
Pharmacokinetic Drug Interaction between Tofacitinib and Voriconazole in Rats


Abstract

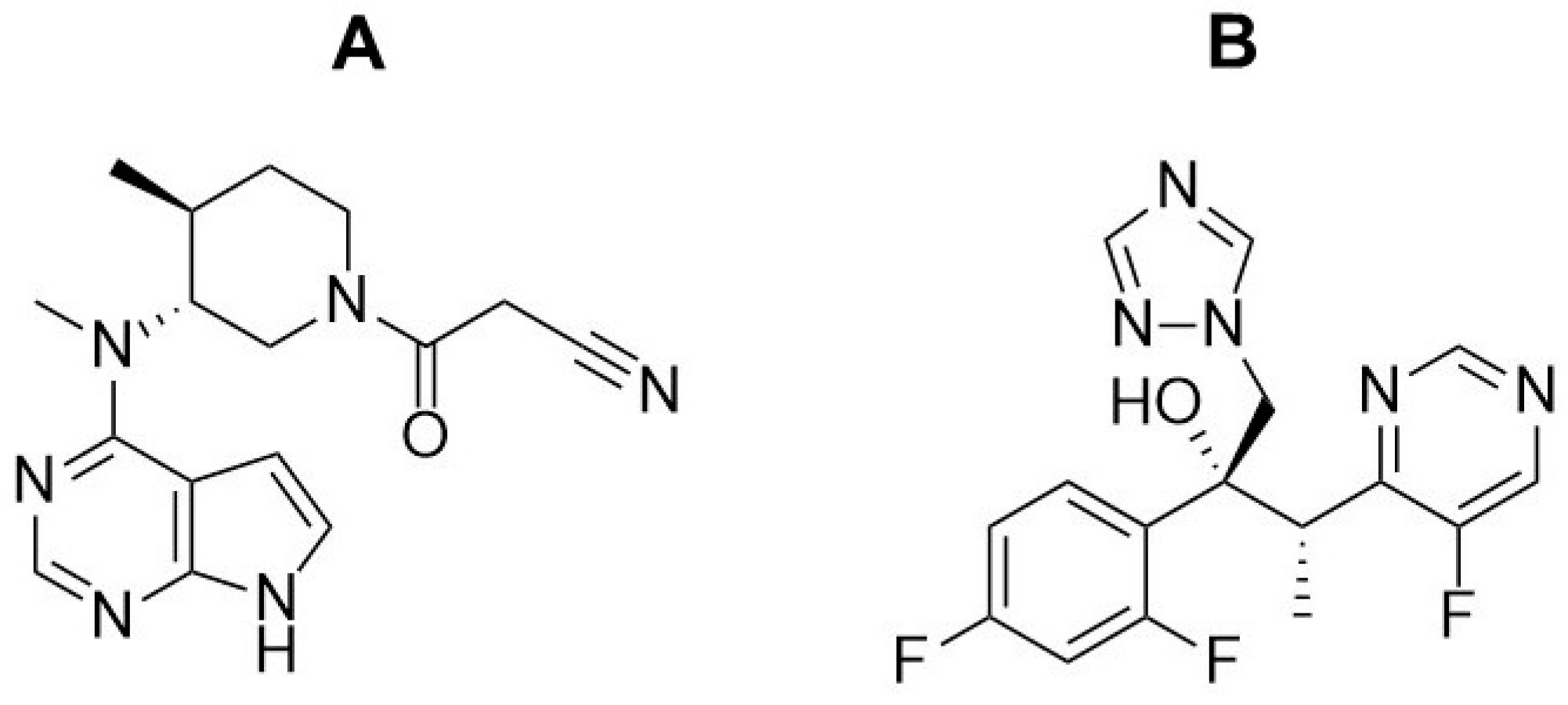
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Intravenous and Oral Administration of Tofacitinib and Voriconazole
2.4. Metabolic Inhibition of Tofacitinib by Voriconazole in Rat Hepatic Microsomes
2.5. Estimation of Enzyme Activity in Rat Hepatic and Intestinal Microsomes
2.6. Estimation of Protein Binding of Tofacitinib to Rat Plasma in the Absence or Presence of Voriconazole
2.7. Measurement of Hepatic and/or Intestinal Concentrations of Voriconazole after Intravenous and Oral Administration of Both Drugs
2.8. Immunoblot Analysis
2.9. HPLC Analysis of Voriconazole and Tofacitinib
2.10. Pharmacokinetic Analysis
2.11. Statistical Analysis
3. Results
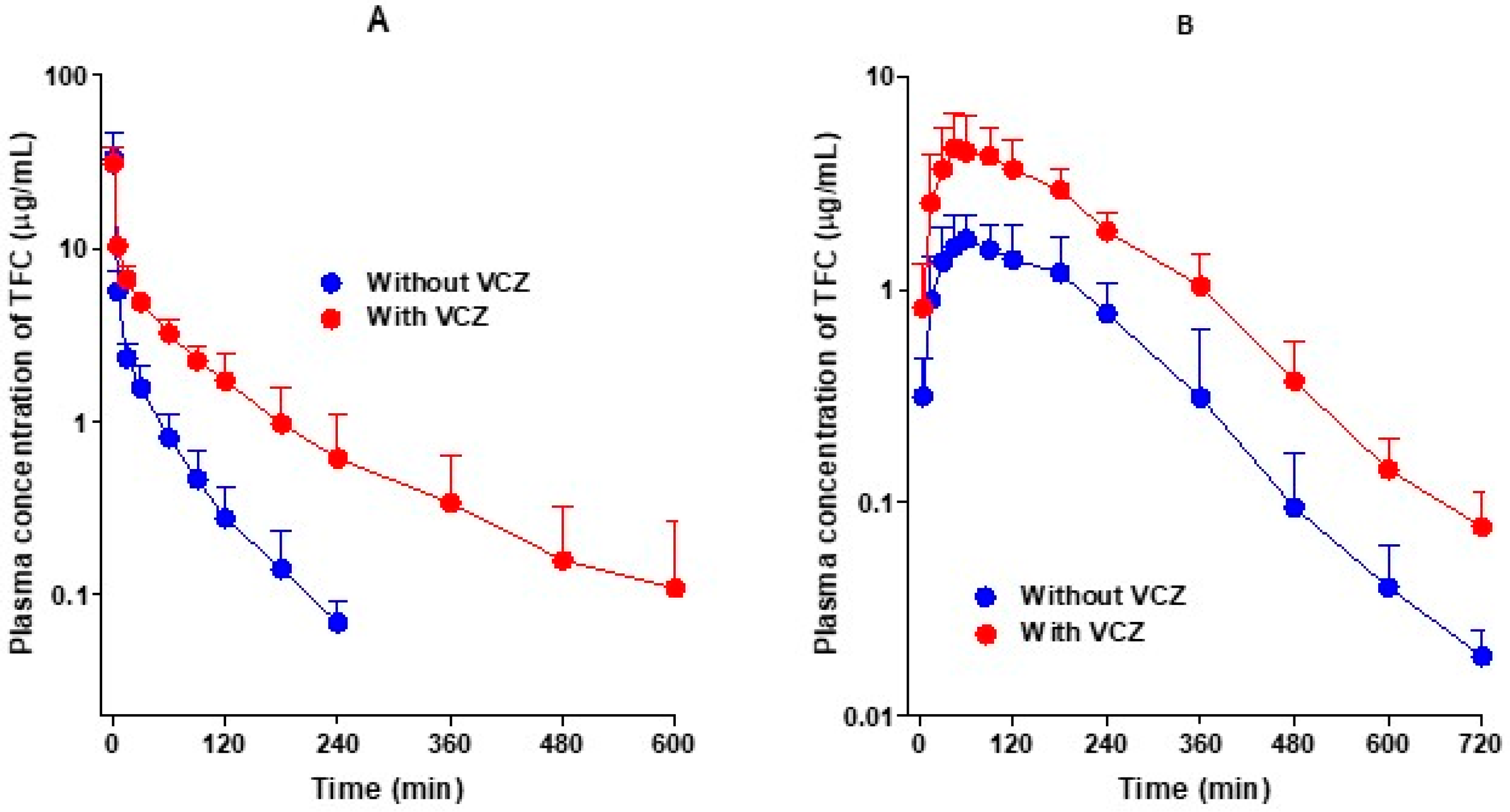
3.1. Pharmacokinetics of Tofacitinib after Intravenous and Oral Administration of Tofacitinib without and with Voriconazole
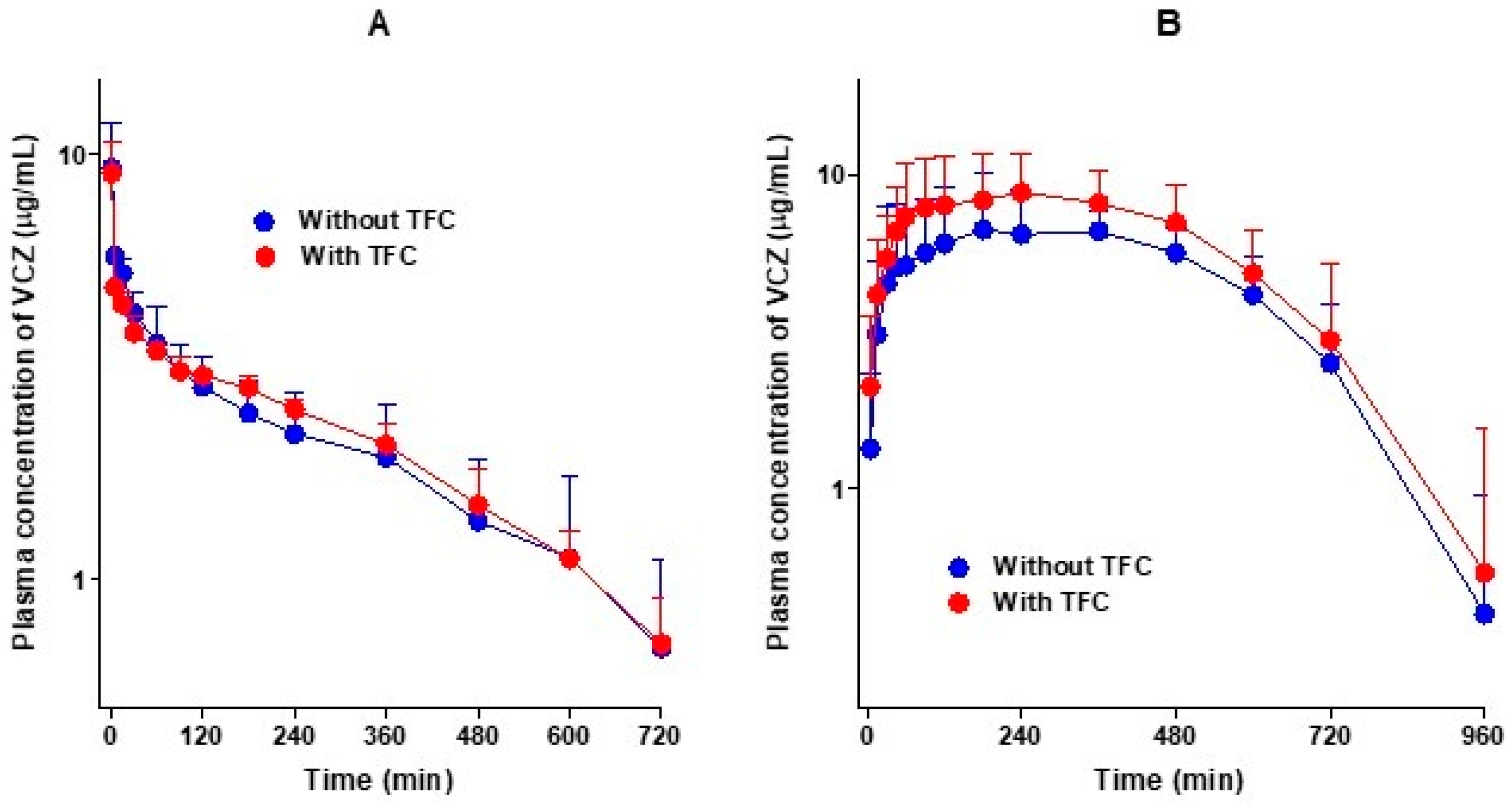
3.2. Pharmacokinetics of Voriconazole after Intravenous and Oral Administration of Voriconazole without and with Tofacitinib
3.3. Non-Competitive Inhibition of Tofacitinib Metabolism by Voriconazole in Rat Hepatic Microsomes
3.4. Vmax, Km and CLint for the Elimination of Tofacitinib in the Absence or Presence of Voriconazole and Apparent Ki of Voriconazole for the Inhibition of Tofacitinib Metabolism in Rat Hepatic and Intestinal Microsomes
3.5. Rat Plasma Protein Binding of Tofacitinib without and with Voriconazole to Fresh Rat Plasma
3.6. Hepatic and Intestinal Concentrations of Voriconazole after Intravenous and Oral Co-Administration of Both Tofacitinib and Voriconazole
3.7. Protein Expression of CYP2C11 and CYP3A1/2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Appendix A. Preparation of Rat Hepaptic and Intestinal Microsomes
References
- Fleischmann, R.; Kremer, J.; Cush, J.; Schulze-Koops, H.; Connell, C.A.; Bradley, J.D.; Gruben, D.; Wallenstein, G.V.; Zwillich, S.H.; Kanik, K.S. ORAL Solo Investigators. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.M.; Jesson, M.I.; Li, X.; Elrick, M.M.; Funckes-Shippy, C.L.; Warner, J.D.; Gross, C.J.; Dowty, M.E.; Ramaiah, S.K.; Hirsch, J.L.; et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J. Inflamm. 2010, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Sanati, H.; Belanger, P.; Fratti, R.; Ghannoum, M. A new triazole, voriconazole (UK-109,496), blocks sterol biosynthesis in Candida albicans and Candida krusei. Antimicrob. Agents Chemother. 1997, 41, 2492–2496. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Ghosh, S.; Panés, J.; Vranic, I.; Su, C.; Rousell, S.; Niezychowski, W. Study A3921063 Investigators. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Su, C.; Sands, B.E.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Danese, S.; Feagan, B.G.; Reinisch, W.; Niezychowski, W.; et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2017, 376, 1723–1736. [Google Scholar] [CrossRef]
- Palasik, B.N.; Wang, H. Tofacitinib, the first oral Janus kinase inhibitor approved for adult ulcerative colitis. J. Pharm. Pract. 2020, 897190020953019. [Google Scholar] [CrossRef] [PubMed]
- Dowty, M.E.; Lin, J.; Ryder, T.F.; Wang, W.; Walker, G.S.; Vaz, A.; Prakash, C. The pharmacokinetics, metabolism, and clearancemechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab. Dispos. 2014, 42, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Cada, D.J.; Demaris, K.; Levien, T.L.; Baker, D.E. Tofacitinib. Hosp. Pharm. 2013, 48, 413–424. [Google Scholar] [CrossRef]
- Scott, L.J. Tofacitinib: A review of its use in adult patients with rheumatoid arthritis. Drugs 2013, 73, 857–874. [Google Scholar] [CrossRef]
- Vallabhaneni, S.; Chiller, T.M. Fungal infections and new biologic therapies. Curr. Rheumatol. Rep. 2016, 18, 29. [Google Scholar] [CrossRef]
- Chen, Y.; Gong, F.Y.; Li, Z.J.; Gong, Z.; Zhou, Z.; Ma, S.Y.; Gao, X.M. A study on the risk of fungal infection with tofacitinib (CP-690550), a novel oral agent for rheumatoid arthritis. Sci. Rep. 2017, 7, 6779. [Google Scholar] [CrossRef]
- Winthrop, K.L.; Park, S.H.; Gul, A.; Cardiel, M.H.; Gomez-Reino, J.J.; Tanake, Y.; Kwok, K.; Lukic, J.; Mortensen, E.; Ponce, D.; et al. Tuberculosis and other opportunistic infections in tofacitinib-treated patients with rheumatoid arthritis. Ann. Rheum. Dis. 2015, 75, 1133–1138. [Google Scholar] [CrossRef]
- Cohen, S.B.; Tanaka, Y.; Mariette, X.; Curtis, J.R.; Lee, E.B.; Nash, P.; Winthrop, K.L.; Charles-Schoeman, C.; Thirunavukkarasu, K.; Demasi, R.; et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: Integrated analysis of data from the global clinical trials. Ann. Rheum. Dis. 2017, 76, 1253–1262. [Google Scholar] [CrossRef]
- Cohen, S.; Curtis, J.R.; DeMasi, R.; Chen, Y.; Fan, H.; Soonasra, A.; Fleischmann, R. Worldwide, 3-year, post-marketing surveillance experience with tofacitinib in rheumatoid arthritis. Rheumatol. Ther. 2018, 5, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Misch, E.A.; Safdar, N. Updated guidelines for the diagnosis and management of aspergillosis. J. Thorac. Dis. 2016, 8, E1771–E1776. [Google Scholar] [CrossRef] [PubMed]
- Rengelshausen, J.; Banfield, M.; Riedel, K.D.; Burhenne, J.; Weiss, J.; Thomsen, T.; Walter-Sack, I.; Haefeli, W.E.; Mikus, G. Opposite effects of short-term and long-term St John’s wort intake on voriconazole pharmacokinetics. Clin. Pharmacol. Ther. 2005, 78, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Levéque, D.; Nivoix, Y.; Jehl, F.; Herbrecht, R. Clinical pharmacokinetics of voriconazole. Int. J. Antimicrob. Agent 2006, 27, 274–284. [Google Scholar] [CrossRef]
- Hohmann, N.; Kocheise, F.; Carls, A.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Mikus, G. Dose-dependent bioavailability and CYP3A inhibition contribute to non-linear pharmacokinetics of voriconazole. Clin. Pharmacokinet. 2016, 55, 1535–1545. [Google Scholar] [CrossRef]
- Howard, A.; Hoffman, J.; Sheth, A. Clinical application of voriconazole concentrations in the treatment of invasive aspergillosis. Ann. Pharmacother. 2008, 42, 1859–1864. [Google Scholar] [CrossRef]
- Scholz, I.; Oberwittler, H.; Riedel, K.D.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Mikus, G. Pharmacokinetics, metabolism and bioavailability of the triazole antifungal agent voriconazole in relation to CYP2C19 genotype. Br. J. Clin. Pharmacol. 2009, 68, 906–915. [Google Scholar] [CrossRef]
- Gwak, E.H.; Yoo, H.Y.; Kim, S.H. Effects of diabetes mellitus on the disposition of tofacitinib, a Janus kinase inhibitor, in rats. Biomol. Ther. 2020, 28, 361–369. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, S.H. Dose-dependent pharmacokinetics of tofacitinib in rats: Influence of hepatic and intestinal first-pass metabolism. Pharmaceutics 2019, 11, 318. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Choi, Y.M.; Lee, M.G. Pharmacokinetics and pharmacodynamics of furosemide in protein-calorie malnutrition. J. Pharmacokinet. Biopharm. 1993, 21, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Michael, C.; Teicher, J.; Preiss, R. Determination of voriconazole in human plasma and saliva using high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B 2008, 865, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, M.Y.; Kim, S.H. Simple determination and quantifcation of tofacitinib, a JAK inhibitor, in rat plasma, urine and tissue homogenates by HPLC and its application to a pharmacokinetic study. J. Pharm. Investig. 2020, 50, 603–612. [Google Scholar] [CrossRef]
- Choi, Y.H.; Lee, U.; Lee, B.K.; Lee, M.G. Pharmacokinetic interaction between itraconazole and metformin in rats: Competitive inhibition of metabolism of each drug by each other via hepatic and intestinal CYP3A1/2. Br. J. Pharmacol. 2010, 161, 815–829. [Google Scholar] [CrossRef]
- Peng, J.Z.; Remmel, R.P.; Sawchuk, R.K. Inhibition of murine cytochrome P4501A by tacrine: In vitro studies. Drug Metab. Dispos. 2004, 32, 805–812. [Google Scholar] [CrossRef]
- Duggleby, R.G. Analysis of enzyme progress curves by nonlinear regression. Methods Enzymol. 1995, 249, 61–90. [Google Scholar]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; Wiley: New York, NY, USA, 1993. [Google Scholar]
- Yang, S.H.; Suh, J.H.; Lee, M.G. Pharmacokinetic interaction between tamoxifen and ondansetron in rats: Non-competitive (hepatic) and competitive (intestinal) inhibition of tamoxifen metabolism by ondansetron via CYP2D subfamily and 3A1/2. Cancer Chemother. Pharmacol. 2010, 65, 407–418. [Google Scholar] [CrossRef]
- Bae, S.H.; Chang, S.Y.; Kim, S.H. Slower elimination of tofacitinib in acute renal failure rat models: Contribution of hepatic metabolism and renal excretion. Pharmaceutics 2020, 12, 714. [Google Scholar] [CrossRef]
- Gibaldi, M.; Perrier, D. Pharmacokinetics, 2nd ed.; Marcel-Dekker: New York, NY, USA, 1982. [Google Scholar]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef]
- Bechmann, K.A.; Lewis, J.D. Predicting inhibitory drug–drug interactions and evaluating drug interaction reports using inhibition constants. Ann. Pharmacother. 2005, 39, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Roffey, S.J.; Cole, S.; Comby, P.; Gibson, D.; Jezequel, S.G.; Nedderman, A.N.R.; Smith, D.A.; Walker, D.K.; Wood, N. The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog, and human. Drug Metab. Dispos. 2003, 31, 931. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.R.; Shand, D.G. A physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 1975, 18, 377–390. [Google Scholar] [CrossRef]
- Lee, M.G.; Chiou, W.L. Evaluation of potential causes for the incomplete bioavailability of furosemide: Gastric first-pass metabolism. J. Pharmacokinet. Biopharm. 1983, 11, 623–640. [Google Scholar] [CrossRef]
- Yau, E.; Petersson, C.; Dolgos, H.; Peters, S.A. A comparative evaluation of models to predict uman intestinal metabolism from nonclinical data. Biopharm. Drug Dispos. 2017, 38, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Paine, M.R.; Hart, H.L.; Ludington, S.S.; Haining, R.L.; Rettie, A.E.; Zeldin, D.C. The human intestinal cytochrome P450 ‘pie’. Drug. Metab. Dispos. 2006, 34, 880–886. [Google Scholar] [CrossRef]
- Amrhein, J.; Drynda, S.; Schlatt, L.; Karst, U.; Lohmann, C.H.; Ciarimboli, G.; Bertrand, J. Tofacitinib and baricitinib are taken up by different uptake mechanisms determining the efficacy of both drugs in RA. Int. J. Mol. Sci. 2020, 21, 6632. [Google Scholar] [CrossRef]
- Shen, J.; Wang, B.; Wang, S.; Chen, F.; Meng, D.; Jiang, H.; Zhou, Y.; Geng, P.; Zhou, Q.; Liu, B. Effects of voriconazole on the pharmacokinetics of vonoprazan in rats. Drug Design Dev. Ther. 2020, 14, 2199–2206. [Google Scholar] [CrossRef]
- Topal, F. Inhibition profiles of voriconazole against acetylcholinesterase, α-glycosidase, and human carbonic anhydrase I and II isoenzymes. J. Biochem. Mol. Toxicol. 2019, 33, e22385. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Chow, V.; Wang, R.; Kaplan, I.; Chan, G.; Alvey, C.; Ni, G.; Ndongo, N.-N.; LaBadie, R.; Krishnaswami, S. Evaluation of the effect of fluconazole and ketoconazole on the pharmacokinetics of tofacitinib in healthy adult subjects. Clin. Pharmacol. Drug Dev. 2013, 3, 72–77. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Intravenous | Oral | ||
|---|---|---|---|---|
| Without VCZ (n = 5) | With VCZ (n = 6) | Without VCZ (n = 6) | With VCZ (n = 7) | |
| Body weight (g) | 277 ± 4.55 | 283 ± 10.4 | 279 ± 6.15 | 282 ± 7.06 |
| AUC (µg·min/mL) | 278 ± 76.6 | 740 ± 154 *** | 396 ± 61.6 | 1074 ± 271 *** |
| Cmax (µg/mL) | 1.98 ± 0.405 | 4.46 ± 2.00 * | ||
| Tmax (min) | 87.5 ± 52.3 | 62.5 ± 29.1 | ||
| Terminal half-life (min) | 43.2 ± 7.45 | 104 ± 30.2 ** | ||
| MRT (min) | 32.8 ± 13.4 | 105 ± 46.7 ** | ||
| CL (mL/min/kg) | 37.9 ± 8.87 | 14.1 ± 3.16 *** | ||
| CLR (mL/min/kg) | 3.61 ± 0.829 | 0.144 ± 0.0678 *** | 0.536 ± 0.135 | 0.310 ± 0.117 ** |
| CLNR (mL/min/kg) | 34.3 ± 8.10 | 13.9 ± 3.18 *** | ||
| Vss (mL/kg) | 1211 ± 489 | 1292 ± 193 | ||
| Ae0–24 h (% of dose) | 9.60 ± 1.00 | 10.9 ± 6.25 | 10.6 ± 3.49 | 16.3 ± 5.74 * |
| GI24 h (% of dose) | 0.411 ± 0.313 | 0.912 ± 0.527 | ||
| F (%) | 71.2 | 72.4 | ||
| Parameters | Intravenous | Oral | ||
|---|---|---|---|---|
| Without TFC (n = 6) | With TFC (n = 5) | Without TFC (n = 6) | With TFC (n = 7) | |
| Body weight (g) | 298 ± 14.9 | 283 ± 11.5 | 274 ± 4.59 | 283 ± 8.25 |
| AUC (µg·min/mL) | 1862 ± 600 | 1804 ± 436 | 3938 ± 416 | 4415 ± 760 |
| Cmax (µg/mL) | 7.82 ± 1.13 | 8.97 ± 1.73 | ||
| Tmax (min) | 263 ± 201 | 283 ± 234 | ||
| Terminal half-life (min) | 314 ± 96.7 | 296 ± 110 | ||
| MRT (min) | 422 ± 132 | 400 ± 149 | ||
| CL (mL/min/kg) | 6.01 ± 2.47 | 5.77 ± 1.18 | ||
| CLR (mL/min/kg) | 0.028 ± 0.015 | 0.022 ± 0.015 | 0.37 ± 0.24 | 0.096 ± 0.046 * |
| CLNR (mL/min/kg) | 5.98 ± 2.46 | 5.74 ± 1.17 | ||
| Vss (mL/kg) | 2275 ± 159 | 5169 ± 248 | ||
| Ae0–24 h (% of dose) | 4.54 ± 1.30 | 3.70 ± 2.43 | 7.48 ± 5.15 | 2.17 ± 1.26 * |
| GI24 h (% of dose) | 0.041 ± 0.017 | 1.03 ± 0.911 * | ||
| F (%) | 106 | 122 | ||
| Parameter | Withou VCZ (n = 3) | With VCZ (n = 3) |
|---|---|---|
| Liver | ||
| Km (μM) | 124 ± 13.7 | 147 ±43.4 |
| Vmax (pmol/min/mg protein) | 2895 ± 319 | 1184 ± 470 *** |
| CLint (mL/min/mg protein) | 0.0233 ± 0.000689 | 0.00805 ± 0.00201 *** |
| Ki (μM) | 6.52 ± 0.828 | |
| Intestine | ||
| Km (μM) | 43.1 ± 11.7 | 30.1 ± 3.29 |
| Vmax (pmol/min/mg protein) | 380 ± 86.1 | 212 ± 27.0 * |
| CLint (mL/min/mg protein) | 0.00894 ± 0.00100 | 0.00705 ± 0.000120 * |
| Ki (μM) | 26.2 ± 8.05 |
| Intravenous | Oral | ||||||
|---|---|---|---|---|---|---|---|
| Time (min) | Liver Concentration (μM) | [I]/Ki | Time (min) | Liver Concentration (μM) | [I]/Ki | Intestine Concentration (μM) | [I]/Ki |
| 1 | 24.0 ± 9.86 | 3.68 | 5 | 31.7 ± 7.91 | 4.86 | 11.7 ± 8.11 | 0.447 |
| 5 | 14.5 ± 8.31 | 2.22 | 15 | 22.1 ± 4.05 | 3.39 | 4.13 ± 1.65 | 0.158 |
| 15 | 16.7 ± 4.08 | 2.56 | 30 | 35.8 ± 5.22 | 5.49 | 10.2 ± 9.13 | 0.389 |
| 30 | 13.7 ± 2.68 | 2.10 | 60 | 27.6 ± 5.07 | 4.23 | 4.63 ± 0.318 | 0.177 |
| 60 | 12.8 ± 4.51 | 1.96 | 120 | 28.9 ± 3.01 | 4.43 | 5.08 ± 0.863 | 0.194 |
| 120 | 15.0 ± 1.01 | 2.30 | 180 | 32.9 ± 3.36 | 5.05 | 4.93 ± 2.21 | 0.188 |
| 240 | 27.5 ± 4.90 | 4.22 | 4.57 ± 0.664 | 0.174 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-S.; Kim, H.-S.; Jung, Y.-S.; Choi, H.-G.; Kim, S.-H. Pharmacokinetic Drug Interaction between Tofacitinib and Voriconazole in Rats. Pharmaceutics 2021, 13, 740. https://doi.org/10.3390/pharmaceutics13050740
Lee J-S, Kim H-S, Jung Y-S, Choi H-G, Kim S-H. Pharmacokinetic Drug Interaction between Tofacitinib and Voriconazole in Rats. Pharmaceutics. 2021; 13(5):740. https://doi.org/10.3390/pharmaceutics13050740
Chicago/Turabian StyleLee, Ji-Sang, Hyo-Sung Kim, Yong-Seob Jung, Hyeon-Gyeom Choi, and So-Hee Kim. 2021. "Pharmacokinetic Drug Interaction between Tofacitinib and Voriconazole in Rats" Pharmaceutics 13, no. 5: 740. https://doi.org/10.3390/pharmaceutics13050740
APA StyleLee, J.-S., Kim, H.-S., Jung, Y.-S., Choi, H.-G., & Kim, S.-H. (2021). Pharmacokinetic Drug Interaction between Tofacitinib and Voriconazole in Rats. Pharmaceutics, 13(5), 740. https://doi.org/10.3390/pharmaceutics13050740