
Advanced Therapy Medicinal Products for the Eye: Definitions and Regulatory Framework

 , ,
, ,
Abstract
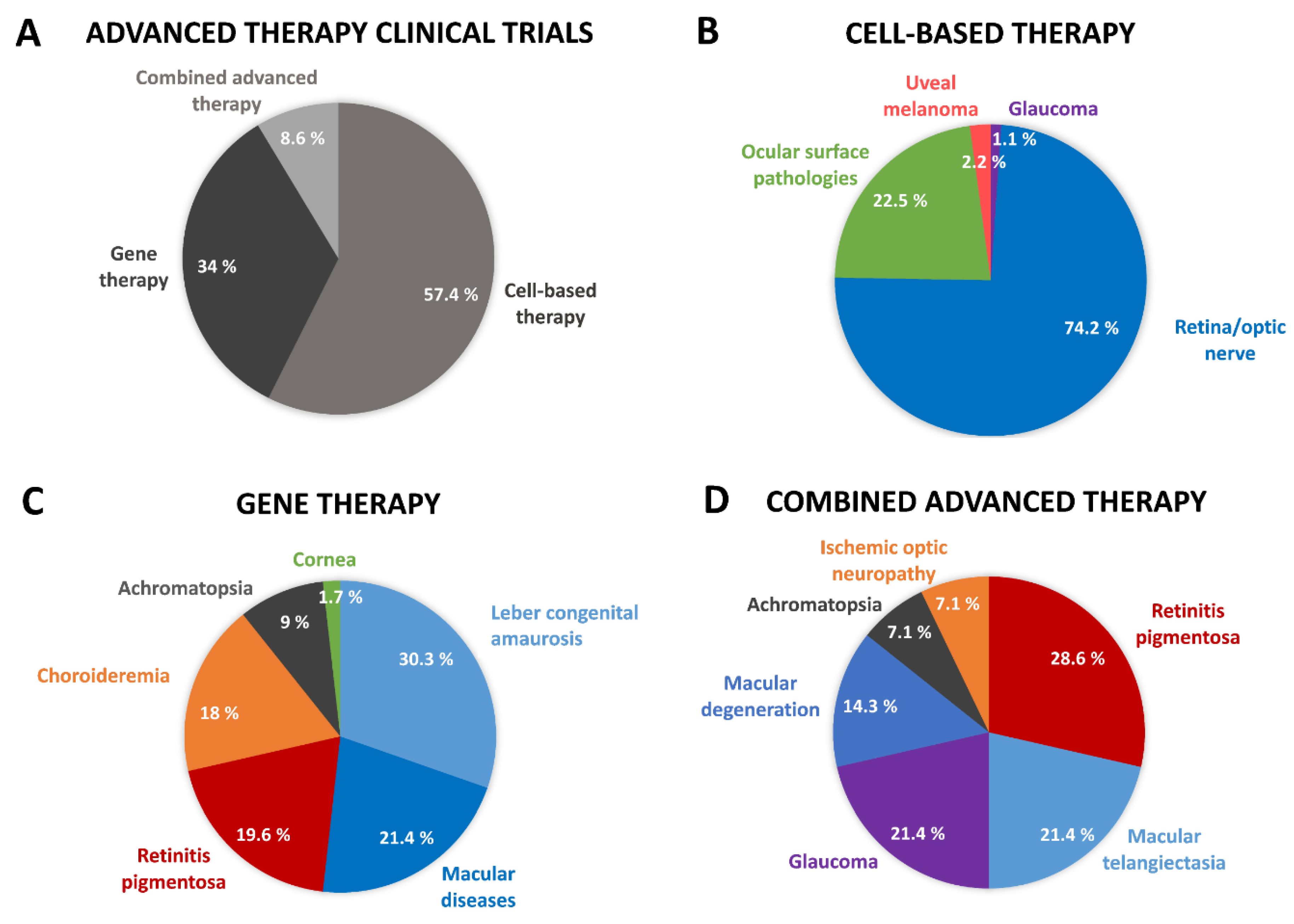
1. Introduction
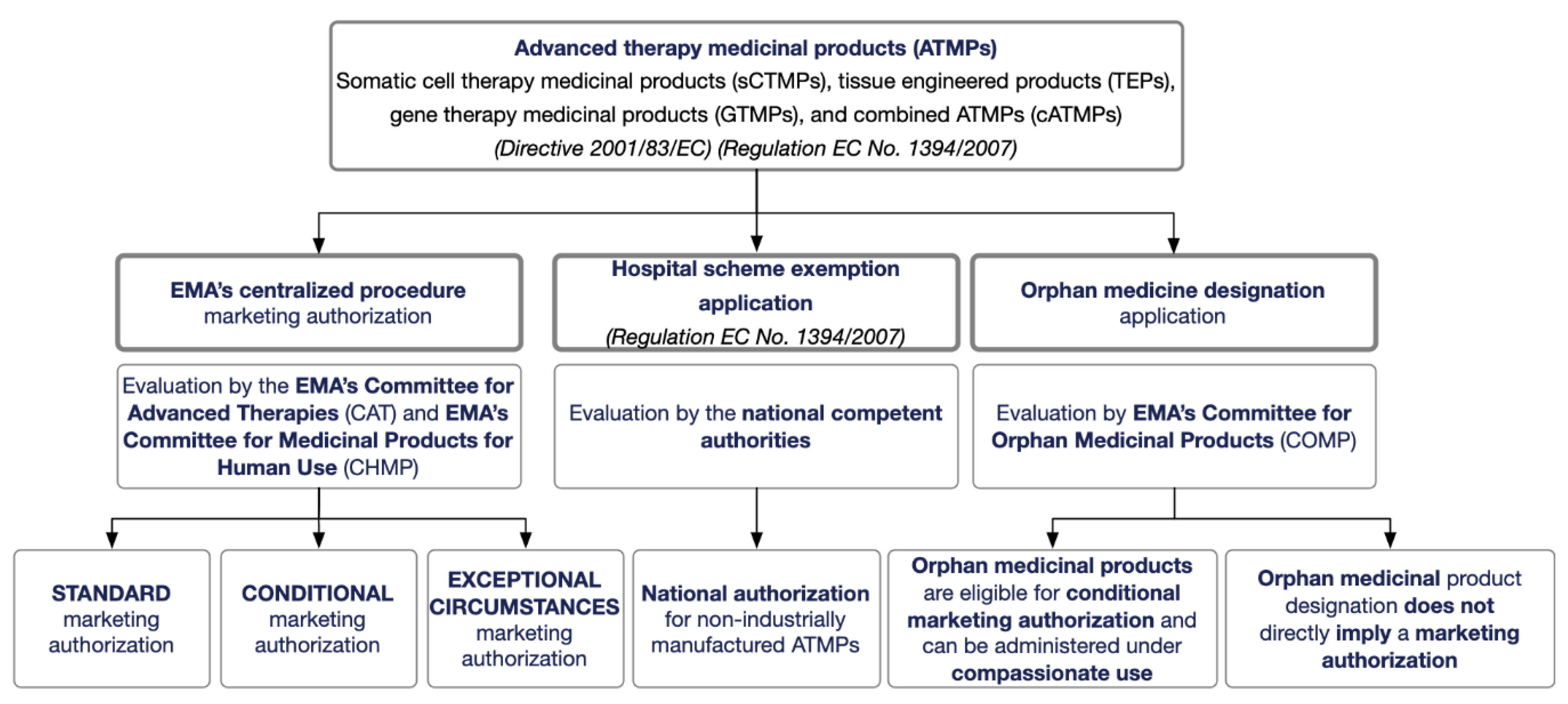
2. ATMP Regulatory Framework
2.1. ATMP Regulatory Framework in the EU
2.2. Regulatory Framework for Cell- and Gene-Based Therapies in Other Jurisdictions
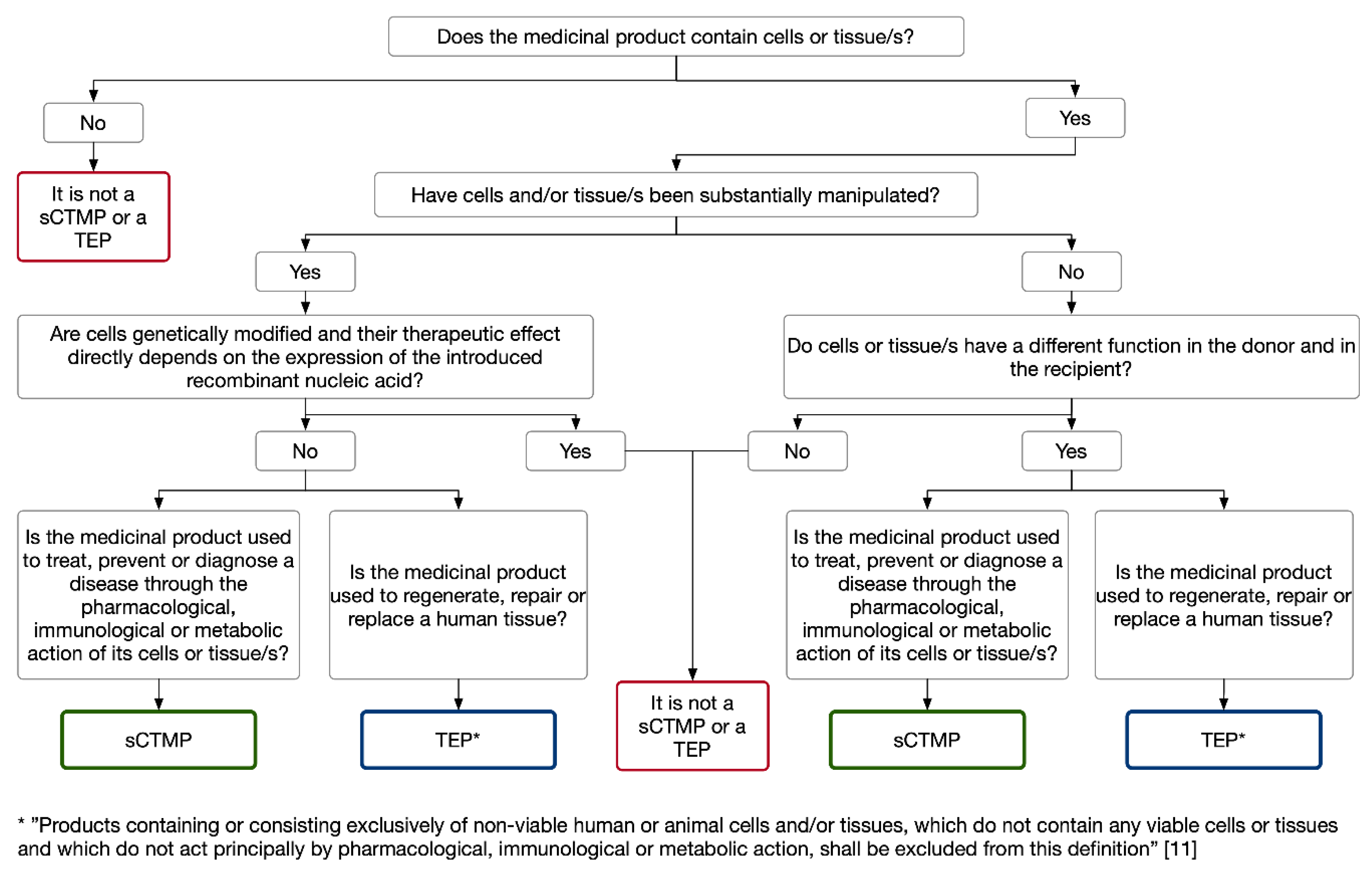
3. Definitions and ATMP Classification Criteria
3.1. Cell-Based Medicinal Products: Somatic Cell Therapy and Tissue-Engineered Medicinal Products
Somatic Cell Therapy and Tissue Engineered Medicinal Products for the Eye
3.2. Gene Therapy Medicinal Products
Gene Therapy Medicinal Products for the Eye
3.3. Combined ATMPs
Combined ATMPs for the Eye
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Regulation (EC) No. 1394/2007 of the European Parliament and of the Council of 13 November 2007 on Advanced Therapy Medicinal Products and Amending Directive 2001/83/EC and Regulation /EC) N0 726/2004. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:324:0121:0137:en:PDF (accessed on 4 February 2021).
- Regulation (EU) No. 536/2014 of the European Parliament and of the Council of 16 April 2014 on Clinical Trials on Medicinal Products for Human Use, and Repealing Directive 2001/20/EC. Available online: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf (accessed on 4 February 2021).
- Commission Directive 2005/28/EC of 8 April 2005 Laying down Principles and Detailed Guidelines for Good Clinical Practice as Regards Investigational Medicinal Products for Human Use, as well as the Requirements for Authorisation of the Manufacturing or Importation of Such Products. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2005:091:0013:0019:en:PDF (accessed on 4 February 2021).
- Commission Directive 2003/94/EC of 8 October 2003 Laying down the Principles and Guidelines of Good Manufacturing Practice in Respect of Medicinal Products for Human Use and Investigational Medicinal Products for Human Use. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2003:262:0022:0026:en:PDF (accessed on 4 February 2021).
- Salmikangas, P.; Schuessler-Lenz, M.; Ruiz, S.; Celis, P.; Reischl, I.; Menezes-Ferreira, M.; Flory, E.; Renner, M.; Ferry, N. Marketing Regulatory Oversight of Advanced Therapy Medicinal Products (ATMPs) in Europe: The EMA/CAT Perspective. Adv. Exp. Med. Biol. 2015, 871, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Committee for Advanced Therapies (CAT); CAT Scientific Secretariat; Schneider, C.; Salmikangas, P.; Jilma, B.; Flamion, B.; Todorova, L.; Paphitou, A.; Haunerova, I.; Maimets, T.; et al. Challenges with advanced therapy medicinal products and how to meet them. Nat. Rev. Drug Discov. 2010, 9, 195–201. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Procedural Advice on the Evaluation of Advanced Therapy Medicinal Product in Accordance with Article 8 of Regulation (EC) No. 1394/2007. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/procedural-advice-evaluation-advanced-therapy-medicinal-product-accordance-article-8-regulation-ec/2007_en.pdf (accessed on 4 February 2021).
- Hanna, E.; Rémuzat, C.; Auquier, P.; Toumi, M. Advanced therapy medicinal products: Current and future perspectives. J. Mark. Access Health Policy 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use. Available online: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf (accessed on 4 February 2021).
- Regulation (EC) No. 726/2004 of the European Parliament and of the Council of 31 March 2004 Laying down Community Procedures for the Authorisation and Supervision of Medicinal Products for Human and Veterinary Use and Establishing a European Medicines Agency. Available online: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2004_726/reg_2004_726_en.pdf (accessed on 4 February 2021).
- Commission Directive 2009/120/EC of 14 September 2009 Amending Directive 2001/83/EC of the European Parliament and of the Council on the Community Code Relating to Medicinal Products for Human Use as Regards Advanced Therapy Medicinal Products. Available online: https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-1/dir_2009_120/dir_2009_120_en.pdf (accessed on 4 February 2021).
- Detela, G.; Lodge, A. EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation. Mol. Ther. Methods Clin. Dev. 2019, 13, 205–232. [Google Scholar] [CrossRef]
- European Medicines Agency, Committee for Advanced Therapies (CAT). Available online: https://www.ema.europa.eu/en/committees/committee-advanced-therapies-cat (accessed on 4 February 2021).
- European Medicines Agency. Reflection Paper on Classification of Advanced Therapy Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-classification-advanced-therapy-medicinal-products_en-0.pdf (accessed on 4 February 2021).
- European Medicines Agency. Procedural Advice on the Evaluation of Combined Advanced Therapy Medicinal Products and the Consultation of Notified Bodies in Accordance with Article 9 of Regulation (EC) No. 1394/2007. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/procedural-advice-consultation-notified-bodies-accordance-article-9-regulation-ec-no-1394/2007_en.pdf (accessed on 4 February 2021).
- Regulation (EC) No. 141/2000 of the European Parliament and of the Council of 16 December 1999 on Orphan Medicinal Products. Available online: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf (accessed on 4 February 2021).
- Galli, M.C.; Serabian, M. Regulatory Aspects of Gene Therapy and Cell Therapy Products; Springer International Publishing: Cham, Switzerland, 2015; Volume 871, ISBN 978-3-319-18617-7. [Google Scholar]
- U.S. Food and Drug Administration. Framework for the Regulation of Regenerative Medicine Products. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/framework-regulation-regenerative-medicine-products (accessed on 4 February 2021).
- U.S. Food and Drug Administration. Cellular & Gene Therapy Products. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products (accessed on 4 February 2021).
- Iglesias-Lopez, C.; Agustí, A.; Obach, M.; Vallano, A. Regulatory Framework for Advanced Therapy Medicinal Products in Europe and United States. Front. Pharmacol. 2019, 10, 921. [Google Scholar] [CrossRef]
- Azuma, K. Regulatory Landscape of Regenerative Medicine in Japan. Curr. Stem Cell Rep. 2015, 1, 118–128. [Google Scholar] [CrossRef]
- Jokura, Y.; Yano, K.; Yamato, M. Comparison of the new Japanese legislation for expedited approval of regenerative medicine products with the existing systems in the USA and European Union. J. Tissue Eng. Regen. Med. 2018, 12, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Biological Medicine. Available online: https://www.ema.europa.eu/en/glossary/biological-medicine (accessed on 4 February 2021).
- Okada, K.; Koike, K.; Sawa, Y. Consideration of and Expectations for the Pharmaceuticals, Medical Devices and Other Therapeutic Products Act in Japan. Regen. Ther. 2015, 1, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Bobba, S.; Di Girolamo, N.; Munsie, M.; Chen, F.; Pébay, A.; Harkin, D.; Hewitt, A.W.; O’Connor, M.; McLenachan, S.; Shadforth, A.M.A.; et al. The current state of stem cell therapy for ocular disease. Exp. Eye Res. 2018, 177, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Sharma, T. Stargardt Disease. Adv. Exp. Med. Biol. 2018, 1085, 139–151. [Google Scholar] [CrossRef]
- European Medicines Agency. EU/3/11/874. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu311874 (accessed on 4 February 2021).
- Dua, H.S.; Azuara-Blanco, A. Limbal Stem Cells of the Corneal Epithelium. Surv. Ophthalmol. 2000, 44, 415–425. [Google Scholar] [CrossRef]
- European Medicines Agency. EU/3/13/1168. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3131168 (accessed on 4 February 2021).
- European Medicines Agency. EU/3/14/1340. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3141340 (accessed on 4 February 2021).
- European Medicines Agency. OraNera. Available online: https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/oranera (accessed on 4 February 2021).
- Pellegrini, G.; Golisano, O.; Paterna, P.; Lambiase, A.; Bonini, S.; Rama, P.; De Luca, M. Location and Clonal Analysis of Stem Cells and Their Differentiated Progeny in the Human Ocular Surface. J. Cell Biol. 1999, 145, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Dellambra, E.; Golisano, O.; Martinelli, E.; Fantozzi, I.; Bondanza, S.; Ponzin, D.; McKeon, F.; De Luca, M. p63 identifies keratinocyte stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3156–3161. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Ranno, R.; Stracuzzi, G.; Bondanza, S.; Guerra, L.; Zambruno, G.; Micali, G.; De Luca, M. The control of epidermal stem cells (holoclones) in the treatment of massive full-thickness burns with autologous keratinocytes cultured on fibrin1. Transplantation 1999, 68, 868–879. [Google Scholar] [CrossRef]
- Pellegrini, G.; Lambiase, A.; Macaluso, C.; Pocobelli, A.; Deng, S.; Cavallini, G.M.; Esteki, R.; Rama, P. From discovery to approval of an advanced therapy medicinal product-containing stem cells, in the EU. Regen. Med. 2016, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. EU/3/08/579. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu308579 (accessed on 4 February 2021).
- Pellegrini, G.; Ardigò, D.; Milazzo, G.; Iotti, G.; Guatelli, P.; Pelosi, D.; De Luca, M. Navigating Market Authorization: The Path Holoclar Took to Become the First Stem Cell Product Approved in the European Union. Stem Cells Transl. Med. 2018, 7, 146–154. [Google Scholar] [CrossRef]
- European Medicines Agency. Holoclar. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/holoclar (accessed on 4 February 2021).
- Pharmaceuticals and Medical Devices Agency. New Regenerative Medical Products. Available online: https://www.pmda.go.jp/english/review-services/reviews/approved-information/0002.html (accessed on 4 February 2021).
- Somia, N.; Verma, I.M. Gene therapy: Trials and tribulations. Nat. Rev. Genet. 2000, 1, 91–99. [Google Scholar] [CrossRef]
- del Pozo-Rodríguez, A.; Rodríguez-Gascón, A.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Gómez-Aguado, I.; Battaglia, L.S.; Solinís, M.Á. Gene Therapy. Adv. Biochem. Eng. Biotechnol. 2020, 171, 321–368. [Google Scholar] [CrossRef]
- Solinís, M.Á.; del Pozo-Rodríguez, A.; Apaolaza, P.S.; Rodríguez-Gascón, A. Treatment of ocular disorders by gene therapy. Eur. J. Pharm. Biopharm. 2015, 95, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.A.; Coster, D.J. Gene therapy for diseases of the cornea a review. Clin. Exp. Ophthalmol. 2009, 38, 93–103. [Google Scholar] [CrossRef]
- Parashar, A. Aptamers in Therapeutics. J. Clin. Diagn. Res. 2016, 10. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 -mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Council Directive 93/42/EEC of 14 June 1993 Concerning Medical Devices. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:1993L0042:20071011:en:PDF (accessed on 4 February 2021).
- Council Directive of 20 June 1990 on the Approximation of the Laws of the Member States Relating to Active Implantable Medical Devices (90/385/EEC). Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:1990L0385:20071011:EN:PDF (accessed on 4 February 2021).
- European Commission. Guidelines on Medical Devices. Available online: http://ec.europa.eu/DocsRoom/documents/17522/attachments/1/translations/ (accessed on 4 February 2021).
- ten Ham, R.M.T.; Hoekman, J.; Hövels, A.M.; Broekmans, A.W.; Leufkens, H.G.M.; Klungel, O.H. Challenges in Advanced Therapy Medicinal Product Development: A Survey among Companies in Europe. Mol. Ther. Methods Clin. Dev. 2018, 11, 121–130. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Closure of EU Manufacturing Site for MACI. Available online: https://www.ema.europa.eu/en/documents/referral/maci-article-20-procedure-closure-eu-manufacturing-site-maci_en.pdf (accessed on 4 February 2021).
- Eldem, T.; Eldem, B. Ocular Drug, Gene and Cellular Delivery Systems and Advanced Therapy Medicinal Products. Türk Oftalmol. Derg. 2018, 48, 132–141. [Google Scholar] [CrossRef]
- Emerich, D.F.; Thanos, C.G. NT-501: An ophthalmic implant of polymer-encapsulated ciliary neurotrophic factor-producing cells. Curr. Opin. Mol. Ther. 2008, 10, 506–515. [Google Scholar] [PubMed]
- European Medicines Agency. Scientific Recommendation on Classification of Advanced Therapy Medicinal Products. Available online: https://www.ema.europa.eu/en/documents/report/scientific-recommendation-classification-advanced-therapy-medicinal-products-human-ciliary_en.pdf (accessed on 4 February 2021).
- Kauper, K.; McGovern, C.; Sherman, S.; Heatherton, P.; Rapoza, R.; Stabila, P.; Dean, B.; Lee, A.; Borges, S.; Bouchard, B.; et al. Two-Year Intraocular Delivery of Ciliary Neurotrophic Factor by Encapsulated Cell Technology Implants in Patients with Chronic Retinal Degenerative Diseases. Investig. Opthalmol. Vis. Sci. 2012, 53, 7484–7491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hopkins, J.J.; Heier, J.S.; Birch, D.G.; Halperin, L.S.; Albini, T.A.; Brown, D.M.; Jaffe, G.J.; Tao, W.; Williams, G.A. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 6241–6245. [Google Scholar] [CrossRef]
- Birch, D.G.; Weleber, R.G.; Duncan, J.L.; Jaffe, G.J.; Tao, W. Randomized Trial of Ciliary Neurotrophic Factor Delivered by Encapsulated Cell Intraocular Implants for Retinitis Pigmentosa. Am. J. Ophthalmol. 2013, 156, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Talcott, K.E.; Ratnam, K.; Sundquist, S.M.; Lucero, A.S.; Lujan, B.J.; Tao, W.; Porco, T.C.; Roorda, A.; Duncan, J.L. Longitudinal Study of Cone Photoreceptors during Retinal Degeneration and in Response to Ciliary Neurotrophic Factor Treatment. Investig. Opthalmol. Vis. Sci. 2011, 52, 2219–2226. [Google Scholar] [CrossRef]
- Pilli, S.; Zawadzki, R.J.; Telander, D.G. The dose-dependent macular thickness changes assessed by fd-oct in patients with retinitis pigmentosa treated with ciliary neurotrophic factor. Retina 2014, 34, 1384–1390. [Google Scholar] [CrossRef]
- Birch, D.G.; Bennett, L.D.; Duncan, J.L.; Weleber, R.G.; Pennesi, M.E. Long-term Follow-up of Patients With Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. Am. J. Ophthalmol. 2016, 170, 10–14. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Public Summary of Opinion on Orphan Designation. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/12/1072-public-summary-opinion-orphan-designation-encapsulated-human-retinal-pigment-epithelial-cell_en.pdf (accessed on 4 February 2021).
- European Medicines Agency. Public Summary of Opinion on Orphan Designation. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/12/1098-public-summary-opinion-orphan-designation-encapsulated-human-retinal-pigment-epithelial-cell_en.pdf (accessed on 4 February 2021).




{kind=link}
{kind=link}
{kind=link}
| Jurisdiction | European Union | United States | Japan |
|---|---|---|---|
| Agency | European Medicines Agency (EMA) | Food and Drug Administration (FDA) | Pharmaceuticals and Medical Devices Agency (PMDA) Ministry of Health, Labour and Welfare (MHLW) |
| Regulatory framework | Directive 2001/83/EC (related to medical products for human use) European Commission 2007_Regulation EC No. 1394/2007 (related to advanced therapy medicinal products) | Federal Food, Drug, and Cosmetic Act (FDCA) and the Public Health Services Act (PHSA) Regenerative Medicine Advanced Therapy (RMAT) designation: section 3033 of the 21st Century Cures Act. | Act on the Safety of Regenerative Medicine (RM Act) and Pharmaceuticals and Medical Devices Act (PMD Act) 1960 Act No. 145 revised by 2013 Act No. 84 |
| Therapy classification | Somatic cell therapy medicinal products (sCTMPs), tissue engineered products (TEPs), gene therapy medicinal products (GTMPs), and combined ATMPs (cATMPs) | Cell therapy and gene therapy products | Gene-, cell-, and tissue-based therapies |
| Product (Commercial Name or Number Designated by EMA 1) | sCTMP 2 or TEP 3 | Manufacturer | Active Substance | Administration Route | Indication | Regulatory Status |
|---|---|---|---|---|---|---|
| EU/3/11/874 | sCTMP, as implanted cells are expected to help retinal function | Astellas Pharma Europe B.V.(Leiden, The Netherlands) | Human embryonic stem-cell-derived retinal pigment epithelial cells | Intravitreal injection | Stargardt’s disease | Orphan medicinal product designation by the EMA in 2011 Orphan medicinal product designation by the FDA 4 for the treatment of Stargardt’s macular dystrophy |
| EU/3/13/1168 | TEP, as implanted cells expected to help corneal regeneration | University of Newcastle. (Newcastle upon Tyne, United Kingdom) | Ex Vivo expanded autologous human corneal epithelium containing stem cells | Transplantation of a cell sheet | Limbal stem cell deficiency | Orphan medicinal product designation by the EMA in 2013 |
| EU/3/14/1340 | TEP, as implanted cells, expected to help corneal regeneration | NHS National Services Scotland, trading as Scottish National Blood Transfusion Service. (Edinburgh, United Kingdom) | Culture allogeneic corneal limbal stem cells | Transplantation of a cell sheet | Limbal stem cell deficiency | Orphan medicinal product designation by the EMA in 2014 |
| OraNera (EMEA/H/C/002443) | TEP, as OraNera, expected to replace damaged corneal cells | CellSeed Europe Ltd.. (London, United Kingdom) | Autologous oral mucosal epithelial cells | Transplantation of a cell sheet | Limbal stem cell deficiency | Application for a marketing authorization withdrawn from the EMA in 2013 |
| Holoclar (EU/3/08/579) | TEP (EMA classification) | Holostem Terapie Avanzate S.R.L.(Modena, Italy) | Ex vivo expan ded autologous human corneal epithelium containing stem cells | Transplantation of a cell sheet | Moderate-severe limbal stem cell deficiency, unilateral or bilateral, due to chemical or physical burns | Orphan medicinal product designation by the EMA in 2008 Conditional marketing authorization by the EMA in 2015. The orphan medicinal product designation was maintained |
| Nepic | Human somatic stem cell-processed products (Japanese PMDA 5 classification) | Japan Tissue Engineering Co., Ltd. (Gamagori, Japan) | Human autologous corneal limbus-derived corneal epithelial cell sheet | Transplantation of a cell sheet | Limbal stem cell deficiency | Orphan regenerative medical product designation by the Japanese PMDA in 2020 |
| Product (Commercial Name or Number Designated by EMA 1) | Manufacturer | Active Substance | Administration Route | Indication | REGULATORY STATUS |
|---|---|---|---|---|---|
| Vitravene EMA/H/C/000244 | Novartis (Basel, Switzerland) | Fomivirsen (antisense PODN 2) | Intravitreal injection | CMV 3 retinitis in HIV 4 infection | Marketing authorization by the FDA 5 1998 and by the EMA 1999. Withdrawn in 2002 in the EU 6 and in 2006 in the US 7 Currently authorized in Switzerland |
| Macugen EMA/671614/2010 | Pfizer (New York, USA) | Pegaptanib (RNA aptamer) | Intravitreal injection | Wet form of AMD 8/Diabetic macular edema | Marketing authorization by the EMA and by the FDA in 2006/ Withdrawn in 2011 to include a new application (diabetic macular edema) |
| Luxturna EU/3/15/1518; EU/3/12/981 | Novartis | Voretigene neparvovec (AAV 9-RPE65) | Subretinal injection | Retinitis pigmentosa/Leber´s congenital amaurosis | Marketing authorization by the EMA in 2018 and by the FDA in 2017 |
| Product (Commercial Name or Number Designated by EMA 1) | Manufacturer | Active Substance | Administration Route | Indication | Regulatory Status |
|---|---|---|---|---|---|
| NT-501 (EMA/COMP/808529/2012) (EMA/COMP/682942/2012) | Neurotech Pharmaceuticals Inc. (Cumberland, RI, USA) / Enpharma Ltd. (Oxford, United Kingdom) | Encapsulated human retinal pigment epithelial cell line transfected with plasmid vector expressing human CNTF 3 | Intravitreal implant | Retinitis pigmentosa/Macular telangiectasia type 2 | Orphan designation by the FDA 2 in 2004 and by the EMA in 2013/Orphan designation by the EMA and the FDA in 2012 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Paniagua, M.; de la Mata, A.; Galindo, S.; Blázquez, F.; Calonge, M.; Nieto-Miguel, T. Advanced Therapy Medicinal Products for the Eye: Definitions and Regulatory Framework. Pharmaceutics 2021, 13, 347. https://doi.org/10.3390/pharmaceutics13030347
López-Paniagua M, de la Mata A, Galindo S, Blázquez F, Calonge M, Nieto-Miguel T. Advanced Therapy Medicinal Products for the Eye: Definitions and Regulatory Framework. Pharmaceutics. 2021; 13(3):347. https://doi.org/10.3390/pharmaceutics13030347
Chicago/Turabian StyleLópez-Paniagua, Marina, Ana de la Mata, Sara Galindo, Francisco Blázquez, Margarita Calonge, and Teresa Nieto-Miguel. 2021. "Advanced Therapy Medicinal Products for the Eye: Definitions and Regulatory Framework" Pharmaceutics 13, no. 3: 347. https://doi.org/10.3390/pharmaceutics13030347
APA StyleLópez-Paniagua, M., de la Mata, A., Galindo, S., Blázquez, F., Calonge, M., & Nieto-Miguel, T. (2021). Advanced Therapy Medicinal Products for the Eye: Definitions and Regulatory Framework. Pharmaceutics, 13(3), 347. https://doi.org/10.3390/pharmaceutics13030347