
Interaction of Remdesivir with Clinically Relevant Hepatic Drug Uptake Transporters

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines Stably Expressing Human Hepatic Uptake Transporters
2.3. Cellular Uptake Studies
2.4. Quantification of Remdesivir by UHPLC-MS-MS
2.5. Statistical Analysis
3. Results
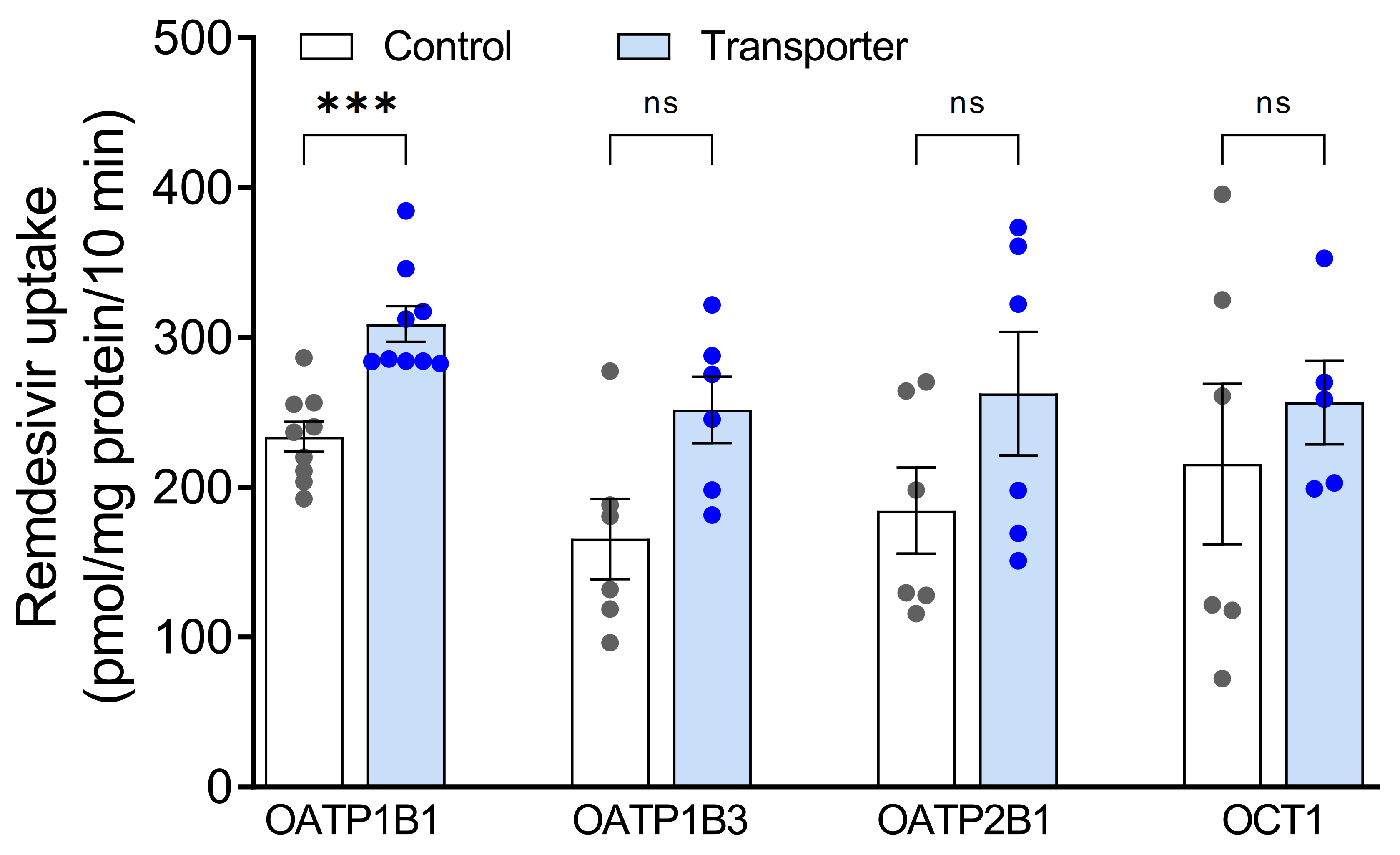
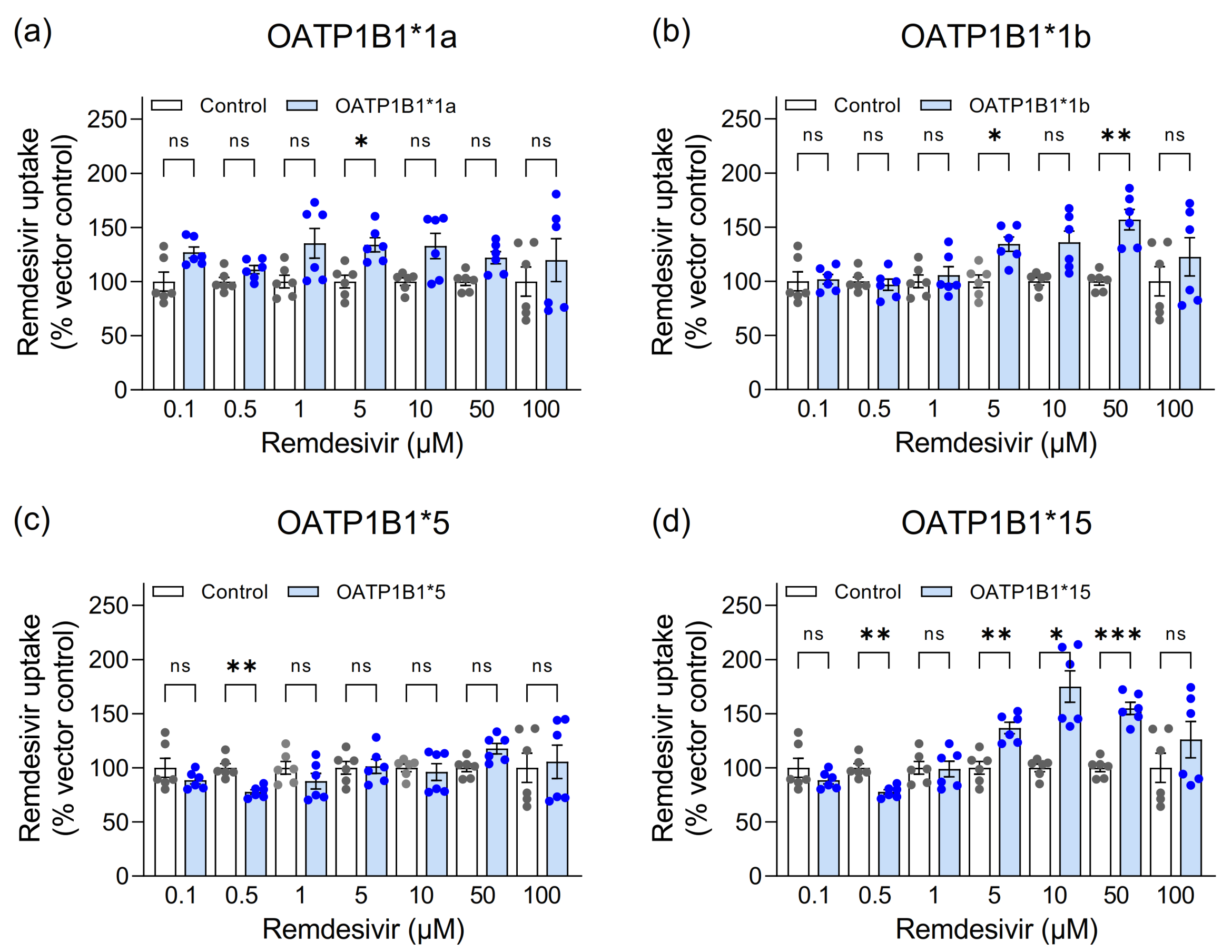
3.1. Assessment of Remdesivir as Transported Substrate of Hepatic Uptake Transporters
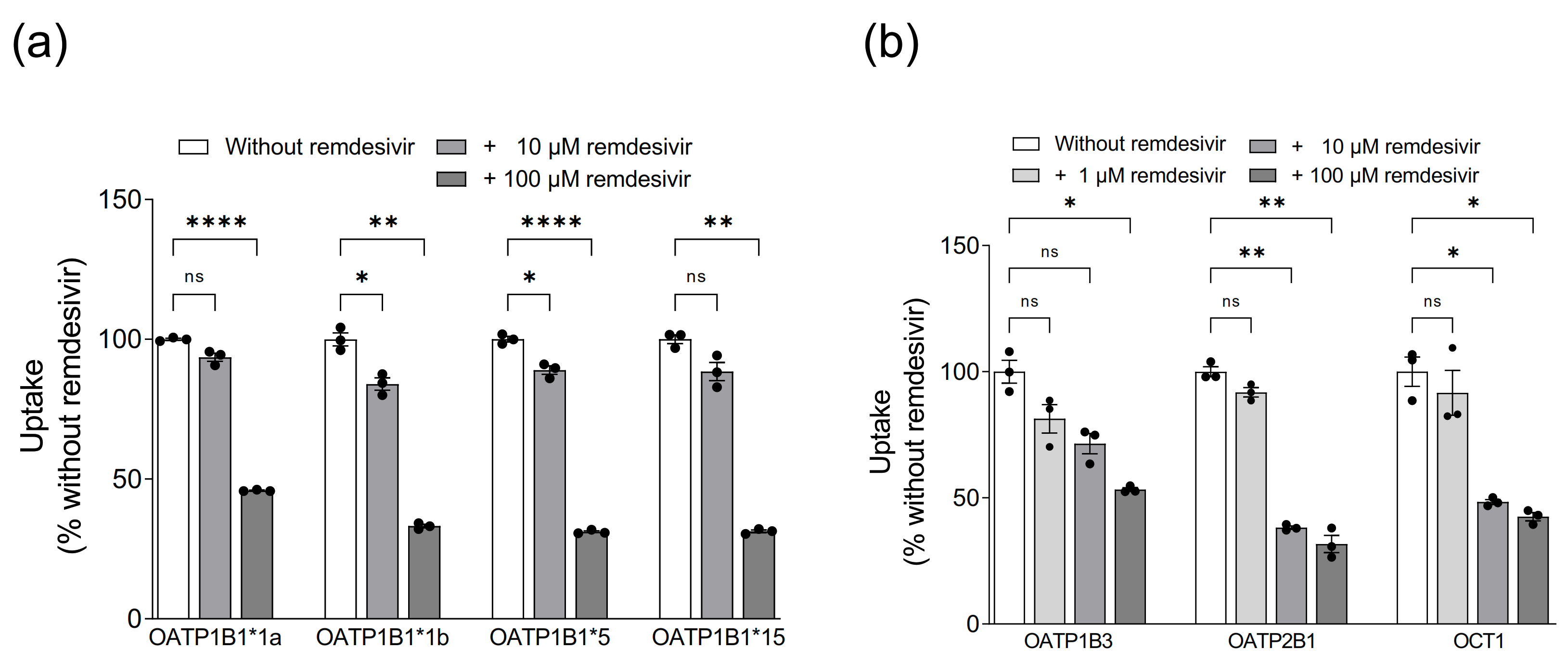
3.2. Assessment of Remdesivir as Inhibitor of Hepatic Uptake Transporters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Xu, Y.; Barauskas, O.; Kim, C.; Babusis, D.; Murakami, E.; Kornyeyev, D.; Lee, G.; Stepan, G.; Perron, M.; Bannister, R.; et al. Off-target in vitro profiling demonstrates that remdesivir is a highly selective antiviral agent. Antimicrob. Agents Chemother. 2021, 65, e02237-20. [Google Scholar] [CrossRef]
- Gilead Sciences. Veklury; Gilead Sciences: Foster City, CA, USA, 2010; [package insert]. [Google Scholar]
- Yang, K. What do we know about remdesivir drug interactions? Clin. Transl. Sci. 2020, 13, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Giacomini, K.M.; Zhang, L. Emerging clinical importance of hepatic organic cation transporter 1 (OCT1) in drug pharmacokinetics, dynamics, pharmacogenetic variability, and drug interactions. Clin. Pharmacol. Ther. 2018, 103, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Gessner, A.; König, J.; Fromm, M.F. Clinical aspects of transporter-mediated drug-drug interactions. Clin. Pharmacol. Ther. 2019, 105, 1386–1394. [Google Scholar] [CrossRef]
- Unger, M.S.; Mudunuru, J.; Schwab, M.; Hopf, C.; Drewes, G.; Nies, A.T.; Zamek-Gliszczynski, M.J.; Reinhard, F. Clinically-relevant OATP2B1 inhibitors in marketed drug space. Mol. Pharm. 2020, 17, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B.; Hagenbuch, B. Organic anion-transporting polypeptides. Curr. Top. Membr. 2014, 73, 205–232. [Google Scholar] [CrossRef]
- Fahrmayr, C.; Fromm, M.F.; König, J. Hepatic OATP and OCT uptake transporters: Their role for drug-drug interactions and pharmacogenetic aspects. Drug Metab. Rev. 2010, 42, 380–401. [Google Scholar] [CrossRef]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. Organic anion transporting polypeptide 1B1: A genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011, 63, 157–181. [Google Scholar] [CrossRef]
- Nies, A.T.; Niemi, M.; Burk, O.; Winter, S.; Zanger, U.M.; Stieger, B.; Schwab, M.; Schaeffeler, E. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 2013, 5, 1. [Google Scholar] [CrossRef]
- Nies, A.T.; Koepsell, H.; Winter, S.; Burk, O.; Klein, K.; Kerb, R.; Zanger, U.M.; Keppler, D.; Schwab, M.; Schaeffeler, E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 2009, 50, 1227–1240. [Google Scholar] [CrossRef]
- Meyer, M.J.; Neumann, V.E.; Friesacher, H.R.; Zdrazil, B.; Brockmöller, J.; Tzvetkov, M.V. Opioids as substrates and inhibitors of the genetically highly variable organic cation transporter OCT1. J. Med. Chem. 2019, 62, 9890–9905. [Google Scholar] [CrossRef]
- Tzvetkov, M.V.; Matthaei, J.; Pojar, S.; Faltraco, F.; Vogler, S.; Prukop, T.; Seitz, T.; Brockmöller, J. Increased systemic exposure and stronger cardiovascular and metabolic adverse reactions to fenoterol in individuals with heritable OCT1 deficiency. Clin. Pharmacol. Ther. 2018, 103, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Taub, M.E.; Chothe, P.P.; Chu, X.; Giacomini, K.M.; Kim, R.B.; Ray, A.S.; Stocker, S.L.; Unadkat, J.D.; Wittwer, M.B.; et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin. Pharmacol. Ther. 2018, 104, 890–899. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Glaeser, H.; Keiser, M.; Mandery, K.; Klotz, U.; Fromm, M.F. Role of organic anion-transporting polypeptides for cellular mesalazine (5-aminosalicylic acid) uptake. Drug Metab. Dispos. 2011, 39, 1097–1102. [Google Scholar] [CrossRef]
- Dickens, D.; Rädisch, S.; Chiduza, G.N.; Giannoudis, A.; Cross, M.J.; Malik, H.; Schaeffeler, E.; Sison-Young, R.L.; Wilkinson, E.L.; Goldring, C.E.; et al. Cellular uptake of the atypical antipsychotic clozapine is a carrier-mediated process. Mol. Pharm. 2018, 15, 3557–3572. [Google Scholar] [CrossRef]
- Nies, A.T.; Hofmann, U.; Resch, C.; Schaeffeler, E.; Rius, M.; Schwab, M. Proton pump inhibitors inhibit metformin uptake by organic cation uptake transporters (OCTs). PLoS ONE 2011, 6, e22163. [Google Scholar] [CrossRef]
- Misaka, S.; Knop, J.; Singer, K.; Hoier, E.; Keiser, M.; Muller, F.; Glaeser, H.; König, J.; Fromm, M.F. The non-metabolized beta-blocker nadolol is a substrate of OCT1, OCT2, MATE1, MATE2-K and P-glycoprotein, but not of OATP1B1 and OATP1B3. Mol. Pharm. 2016, 13, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Avataneo, V.; de Nicolò, A.; Cusato, J.; Antonucci, M.; Manca, A.; Palermiti, A.; Waitt, C.; Walimbwa, S.; Lamorde, M.; Di Perri, G.; et al. Development and validation of a UHPLC-MS/MS method for quantification of the prodrug remdesivir and its metabolite GS-441524: A tool for clinical pharmacokinetics of SARS-CoV-2/COVID-19 and Ebola virus disease. J. Antimicrob. Chemother. 2020, 75, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, K.L.; Keppler, D.; Hoffmaster, K.A.; Bow, D.A.; Cheng, Y.; Lai, Y.; Palm, J.E.; Stieger, B.; Evers, R. In vitro methods to support transporter evaluation in drug discovery and development. Clin. Pharmacol. Ther. 2013, 94, 95–112. [Google Scholar] [CrossRef]
- FDA Guidance. In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. 2020. Available online:https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 18 February 2020).
- Jorgensen, S.C.J.; Kebriaei, R.; Dresser, L.D. Remdesivir: Review of pharmacology, pre-clinical data, and emerging clinical experience for COVID-19. Pharmacotherapy 2020, 40, 659–671. [Google Scholar] [CrossRef] [PubMed]
- EMA. Summary on Compassionate Use. Remdesivir Gilead. Procedure No. EMEA/H/K/5622/CU. 2020. Available online: https://www.ema.europa.eu/en/documents/other/summary-compassionate-use-remdesivir-gilead_en.pdf (accessed on 6 May 2020).
- Telbisz, Á.; Ambrus, C.; Mózner, O.; Szabó, E.; Várady, G.; Bakos, É.; Sarkadi, B.; Özvegy-Laczka, C. Interactions of potential anti-COVID-19 compounds with multispecific ABC and OATP drug transporters. Pharmaceutics 2021, 13, 81. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nies, A.T.; König, J.; Hofmann, U.; Kölz, C.; Fromm, M.F.; Schwab, M. Interaction of Remdesivir with Clinically Relevant Hepatic Drug Uptake Transporters. Pharmaceutics 2021, 13, 369. https://doi.org/10.3390/pharmaceutics13030369
Nies AT, König J, Hofmann U, Kölz C, Fromm MF, Schwab M. Interaction of Remdesivir with Clinically Relevant Hepatic Drug Uptake Transporters. Pharmaceutics. 2021; 13(3):369. https://doi.org/10.3390/pharmaceutics13030369
Chicago/Turabian StyleNies, Anne T., Jörg König, Ute Hofmann, Charlotte Kölz, Martin F. Fromm, and Matthias Schwab. 2021. "Interaction of Remdesivir with Clinically Relevant Hepatic Drug Uptake Transporters" Pharmaceutics 13, no. 3: 369. https://doi.org/10.3390/pharmaceutics13030369
APA StyleNies, A. T., König, J., Hofmann, U., Kölz, C., Fromm, M. F., & Schwab, M. (2021). Interaction of Remdesivir with Clinically Relevant Hepatic Drug Uptake Transporters. Pharmaceutics, 13(3), 369. https://doi.org/10.3390/pharmaceutics13030369