TP53 (p53), XPC |
| Unfolded Protein Response ATF4, ATF6, ATF6B, BBC3 (PUMA), BID, CALR, DDIT3 (GADD153, CHOP), DNAJC3, HSP90AA1, HSP90B1, HSPA4 (HSP70), HSPA5 (GRP78) |
| Inflammatory Response CCL2 (MCP-1), CD40LG, CRP, CXCL8 (IL8), IFNG, IL1A, IL1B, IL6, TLR4, TNF |
| a. Oxidative stress | ||||
| PDT-treated vs. non-treated HT29 tumor cells at 24 h post-PDT | ||||
| Gene | Adhered cells | Detached cells | ||
| FR | pvalue | FR | pvalue | |
| HMOX1 | 8.87 | <0.05 | 7.91 | <0.01 |
| FTH1 | 2.38 | <0.05 | 5.41 | <0.01 |
| GCLC | 3.36 | <0.05 | 2.74 | <0.01 |
| GCLM | 2.85 | <0.05 | 3.96 | <0.001 |
| GSR | 2.01 | <0.01 | 2.85 | <0.001 |
| SQSTM1 | 4.98 | <0.001 | 7.49 | <0.001 |
| PRDX1 | ns | ns | 4.18 | <0.001 |
| NQO1 | ns | ns | 3.02 | <0.01 |
| TXN | ns | ns | 3.29 | 0,0.05 |
| GSTP1 | ns | ns | 2.41 | <0.01 |
| TXNRD1 | 4.16 | <0.001 | ns | ns |
| b. Hypoxia signaling | ||||
| PDT-treated vs. non-treated HT29 tumor cells at 24 h post-PDT | ||||
| Gene | Adhered cells | Detached cells | ||
| FR | pvalue | FR | pvalue | |
| HMOX1 | 8.87 | <0.05 | 7.91 | <0.01 |
| SERPINE1 | 9.78 | <0.001 | 10.74 | <0.001 |
| ADM | 4.60 | <0.01 | 14.27 | <0.001 |
| ARNT | ns | ns | 2.36 | <0.05 |
| VEGFA | 1.81 | ns | 2.11 | ≤0.001 |
| BNIP3L | 2.02 | ns | 2.20 | <0.05 |
| c. Cell death | ||||
| PDT-treated vs. non-treated HT29 tumor cells at 24 h post-PDT | ||||
| Gene | Adhered cells | Detached cells | ||
| FR | pvalue | FR | pvalue | |
| Apoptosis | ||||
| TNFRSF10B (DR5/TRAILR2) | 3.09 | <0.001 | 2.86 | <0.001 |
| BID | 1.75 | ns | 3.96 | <0.05 |
| BNIP3L | 2.02 | ns | 2.20 | <0.05 |
| BBC3 | 4.19 | <0.05 | 5.30 | ns |
| Necrosis | ||||
| RIPK1 | 1.78 | <0.01 | ns | ns |
| Autophagy | ||||
| ATG12 | 1.85 | <0.05 | 3.29 | <0.01 |
| SQSTM1 | 4.98 | <0.001 | 7.49 | <0.001 |
| d. DNA damage responses | ||||
| PDT-treated vs. non-treated HT29 tumor cells at 24 h post-PDT | ||||
| Gene | Adhered cells | Detached cells | ||
| FR | pvalue | FR | pvalue | |
| Cell cycle arrest | ||||
| CDKN1A | 2.74 | <0.05 | 5.29 | <0.01 |
| GADD45A | 3.01 | <0.001 | 2.35 | <0.001 |
| DDIT3 (GADD153) | 4.77 | <0.01 | 12.86 | <0.01 |
| GADD45G | 3.01 | ns | 14.77 | <0.05 |
| CHEK2 | ns | ns | 1.71 | <0.05 |
| HUS1 | 1.62 | <0.01 | ns | ns |
| Other DNA damage responses | ||||
| DDB2 | 1.51 | <0.05 | 2.63 | <0.01 |
| XPC | 2.20 | <0.05 | 1.67 | ns |
| e. Unfolded protein response | ||||
| PDT-treated vs. non-treated HT29 tumor cells at 24 h post-PDT | ||||
| Gene | Adhered cells | Detached cells | ||
| FR | pvalue | FR | pvalue | |
| HSP90AA1 | 16.52 | <0.05 | 9.26 | <0.01 |
| DDIT3 (CHOP) | 4.77 | <0.01 | 12.86 | <0.01 |
| BBC3 | 4.19 | <0.05 | 5.30 | ns |
| BID | 1.75 | ns | 3.96 | <0.05 |
| HSPA4 (HSP70) | 2.96 | ns | 2.95 | <0.05 |
| HSPA5 (GRP78) | −2.27 | <0.01 | −2.42 | <0.001 |
| DNAJC3 | −1.59 | <0.05 | −4.07 | <0.001 |
| HSP90B1 | −1.77 | ns | −3.59 | <0.001 |
| CALR | −1.89 | <0.001 | ns | ns |
| f. Inflammation | ||||
| PDT-treated vs. non-treated HT29 tumor cells at 24 h post-PDT | ||||
| Gene | Adhered cells | Detached cells | ||
| FR | pvalue | FR | pvalue | |
| CXCL8 | 8.09 | ≤0.01 | 6.46 | <0.01 |
| IL1B | 3.28 | <0.001 | 2.15 | ns |
| IL1A | ns | ns | −2.90 | ≤0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobre, M.; Boscencu, R.; Neagoe, I.V.; Surcel, M.; Milanesi, E.; Manda, G. Insight into the Web of Stress Responses Triggered at Gene Expression Level by Porphyrin-PDT in HT29 Human Colon Carcinoma Cells. Pharmaceutics 2021, 13, 1032. https://doi.org/10.3390/pharmaceutics13071032
Dobre M, Boscencu R, Neagoe IV, Surcel M, Milanesi E, Manda G. Insight into the Web of Stress Responses Triggered at Gene Expression Level by Porphyrin-PDT in HT29 Human Colon Carcinoma Cells. Pharmaceutics. 2021; 13(7):1032. https://doi.org/10.3390/pharmaceutics13071032
Chicago/Turabian StyleDobre, Maria, Rica Boscencu, Ionela Victoria Neagoe, Mihaela Surcel, Elena Milanesi, and Gina Manda. 2021. "Insight into the Web of Stress Responses Triggered at Gene Expression Level by Porphyrin-PDT in HT29 Human Colon Carcinoma Cells" Pharmaceutics 13, no. 7: 1032. https://doi.org/10.3390/pharmaceutics13071032
APA StyleDobre, M., Boscencu, R., Neagoe, I. V., Surcel, M., Milanesi, E., & Manda, G. (2021). Insight into the Web of Stress Responses Triggered at Gene Expression Level by Porphyrin-PDT in HT29 Human Colon Carcinoma Cells. Pharmaceutics, 13(7), 1032. https://doi.org/10.3390/pharmaceutics13071032