
Estimating the Oral Absorption from Self-Nanoemulsifying Drug Delivery Systems Using an In Vitro Lipolysis-Permeation Method



Abstract
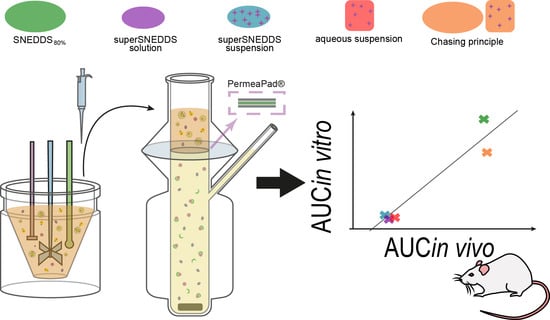
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Media Preparation
2.2.2. Preparation of Cinnarizine Formulations
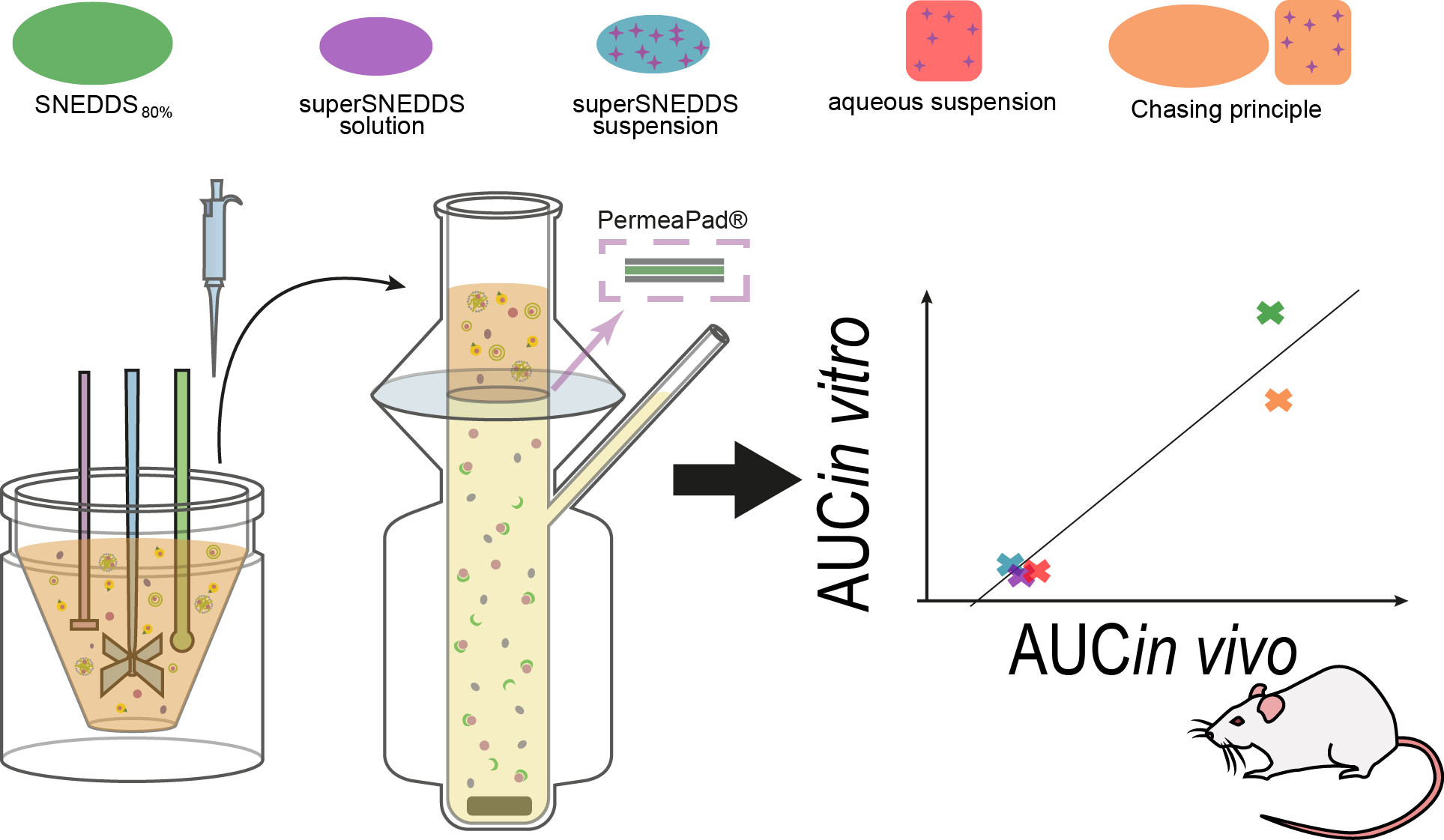
2.2.3. The Lipolysis-Permeation Method
Lipolysis Step
Sample Treatment
Permeation Step
Stability of the PermeaPad® Barrier
Cinnarizine Solubility
2.2.4. Quantification Methods
2.2.5. Data Processing
3. Results and Discussions
3.1. Reproducing In Vitro Lipolysis Results
3.2. Permeation Barrier Integrity
3.3. Permeation Profiles
3.4. In Vivo–In Vitro Correlation
3.5. Absorption Surface Area to Donor Volume Ratio and Sink Conditions of the Lipolysis-Permeation Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruiz-Garcia, A.; Bermejo, M.; Moss, A.; Casabo, V.G. Pharmacokinetics in drug discovery. J. Pharm. Sci. 2008, 97, 654–690. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, J.H.; Bergström, C.A.S. Intestinal solubility and absorption of poorly water soluble compounds: Predictions, challenges and solutions. Ther. Deliv. 2015, 6, 935–959. [Google Scholar] [CrossRef] [PubMed]
- Kostewicz, E.S.; Abrahamsson, B.; Brewster, M.; Brouwers, J.; Butler, J.; Carlert, S.; Dickinson, P.A.; Dressman, J.; Holm, R.; Klein, S.; et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 342–366. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.M.; Artursson, P.; Bergstroöm, C.A.S. A Modified physiological BCS for prediction of intestinal absorption in drug discovery. Mol. Pharm. 2010, 7, 1478–1487. [Google Scholar] [CrossRef]
- Porter, C.J.; Charman, W.N. In vitro assessment of oral lipid based formulations. Adv. Drug Deliv. Rev. 2001, 50, S127–S147. [Google Scholar] [CrossRef]
- Carrière, F. Impact of gastrointestinal lipolysis on oral lipid-based formulations and bioavailability of lipophilic drugs. Biochimie 2016, 125, 297–305. [Google Scholar] [CrossRef]
- Porter, C.J.; Pouton, C.W.; Cuine, J.F.; Charman, W.N. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar] [CrossRef]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.A.; Porter, C.J. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef]
- Christophersen, P.C.; Christiansen, M.L.; Holm, R.; Kristensen, J.; Jacobsen, J.; Abrahamsson, B.; Müllertz, A. Fed and fasted state gastro-intestinal in vitro lipolysis: In vitro in vivo relations of a conventional tablet, a SNEDDS and a solidified SNEDDS. Eur. J. Pharm. Sci. 2014, 57, 232–239. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: The ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik/Eur. J. Pharm. Biopharm. 2007, 67, 96–105. [Google Scholar] [CrossRef]
- Larsen, A.T.; Ohlsson, A.G.; Polentarutti, B.; Barker, R.A.; Phillips, A.R.; Abu-Rmaileh, R.; Dickinson, P.A.; Abrahamsson, B.; Østergaard, J.; Müllertz, A. Oral bioavailability of cinnarizine in dogs: Relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur. J. Pharm. Sci. 2013, 48, 339–350. [Google Scholar] [CrossRef]
- Berthelsen, R.; Klitgaard, M.; Rades, T.; Müllertz, A. In vitro digestion models to evaluate lipid based drug delivery systems; present status and current trends. Adv. Drug Deliv. Rev. 2019, 142, 35–49. [Google Scholar] [CrossRef]
- Berthelsen, R.; Sassene, P.; Rades, T.; Müllertz, A. Evaluating oral drug delivery systems: Digestion models. In Analytical Techniques in the Pharmaceutical Sciences; Müllertz, A., Perrie, Y., Rades, T., Eds.; Springer: New York, NY, USA, 2016; pp. 773–790. [Google Scholar]
- Williams, H.D.; Sassene, P.; Kleberg, K.; Bakala-N’Goma, J.-C.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; Jule, E.; et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: Method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J. Pharm. Sci. 2012, 101, 3360–3380. [Google Scholar] [CrossRef]
- Verger, R. Pancreatic lipases. In Lipases; Borgström, B., Brockman, H.L., Eds.; Elsevier: New York, NY, USA, 1984; pp. 84–150. [Google Scholar]
- Capolino, P.; Guérin, C.; Paume, J.; Giallo, J.; Ballester, J.-M.; Cavalier, J.-F.; Carrière, F. In vitro gastrointestinal lipolysis: Replacement of human digestive lipases by a combination of rabbit gastric and porcine pancreatic extracts. Food Dig. 2011, 2, 43–51. [Google Scholar] [CrossRef]
- Palin, K.J.; Wilson, C.G. The effect of different oils on the absorption of probucol in the rat. J. Pharm. Pharmacol. 1984, 36, 641–643. [Google Scholar] [CrossRef]
- Larsen, A.; Holm, R.; Pedersen, M.L.; Müllertz, A. Lipid-based formulations for danazol containing a digestible surfactant, labrafil M2125CS: In vivo bioavailability and dynamic in vitro lipolysis. Pharm. Res. 2008, 25, 2769–2777. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. Use of a dynamic in vitro lipolysis model to rationalize oral formulation development for poor water soluble drugs: Correlation with in vivo data and the relationship to intra-enterocyte processes in rats. Pharm. Res. 2006, 23, 2165–2174. [Google Scholar] [CrossRef]
- Zangenberg, N.H.; Mullertz, A.; Kristensen, H.G.; Hovgaard, L. A dynamic in vitro lipolysis model. I. Controlling the rate of lipolysis by continuous addition of calcium. Eur. J. Pharm. Sci. 2001, 14, 115–122. [Google Scholar] [CrossRef]
- Lee, K.W.Y.; Porter, C.J.H.; Boyd, B.J. The effect of administered dose of lipid-based formulations on the in vitro and in vivo performance of cinnarizine as a model poorly water-soluble drug. J. Pharm. Sci. 2013, 102, 565–578. [Google Scholar] [CrossRef]
- Griffin, B.T.; Kuentz, M.; Vertzoni, M.; Kostewicz, E.S.; Fei, Y.; Faisal, W.; Stillhart, C.; O’Driscoll, C.M.; Reppas, C.; Dressman, J.B. Comparison of in vitro tests at various levels of complexity for the prediction of in vivo performance of lipid-based formulations: Case studies with fenofibrate. Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik/Eur. J. Pharm. Biopharm. 2014, 86, 427–437. [Google Scholar] [CrossRef]
- Stillhart, C.; Kuentz, M. Trends in the assessment of drug supersaturation and precipitation in vitro using lipid-based delivery systems. J. Pharm. Sci. 2016, 105, 2468–2476. [Google Scholar] [CrossRef]
- Alskär, L.C.; Bergström, C.A.S. Models for predicting drug absorption from oral lipid-based formulations. Curr. Mol. Biol. Rep. 2015, 1, 141–147. [Google Scholar] [CrossRef]
- Stillhart, C.; Imanidis, G.; Kuentz, M. Insights into drug precipitation kinetics during in vitro digestion of a lipid-based drug delivery system using in-line raman spectroscopy and mathematical modeling. Pharm. Res. 2013, 30, 3114–3130. [Google Scholar] [CrossRef]
- Bevernage, J.; Brouwers, J.; Annaert, P.; Augustijns, P. Drug precipitation–permeation interplay: Supersaturation in an absorptive environment. Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik/Eur. J. Pharm. Biopharm. 2012, 82, 424–428. [Google Scholar] [CrossRef]
- Thomas, N.; Holm, R.; Müllertz, A.; Rades, T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J. Control. Release 2012, 160, 25–32. [Google Scholar] [CrossRef]
- Buckley, S.T.; Fischer, S.M.; Fricker, G.; Brandl, M. In vitro models to evaluate the permeability of poorly soluble drug entities: Challenges and perspectives. Eur. J. Pharm. Sci. 2012, 45, 235–250. [Google Scholar] [CrossRef]
- Hens, B.; Brouwers, J.; Corsetti, M.; Augustijns, P. Gastrointestinal behavior of nano- and microsized fenofibrate: In vivo evaluation in man and in vitro simulation by assessment of the permeation potential. Eur. J. Pharm. Sci. 2015, 77, 40–47. [Google Scholar] [CrossRef]
- Keemink, J.; Bergström, C.A.S. Caco-2 cell conditions enabling studies of drug absorption from digestible lipid-based formulations. Pharm. Res. 2018, 35, 1–11. [Google Scholar] [CrossRef]
- Keemink, J.; Mårtensson, E.; Bergström, C.A.S. Lipolysis-permeation setup for simultaneous study of digestion and ab-sorption in vitro. Mol. Pharm. 2019, 16, 921–930. [Google Scholar] [CrossRef]
- Alskär, L.C.; Parrow, A.; Keemink, J.; Johansson, P.; Abrahamsson, B.; Bergström, C.A. Effect of lipids on absorption of carvedilol in dogs: Is coadministration of lipids as efficient as a lipid-based formulation? J. Control. Release 2019, 304, 90–100. [Google Scholar] [CrossRef]
- Mandagere, A.K.; Thompson, T.N.; Hwang, K.-K. Graphical model for estimating oral bioavailability of drugs in humans and other species from their caco-2 permeability and in vitro liver enzyme metabolic stability rates. J. Med. Chem. 2002, 45, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Florence, A.T. Physicochemical Principles of Pharmacy, 5th ed.; Pharmaceutical Press: London, UK, 2011. [Google Scholar]
- Berben, P.; Bauer-Brandl, A.; Brandl, M.; Faller, B.; Flaten, G.E.; Jacobsen, A.-C.; Brouwers, J.; Augustijns, P. Drug permeability profiling using cell-free permeation tools: Overview and applications. Eur. J. Pharm. Sci. 2018, 119, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Bibi, H.A.; Holm, R.; Bauer-Brandl, A. Simultaneous lipolysis/permeation in vitro model, for the estimation of bioavailability of lipid based drug delivery systems. Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik/Eur. J. Pharm. Biopharm. 2017, 117, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Sironi, D.; Christensen, M.; Rosenberg, J.; Bauer-Brandl, A.; Brandl, M. Evaluation of a dynamic dissolution/permeation model: Mutual influence of dissolution and barrier-flux under non-steady state conditions. Int. J. Pharm. 2017, 522, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Mudie, D.M.; Shi, Y.; Ping, H.; Gao, P.; Amidon, G.L.; Amidon, G.E. Mechanistic analysis of solute transport in anin vitrophysiological two-phase dissolution apparatus. Biopharm. Drug Dispos. 2012, 33, 378–402. [Google Scholar] [CrossRef]
- Siqueira, S.D.; Müllertz, A.; Gräeser, K.; Kasten, G.; Mu, H.; Rades, T. Influence of drug load and physical form of cinnarizine in new SNEDDS dosing regimens: In vivo and in vitro evaluations. AAPS J. 2017, 19, 587–594. [Google Scholar] [CrossRef]
- Naderkhani, E.; Isaksson, J.; Ryzhakov, A.; Flaten, G.E. Development of a biomimetic phospholipid vesicle-based permeation assay for the estimation of intestinal drug permeability. J. Pharm. Sci. 2014, 103, 1882–1890. [Google Scholar] [CrossRef]
- Flaten, G.E.; Dhanikula, A.B.; Luthman, K.; Brandl, M. Drug permeability across a phospholipid vesicle based barrier: A novel approach for studying passive diffusion. Eur. J. Pharm. Sci. 2006, 27, 80–90. [Google Scholar] [CrossRef]
- Sassene, P.; Kleberg, K.; Williams, H.D.; Bakala-N’Goma, J.-C.; Carriere, F.; Calderone, M.; Jannin, V.; Igonin, A.; Partheil, A.; Marchaud, D.; et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 6: Effects of varying pancreatin and calcium levels. AAPS J. 2014, 16, 1344–1357. [Google Scholar] [CrossRef]
- Mosgaard, M.D.; Sassene, P.; Mu, H.; Rades, T.; Müllertz, A. Development of a high-throughput in vitro intestinal lipolysis model for rapid screening of lipid-based drug delivery systems. Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik/Eur. J. Pharm. Biopharm. 2015, 94, 493–500. [Google Scholar] [CrossRef]
- Khan, J.; Rades, T.; Boyd, B.J. Lipid-based formulations can enable the model poorly water-soluble weakly basic drug cinnarizine to precipitate in an amorphous-salt form during in vitro digestion. Mol. Pharm. 2016, 13, 3783–3793. [Google Scholar] [CrossRef]
- Gu, C.; Rao, D.; Gandhi, R.B.; Hilden, J.; Raghavan, K. Using a novel multicompartment dissolution system to predict the effect of gastric pH on the oral absorption of weak bases with poor intrinsic solubility. J. Pharm. Sci. 2005, 94, 199–208. [Google Scholar] [CrossRef]
- Berthelsen, R.; Sjögren, E.; Jacobsen, J.; Kristensen, J.; Holm, R.; Abrahamsson, B.; Müllertz, A. Combining in vitro and in silico methods for better prediction of surfactant effects on the absorption of poorly water soluble drugs—A fenofibrate case example. Int. J. Pharm. 2014, 473, 356–365. [Google Scholar] [CrossRef]
- Di Cagno, M.; Bibi, H.A.; Bauer-Brandl, A. New biomimetic barrier Permeapad™ for efficient investigation of passive permeability of drugs. Eur. J. Pharm. Sci. 2015, 73, 29–34. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M. The solubility–Permeability interplay and its implications in formulation design and development for poorly soluble drugs. AAPS J. 2012, 14, 244–251. [Google Scholar] [CrossRef]
- Bibi, H.A.; Di Cagno, M.; Holm, R.; Bauer-Brandl, A. Permeapad™ for investigation of passive drug permeability: The effect of surfactants, co-solvents and simulated intestinal fluids (FaSSIF and FeSSIF). Int. J. Pharm. 2015, 493, 192–197. [Google Scholar] [CrossRef]
- Michaelsen, M.H.; Wasan, K.M.; Sivak, O.; Müllertz, A.; Rades, T. The effect of digestion and drug load on halofantrine absorption from self-nanoemulsifying drug delivery system (SNEDDS). AAPS J. 2015, 18, 180–186. [Google Scholar] [CrossRef]
- Michaelsen, M.H.; Jørgensen, S.D.S.; Abdi, I.M.; Wasan, K.M.; Rades, T.; Müllertz, A. Fenofibrate oral absorption from SNEDDS and super-SNEDDS is not significantly affected by lipase inhibition in rats. Eur. J. Pharm. Biopharm. 2019, 142, 258–264. [Google Scholar] [CrossRef]
- Klitgaard, M.; Beilles, S.; Sassene, P.J.; Berthelsen, R.; Müllertz, A. Adding a gastric step to the intestinal in vitro digestion model improves the prediction of pharmacokinetic data in beagle dogs of two lipid-based drug delivery systems. Mol. Pharm. 2020, 17, 3214–3222. [Google Scholar] [CrossRef]
- Tran, T.; Siqueira, S.D.; Amenitsch, H.; Rades, T.; Müllertz, A. Monoacyl phosphatidylcholine inhibits the formation of lipid multilamellar structures during in vitro lipolysis of self-emulsifying drug delivery systems. Eur. J. Pharm. Sci. 2017, 108, 62–70. [Google Scholar] [CrossRef]
- Tran, T.; Fatouros, D.G.; Vertzoni, M.; Reppas, C.; Müllertz, A. Mapping the intermediate digestion phases of human healthy intestinal contents from distal ileum and caecum at fasted and fed state conditions. J. Pharm. Pharmacol. 2017, 69, 265–273. [Google Scholar] [CrossRef]
- Li, S.; He, H.; Parthiban, L.J.; Yin, H.; Serajuddin, A.T. IV-IVC considerations in the development of immediate-release oral dosage form. J. Pharm. Sci. 2005, 94, 1396–1417. [Google Scholar] [CrossRef]
- Buch, P.; Langguth, P.; Kataoka, M.; Yamashita, S. IVIVC in oral absorption for fenofibrate immediate release tablets using a dissolution/permeation system. J. Pharm. Sci. 2009, 98, 2001–2009. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Concentration (mM) |
|---|---|
| Bovine Bile | 2.95 |
| Phospholipids | 0.26 |
| NaCl | 146.8 |
| Tris | 2 |
| Maleic acid | 2 |
| CaCl2·2H2O | 1.4 |
| Component | Ratio (% w/w) |
|---|---|
| Soybean oil: Maisine 35-1 (1:1, weight ratio) | 55 |
| Kolliphor RH 40 | 35 |
| Ethanol | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klitgaard, M.; Müllertz, A.; Berthelsen, R. Estimating the Oral Absorption from Self-Nanoemulsifying Drug Delivery Systems Using an In Vitro Lipolysis-Permeation Method. Pharmaceutics 2021, 13, 489. https://doi.org/10.3390/pharmaceutics13040489
Klitgaard M, Müllertz A, Berthelsen R. Estimating the Oral Absorption from Self-Nanoemulsifying Drug Delivery Systems Using an In Vitro Lipolysis-Permeation Method. Pharmaceutics. 2021; 13(4):489. https://doi.org/10.3390/pharmaceutics13040489
Chicago/Turabian StyleKlitgaard, Mette, Anette Müllertz, and Ragna Berthelsen. 2021. "Estimating the Oral Absorption from Self-Nanoemulsifying Drug Delivery Systems Using an In Vitro Lipolysis-Permeation Method" Pharmaceutics 13, no. 4: 489. https://doi.org/10.3390/pharmaceutics13040489
APA StyleKlitgaard, M., Müllertz, A., & Berthelsen, R. (2021). Estimating the Oral Absorption from Self-Nanoemulsifying Drug Delivery Systems Using an In Vitro Lipolysis-Permeation Method. Pharmaceutics, 13(4), 489. https://doi.org/10.3390/pharmaceutics13040489