
Tumor Microenvironment-Responsive Shell/Core Composite Nanoparticles for Enhanced Stability and Antitumor Efficiency Based on a pH-Triggered Charge-Reversal Mechanism


Abstract
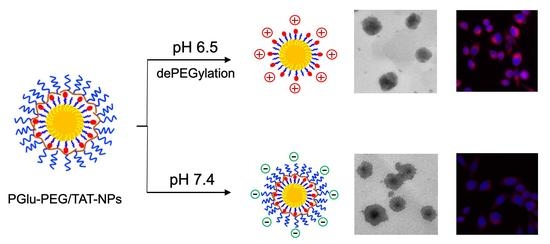
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines and Cell Cultures
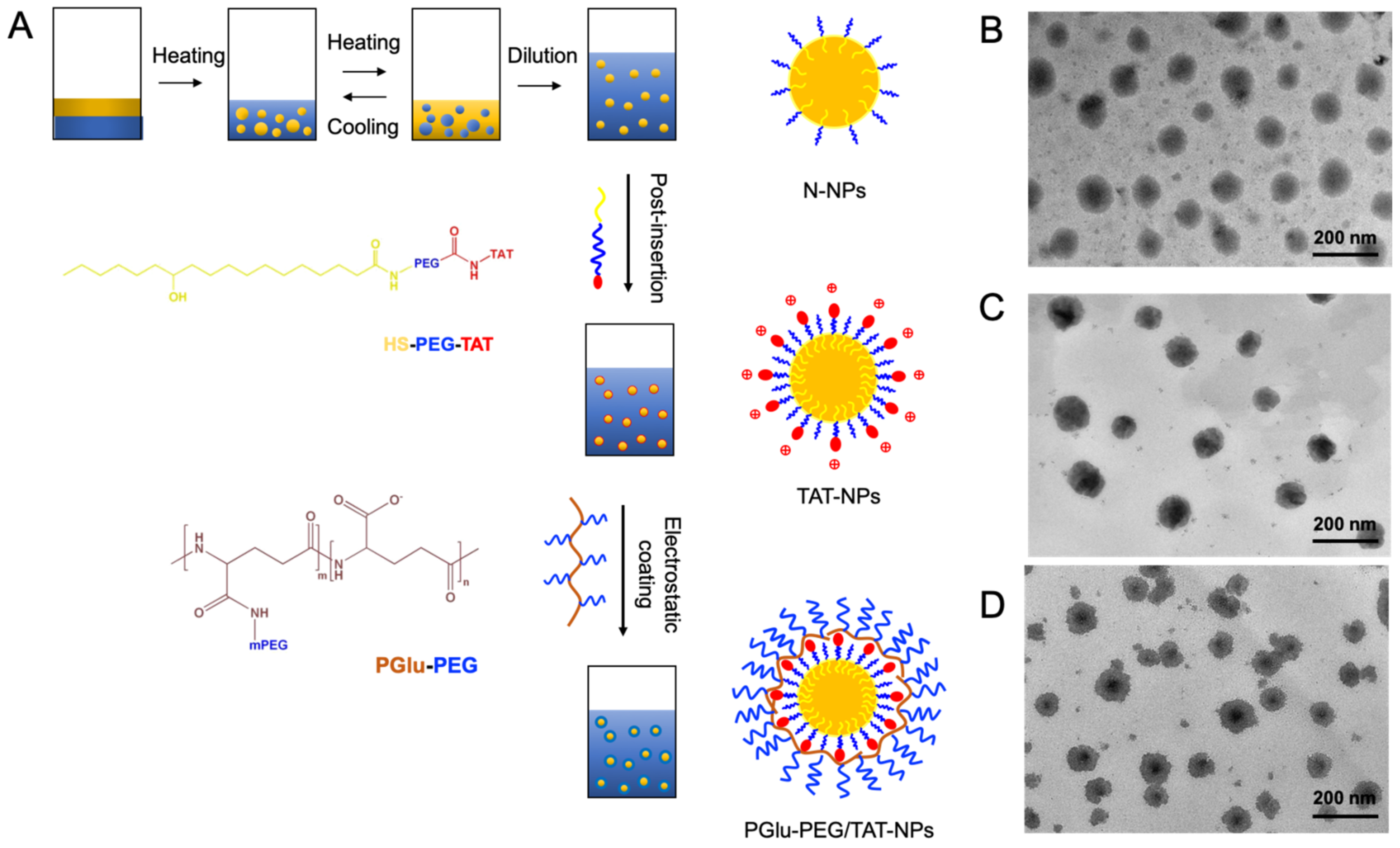
2.3. Preparation and Characteristics of DSF-Loaded Nanoparticles
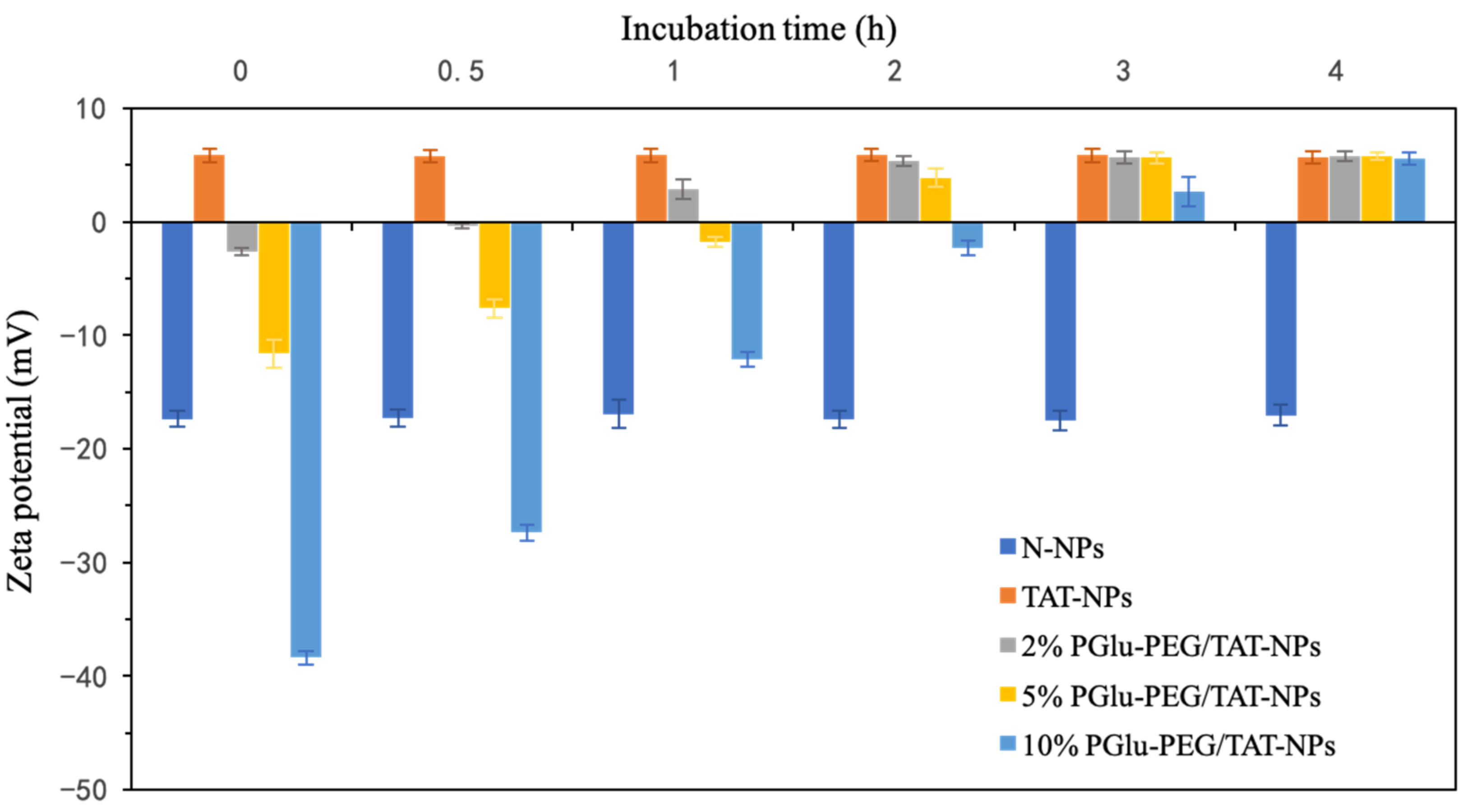
2.4. pH-Responsive Charge Conversion Study
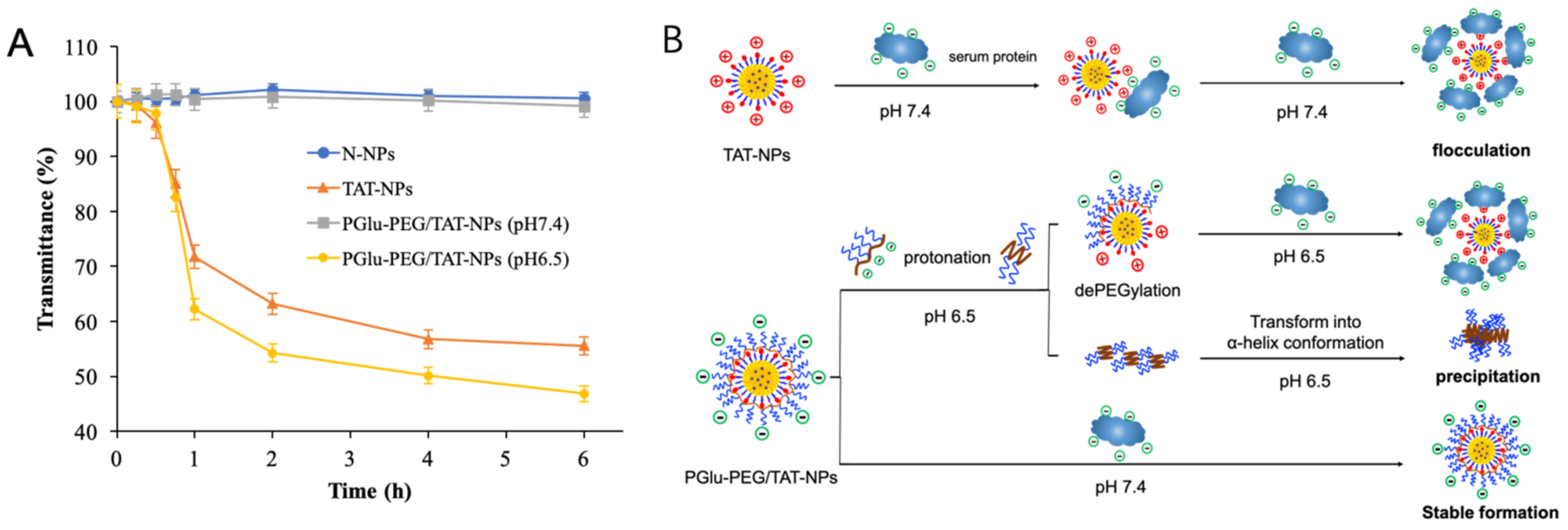
2.5. Plasma Stability Study
2.6. pH-Responsive Intracellular Uptake Study
2.7. Cytotoxicity Study
2.8. In Vivo Antitumor Efficiency and Toxicity
2.9. Statistical Analysis
3. Results and Discussion
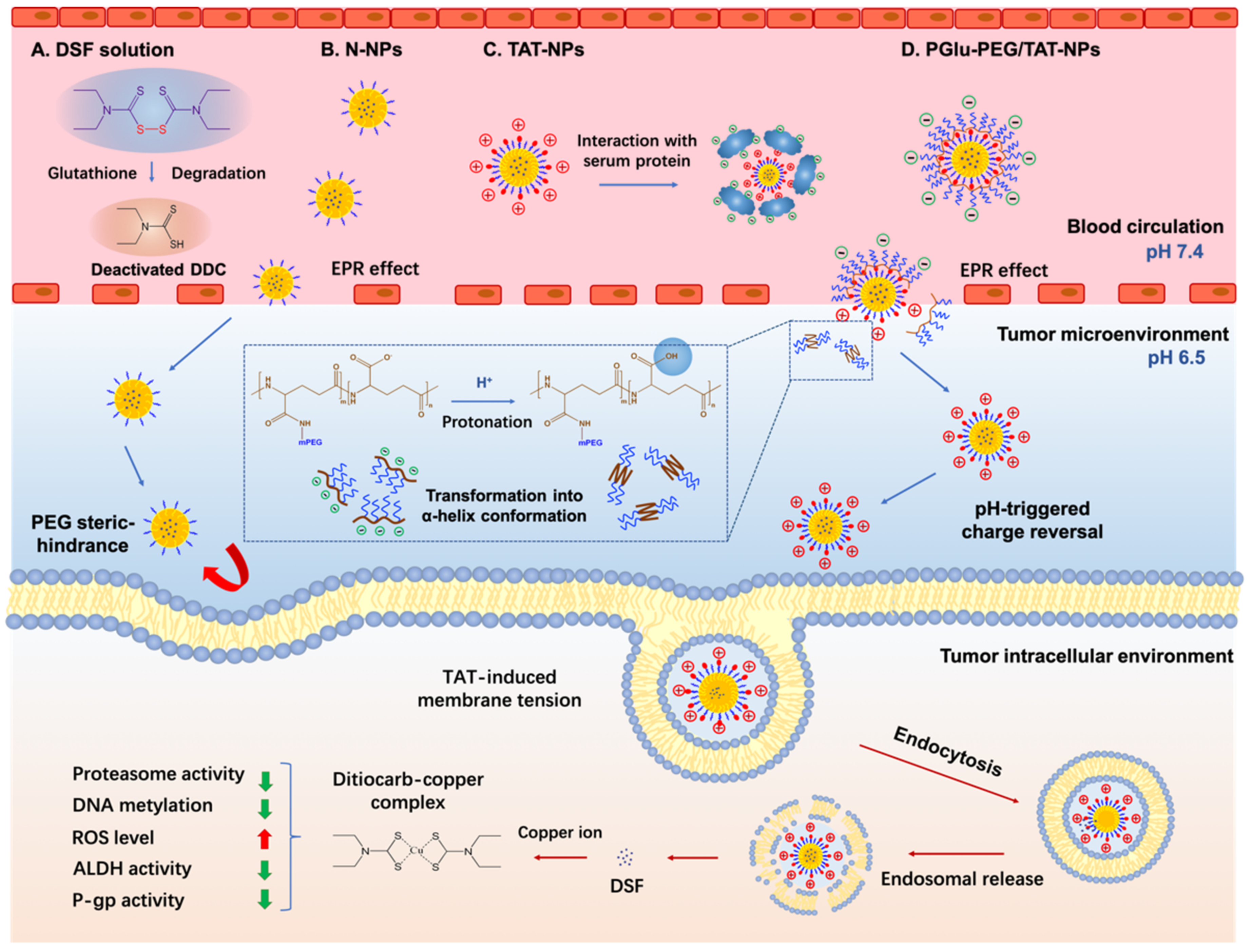
3.1. Preparation and Characterization of DSF-Loaded Nanoparticles
3.2. pH-Responsive Charge Reversal of Shell/Core Composite Nanoparticles
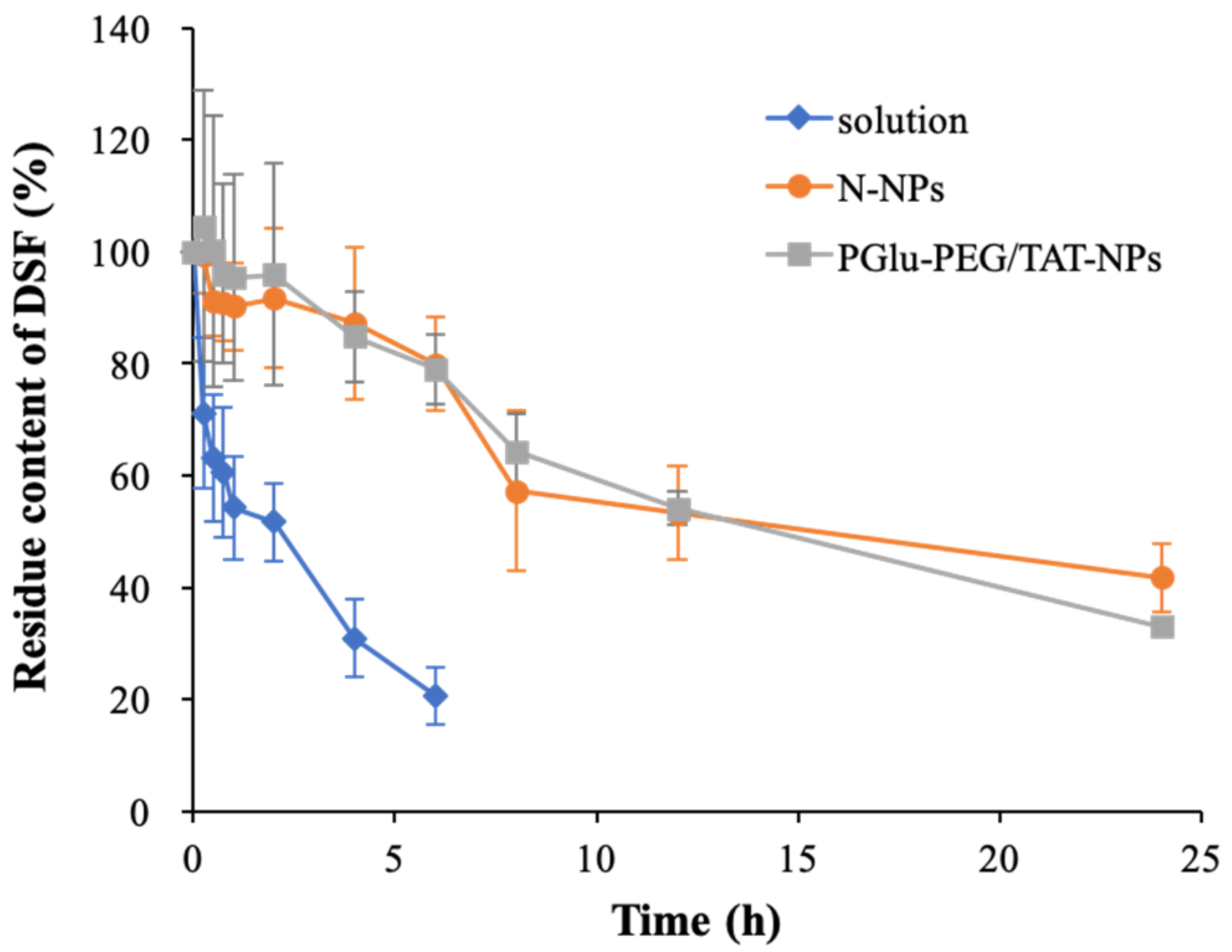
3.3. Plasma Stability Study
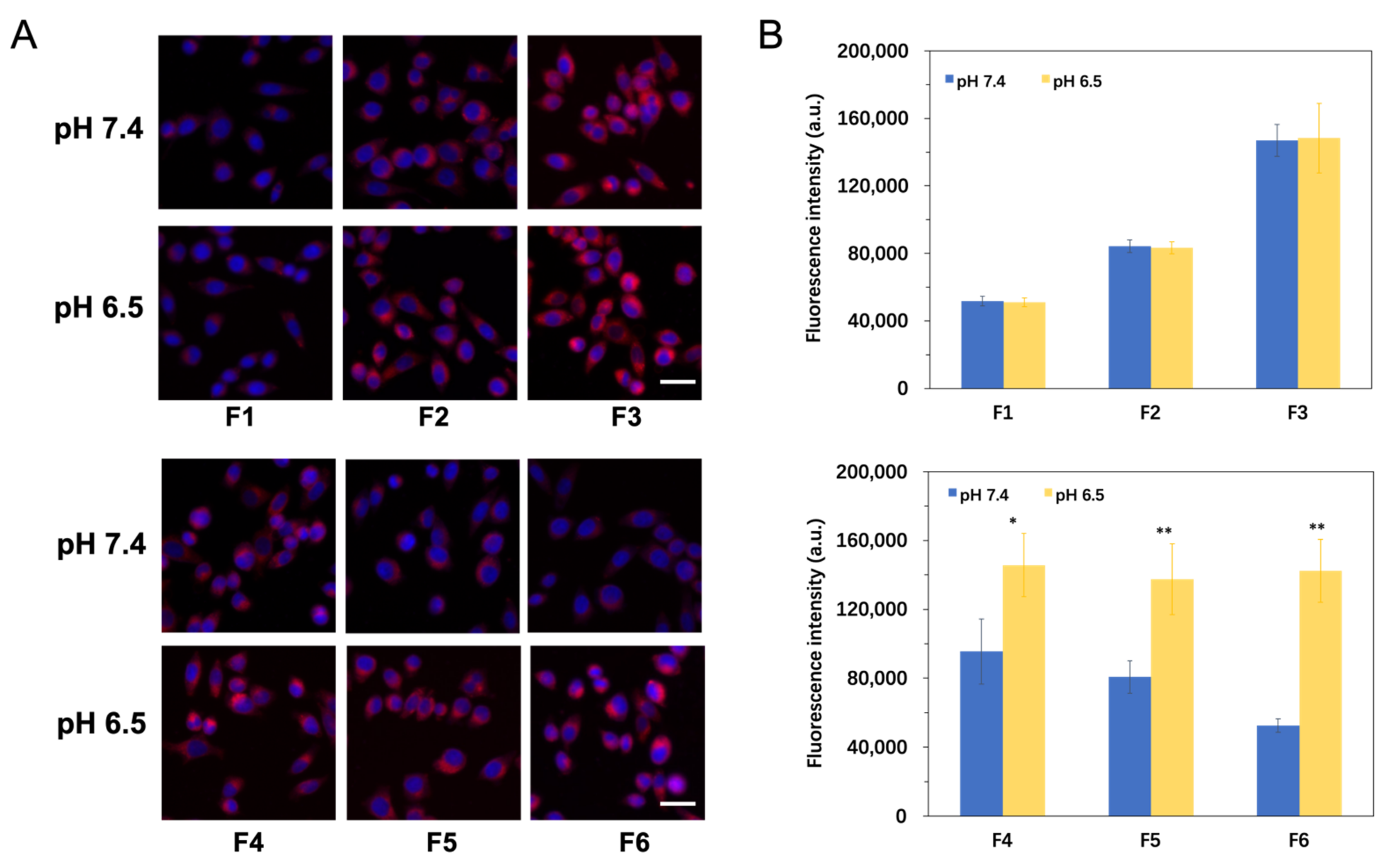
3.4. pH-Responsive Cellular Uptake Study
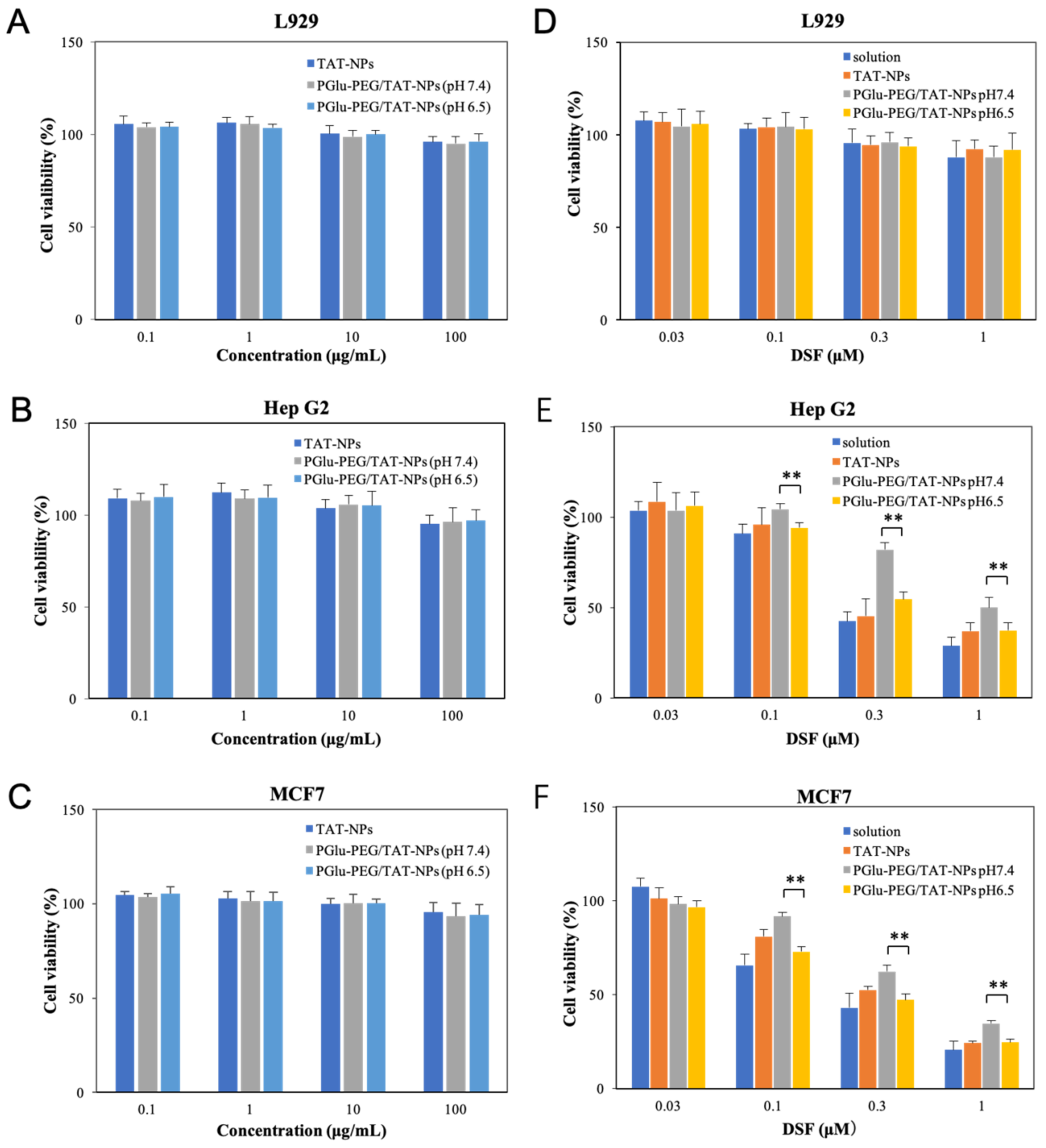
3.5. Cytotoxicity Study
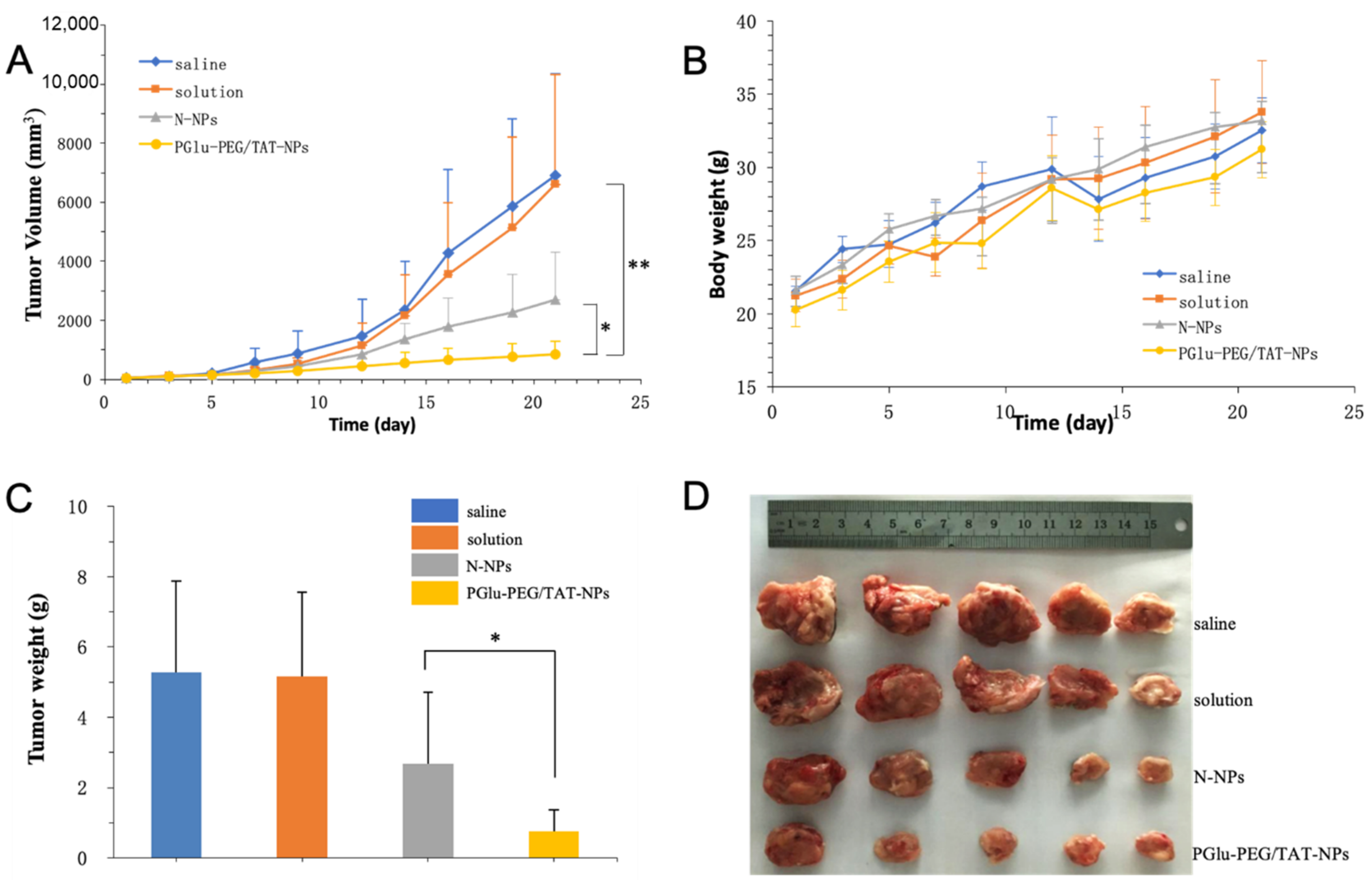
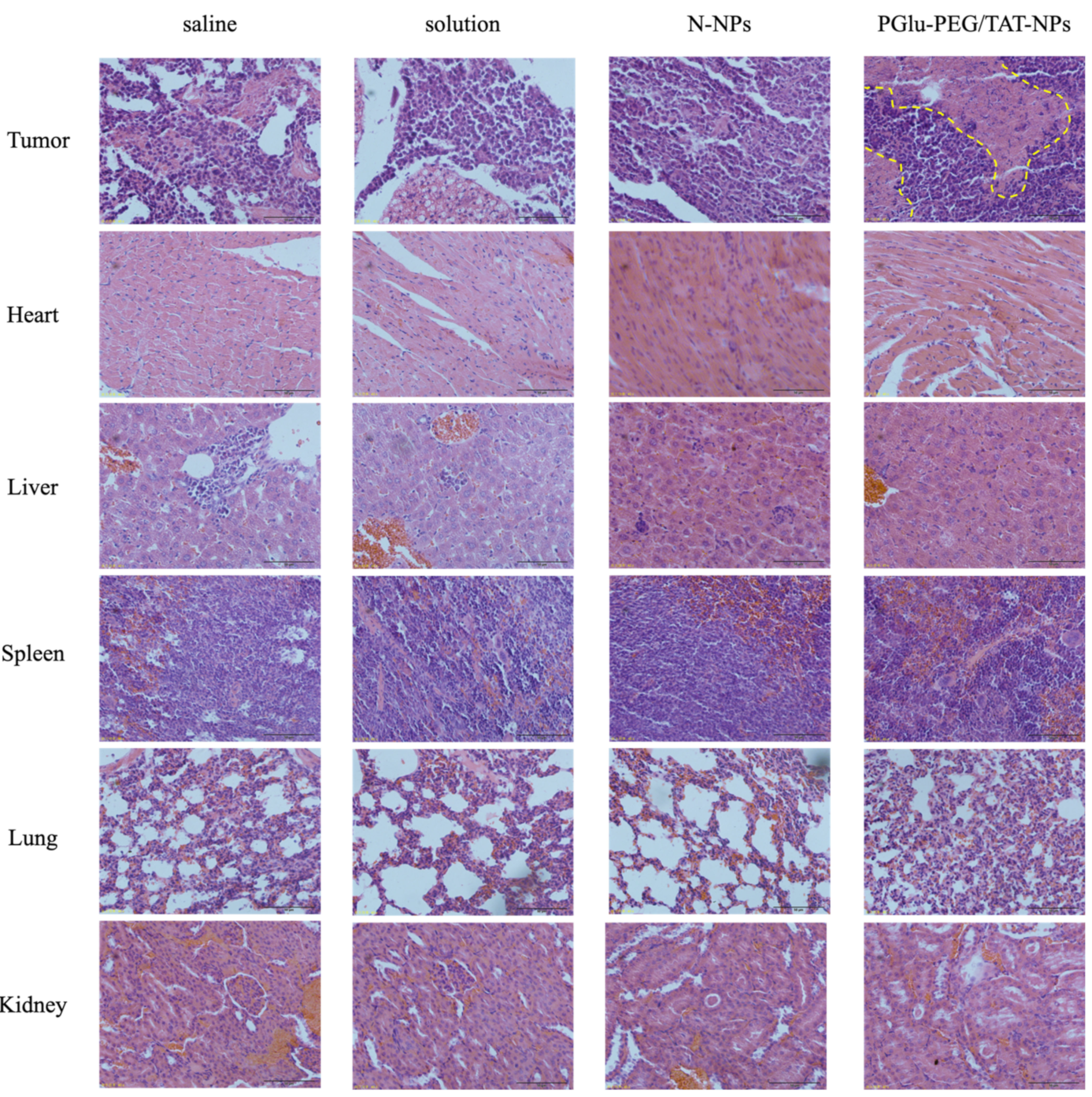
3.6. In Vivo Antitumor Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NDDS | nanoscale drug delivery systems |
| DSF | disulfiram |
| TME | tumor microenvironment |
| PGlu-PEG | poly (glutamate acid)-graft-poly (ethylene glycol) |
| HS-PEG-TAT | 12-hydroxystearic-poly(ethylene glycol)-TAT |
| N-NPs | naked nanoparticles |
| TAT-NPs | TAT-inserted nanoparticles |
| PGlu-PEG/TAT-NPs | shell/core composite nanoparticles |
| TEM | transmission electron microscopy |
| PBS | phosphate buffer |
| TIR | tumor inhibition rate |
| H&E | hematoxylin and eosin |
References
- Butcher, N.J.; Mortimer, G.M.; Minchin, R.F. Drug delivery: Unravelling the stealth effect. Nat. Nanotechnol. 2016, 11, 310–311. [Google Scholar] [CrossRef]
- Song, J.; Ju, Y.; Amarasena, T.H.; Lin, Z.; Mettu, S.; Zhou, J.; Rahim, M.A.; Ang, C.S.; Cortez-Jugo, C.; Kent, S.J.; et al. Influence of poly(ethylene glycol) molecular architecture on particle assembly and ex vivo particle-immune cell interactions in human blood. ACS Nano 2021. [Google Scholar] [CrossRef] [PubMed]
- Von Baeckmann, C.; Kahlig, H.; Linden, M.; Kleitz, F. On the importance of the linking chemistry for the PEGylation of mesoporous silica nanoparticles. J. Colloid Interface Sci. 2021, 589, 453–461. [Google Scholar] [CrossRef]
- Wang, J.; Ni, Q.; Wang, Y.; Zhang, Y.; He, H.; Gao, D.; Ma, X.; Liang, X.J. Nanoscale drug delivery systems for controllable drug behaviors by multi-stage barrier penetration. J. Control. Release 2021, 331, 282–295. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Raposo, L.R.; Valente, R.; Fernandes, A.R.; Baptista, P.V. Combined cancer therapeutics-Tackling the complexity of the tumor microenvironment. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2021, e1704. [Google Scholar] [CrossRef]
- Kong, L.; Campbell, F.; Kros, A. DePEGylation strategies to increase cancer nanomedicine efficacy. Nanoscale Horiz. 2019, 4, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yang, Q.; Shi, K.; Xiao, Y.; Wei, X.; Qian, Z. Intratumoral fate of functional nanoparticles in response to microenvironment factor: Implications on cancer diagnosis and therapy. Adv. Drug Deliv. Rev. 2019, 143, 37–67. [Google Scholar] [CrossRef]
- Jia, X.; He, J.; Shen, L.; Chen, J.; Wei, Z.; Qin, X.; Niu, D.; Li, Y.; Shi, J. Gradient redox-responsive and two-stage rocket-mimetic drug delivery system for improved tumor accumulation and safe Chemotherapy. Nano Lett. 2019, 19, 8690–8700. [Google Scholar] [CrossRef] [PubMed]
- Juang, V.; Chang, C.H.; Wang, C.S.; Wang, H.E.; Lo, Y.L. pH-Responsive PEG-shedding and targeting peptide-modified nanoparticles for dual-delivery of irinotecan and microRNA to enhance tumor-specific therapy. Small 2019, 15, e1903296. [Google Scholar] [CrossRef]
- Das, S.S.; Bharadwaj, P.; Bilal, M.; Barani, M.; Rahdar, A.; Taboada, P.; Bungau, S.; Kyzas, G.Z. Stimuli-responsive polymeric nanocarriers for drug delivery, imaging, and ttheragnosis. Polymers 2020, 12, 1397. [Google Scholar] [CrossRef]
- Niu, Z.; Samaridou, E.; Jaumain, E.; Coëne, J.; Ullio, G.; Shrestha, N.; Garcia, J.; Durán-Lobato, M.; Tovar, S.; Santander-Ortega, M.J.; et al. PEG-PGA enveloped octaarginine-peptide nanocomplexes: An oral peptide delivery strategy. J. Control. Release 2018, 276, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Shao, L.; Lu, J.; Deng, X.; Wu, Y. Tumor acidity-induced sheddable polyethylenimine-poly(trimethylene carbonate)/DNA/polyethylene glycol-2,3-dimethylmaleicanhydride ternary complex for efficient and safe gene delivery. ACS Appl. Mater. Interfaces 2016, 8, 6400–6410. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Wang, Y.; Zhou, X.; Gui, H.; Xu, N.; Wu, S.; He, C.; Zhao, Z. Tumor microenvironment targeting with dual stimuli-responsive nanoparticles based on small heat shock proteins for antitumor drug delivery. Acta Biomater. 2020, 114, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ruan, L.; Zheng, T.; Wang, D.; Zhou, M.; Lu, H.; Gao, J.; Chen, J.; Hu, Y. A surface convertible nanoplatform with enhanced mitochondrial targeting for tumor photothermal therapy. Colloids Surf. B Biointerfaces 2020, 189, 110854. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, Z.; Jiao, Z.; Lin, L.; Xu, C.; Tian, H.; Chen, X. Poly(l-glutamic acid)-based zwitterionic polymer in a charge conversional shielding system for gene therapy of malignant tumors. ACS Appl. Mater. Inter. 2020, 12, 19295–19306. [Google Scholar] [CrossRef]
- Xu, H.; Cai, C.; Gou, J.; Sui, B.; Jin, J.; Xu, H.; Zhang, Y.; Wang, L.; Zhai, Y.; Tang, X. Self-assembled monomethoxy (polyethylene glycol)-b-p(D,L-lactic-co-glycolic acid)-b-p(L-glutamic acid) hybrid-core nanoparticles for intracellular pH-triggered release of doxorubicin. J. Biomed. Nanotechnol. 2015, 11, 1354–1369. [Google Scholar] [CrossRef]
- Weng, H.; Bejjanki, N.K.; Zhang, J.; Miao, X.; Zhong, Y.; Li, H.; Xie, H.; Wang, S.; Li, Q.; Xie, M. TAT peptide-modified cisplatin-loaded iron oxide nanoparticles for reversing cisplatin-resistant nasopharyngeal carcinoma. Biochem. Bioph. Res. Commun. 2019, 511, 597–603. [Google Scholar] [CrossRef]
- Xu, J.; Khan, A.R.; Fu, M.; Wang, R.; Ji, J.; Zhai, G. Cell-penetrating peptide: A means of breaking through the physiological barriers of different tissues and organs. J. Control. Release 2019, 309, 106–124. [Google Scholar] [CrossRef]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef]
- Ekinci, E.; Rohondia, S.; Khan, R.; Dou, Q.P. Repurposing disulfiram as an anti-cancer agent: Updated review on literature and patents. Recent Pat. Anti Cancer Drug Discov. 2019, 14, 113–132. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Hu, X.; Lin, X.; Zhang, Y.; Tang, X. Formulation and preparation of a stable intravenous disulfiram-loaded lipid emulsion. Eur. J. Lipid. Sci. Tech. 2015, 117, 869–878. [Google Scholar] [CrossRef]
- Najlah, M.; Said Suliman, A.; Tolaymat, I.; Kurusamy, S.; Kannappan, V.; Elhissi, A.M.A.; Wang, W. Development of injectable PEGylated liposome encapsulating disulfiram for colorectal cancer treatment. Pharmaceutics 2019, 11, 610. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.; Lei, T.; Miao, L.; Chu, W.; Li, X.; Luo, L.; Gou, J.; Zhang, Y.; Yin, T.; He, H.; et al. Disulfiram-loaded mixed nanoparticles with high drug-loading and plasma stability by reducing the core crystallinity for intravenous delivery. J. Colloid Interf. Sci. 2018, 529, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Su, J.; Zhuo, X.; Luo, L.; Kong, Y.; Gou, J.; Yin, T.; Zhang, Y.; He, H.; Tang, X. mPEG5k-b-PLGA2k/PCL3.4k/MCT mixed micelles as carriers of disulfiram for improving plasma stability and antitumor effect in vivo. Mol. Pharmaceut. 2018, 15, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, B.; Ao, H.; Fu, J.; Wang, Y.; Feng, Y.; Guo, Y.; Wang, X. Soybean lecithin stabilizes disulfiram nanosuspensions with a high drug-loading content: Remarkably improved antitumor efficacy. J. Nanobiotechnol. 2020, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Fasehee, H.; Dinarvand, R.; Ghavamzadeh, A.; Esfandyari-Manesh, M.; Moradian, H.; Faghihi, S.; Ghaffari, S.H. Delivery of disulfiram into breast cancer cells using folate-receptor-targeted PLGA-PEG nanoparticles: In vitro and in vivo investigations. J. Nanobiotechnol. 2016, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, J.; Fu, L.; Ran, R.; Liu, Y.; Yuan, M.; He, Q. A pH-responsive α-helical cell penetrating peptide-mediated liposomal delivery system. Biomaterials 2013, 34, 7980–7993. [Google Scholar] [CrossRef]
- Tao, X.; Gou, J.; Zhang, Q.; Tan, X.; Ren, T.; Yao, Q.; Tian, B.; Kou, L.; Zhang, L.; Tang, X. Synergistic breast tumor cell killing achieved by intracellular co-delivery of doxorubicin and disulfiram via core-shell-corona nanoparticles. Biomater. Sci. 2018, 6, 1869–1881. [Google Scholar] [CrossRef]
- Perrier, T.; Saulnier, P.; Fouchet, F.; Lautram, N.; Benoît, J.-P. Post-insertion into lipid nanocapsules (LNCs): From experimental aspects to mechanisms. Int. J. Pharm. 2010, 396, 204–209. [Google Scholar] [CrossRef]
- Rizzuti, M.; Nizzardo, M.; Zanetta, C.; Ramirez, A.; Corti, S. Therapeutic applications of the cell-penetrating HIV-1 Tat peptide. Drug Discov. Today 2015, 20, 76–85. [Google Scholar] [CrossRef]
- Gou, J.; Liang, Y.; Miao, L.; Guo, W.; Chao, Y.; He, H.; Zhang, Y.; Yang, J.; Wu, C.; Yin, T.; et al. Improved tumor tissue penetration and tumor cell uptake achieved by delayed charge reversal nanoparticles. Acta Biomater. 2017, 62, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B. A review of the pharmacokinetics and pharmacodynamics of disulfiram and its metabolites. Acta Psychiat. Scand. 1992, 369, 15–26. [Google Scholar] [CrossRef]
- Sun, H.; Liu, S.; Gao, X.; Xiong, Z.; He, Z.; Zhao, L. Study on degradation kinetics of epalrestat in aqueous solutions and characterization of its major degradation products under stress degradation conditions by UHPLC-PDA-MS/MS. J. Pharm. Anal. 2019, 9, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Therapeut 2015, 154, 78–86. [Google Scholar] [CrossRef]
- Lo, Y.L.; Chang, C.H.; Wang, C.S.; Yang, M.H.; Lin, A.M.; Hong, C.J.; Tseng, W.H. PEG-coated nanoparticles detachable in acidic microenvironments for the tumor-directed delivery of chemo- and gene therapies for head and neck cancer. Theranostics 2020, 10, 6695–6714. [Google Scholar] [CrossRef]
- Wu, W.; Yu, L.; Jiang, Q.; Huo, M.; Lin, H.; Wang, L.; Chen, Y.; Shi, J. Enhanced tumor-specific disulfiram chemotherapy by in situ Cu(2+) chelation-initiated nontoxicity-to-toxicity transition. J. Am. Chem. Soc. 2019, 141, 11531–11539. [Google Scholar] [CrossRef] [PubMed]
- Falls-Hubert, K.C.; Butler, A.L.; Gui, K.; Anderson, M.; Li, M.; Stolwijk, J.M.; Rodman, S.N., 3rd; Solst, S.R.; Tomanek-Chalkley, A.; Searby, C.C.; et al. Disulfiram causes selective hypoxic cancer cell toxicity and radio-chemo-sensitization via redox cycling of copper. Free Radic Biol. Med. 2020, 150, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, Y.; Jing, G.; Tang, Y.; Chen, X.; Yang, D.; Zhang, Y.; Tang, X. A novel UPLC-ESI-MS/MS method for the quantitation of disulfiram, its role in stabilized plasma and its application. J. Chromatogr. B 2013, 937, 54–59. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | F1 | F2 | F3 | F4 | F5 | F6 |
|---|---|---|---|---|---|---|
| TAT a (%) | 0 | 0.6 | 1.2 | 1.2 | 1.2 | 1.2 |
| PGlu-PEG b (%) | 0 | 0 | 0 | 2 | 5 | 10 |
| Particle size (nm) | 58.5 ± 1.2 | 63.3 ± 1.8 | 65.4 ± 2.7 | 68.3 ± 3.4 | 74.2 ± 2.1 | 87.4 ± 5.8 |
| Zeta potential (mV) | −16.43 ± 1.22 | 3.03 ± 2.43 | 5.85 ± 2.28 | −2.64 ± 2.16 | −11.63 ± 3.48 | −38.38 ± 2.15 |
| DL (%) | 5.16 ± 0.37 | 5.13 ± 0.44 | 5.26 ± 0.52 | 4.81 ± 0.45 | 4.93 ± 0.56 | 4.87 ± 0.24 |
| EE (%) | 94.7 ± 4.6 | 93.5 ± 5.7 | 93.7 ± 4.2 | 94.2 ± 3.9 | 94.6 ± 5.8 | 94.2 ± 4.0 |
| Formulation | First-Order Kinetic Equation | R | K (h−1) | t1/2 (h) |
|---|---|---|---|---|
| Solution a | ln(ct/c0) = −0.0979t + 1.89 | 0.970 | 0.225 | 3.07 |
| N-NPs b | ln(ct/c0) = −0.162t + 1.98 | 0.977 | 0.037 | 18.6 |
| PGlu-PEG/TAT-NPs c | ln(ct/c0) = −0.0208t + 2.00 | 0.993 | 0.048 | 14.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Q.; Shi, W.; Wang, P.; Zhang, Y.; Meng, J.; Zhang, L. Tumor Microenvironment-Responsive Shell/Core Composite Nanoparticles for Enhanced Stability and Antitumor Efficiency Based on a pH-Triggered Charge-Reversal Mechanism. Pharmaceutics 2021, 13, 895. https://doi.org/10.3390/pharmaceutics13060895
Luo Q, Shi W, Wang P, Zhang Y, Meng J, Zhang L. Tumor Microenvironment-Responsive Shell/Core Composite Nanoparticles for Enhanced Stability and Antitumor Efficiency Based on a pH-Triggered Charge-Reversal Mechanism. Pharmaceutics. 2021; 13(6):895. https://doi.org/10.3390/pharmaceutics13060895
Chicago/Turabian StyleLuo, Qiuhua, Wen Shi, Puxiu Wang, Yu Zhang, Jia Meng, and Ling Zhang. 2021. "Tumor Microenvironment-Responsive Shell/Core Composite Nanoparticles for Enhanced Stability and Antitumor Efficiency Based on a pH-Triggered Charge-Reversal Mechanism" Pharmaceutics 13, no. 6: 895. https://doi.org/10.3390/pharmaceutics13060895
APA StyleLuo, Q., Shi, W., Wang, P., Zhang, Y., Meng, J., & Zhang, L. (2021). Tumor Microenvironment-Responsive Shell/Core Composite Nanoparticles for Enhanced Stability and Antitumor Efficiency Based on a pH-Triggered Charge-Reversal Mechanism. Pharmaceutics, 13(6), 895. https://doi.org/10.3390/pharmaceutics13060895