
Patient-Derived Induced Pluripotent Stem Cells (iPSCs) and Cerebral Organoids for Drug Screening and Development in Autism Spectrum Disorder: Opportunities and Challenges




Abstract
1. Introduction
2. iPSC Models of ASDs
2.1. iPSCs-Derived Neural Progenitor Cells
2.2. iPSCs-Derived Neurons
2.3. iPSCs-Derived Astrocytes
2.4. iPSCs-Derived Oligodendrocytes
2.5. Current Challenges for the Applicability of iPSCs in ASD Modeling
3. Brain Organoid Models of ASDs
Current Challenges for the Applicability of Brain Organoids in ASD Modeling
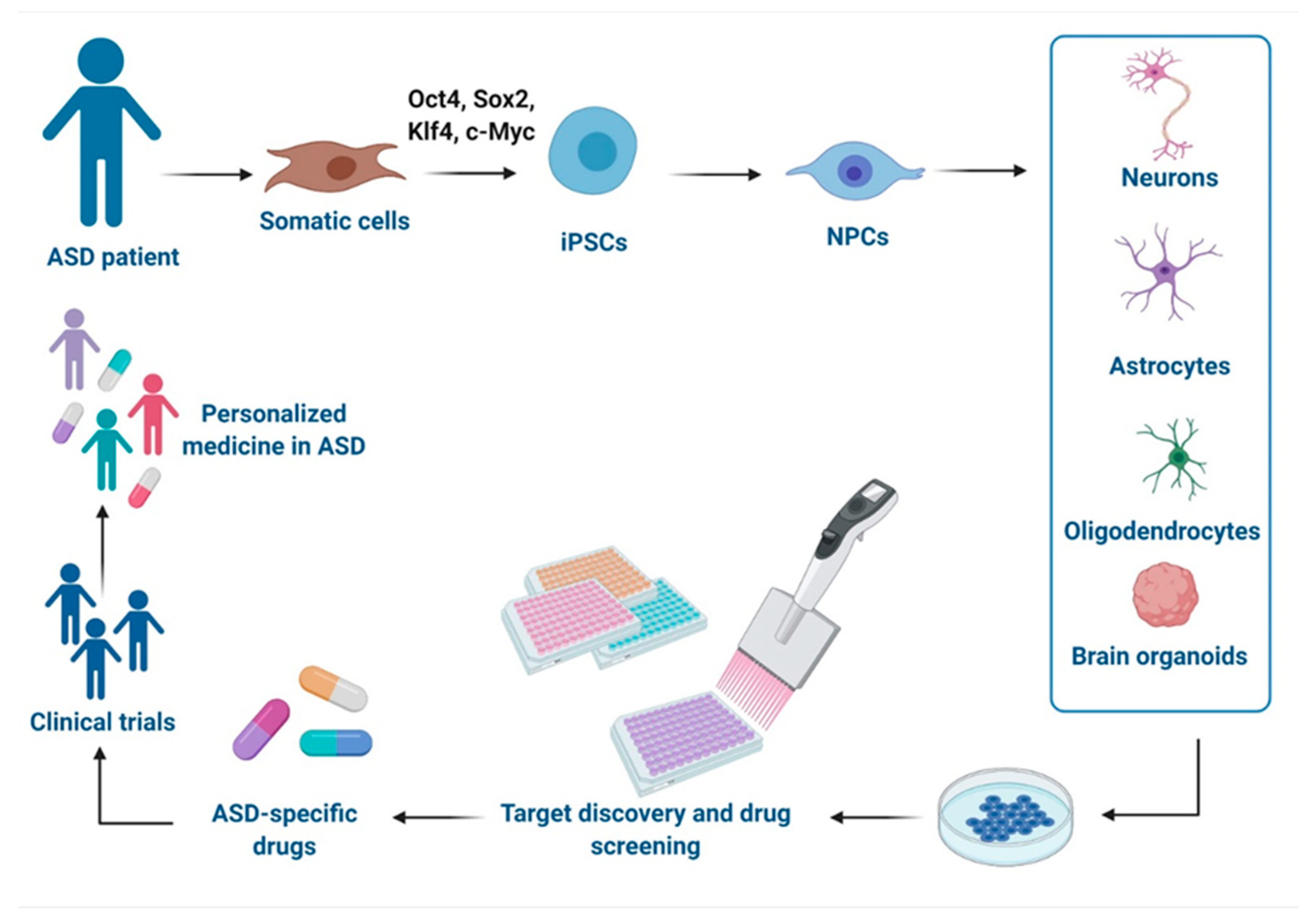
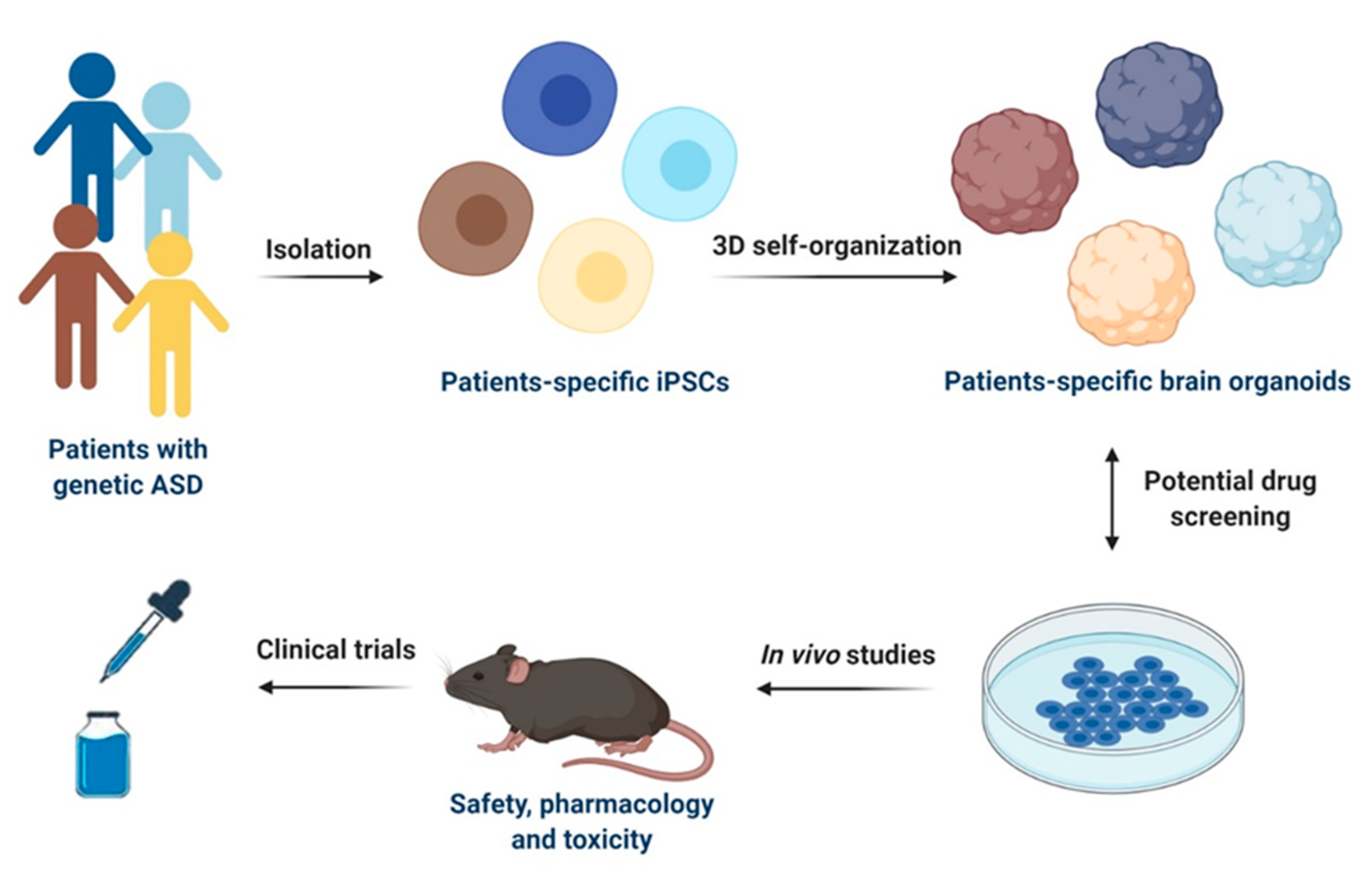
4. Use of ASD Models for Drug Discovery and Development
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, H.I.S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Nam, K.H.; Yi, S.A.; Jang, H.J.; Han, J.-W.; Lee, J. In Vitro modeling for inherited neurological diseases using induced pluripotent stem cells: From 2D to organoid. Arch. Pharmacal Res. 2020, 43, 877–889. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Machairaki, V. Human Pluripotent Stem Cells as In Vitro Models of Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2020, 1195, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nat. Cell Biol. 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- McCauley, H.A.; Wells, J.M. Pluripotent stem cell-derived organoids: Using principles of developmental biology to grow human tissues in a dish. Development 2017, 144, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Doernberg, E.; Hollander, E. Neurodevelopmental Disorders (ASD and ADHD): DSM-5, ICD-10, and ICD-11. CNS Spectrums 2016, 21, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.J.H.; Jones, R.M.; Pickles, A.; State, M.W.; et al. Autism spectrum disorder. Nat. Rev. Dis. Prim. 2020, 6, 1–23. [Google Scholar] [CrossRef]
- Liu, X.; Campanac, E.; Cheung, H.-H.; Ziats, M.N.; Canterel-Thouennon, L.; Raygada, M.; Baxendale, V.; Pang, A.L.-Y.; Yang, L.; Swedo, S.; et al. Idiopathic Autism: Cellular and Molecular Phenotypes in Pluripotent Stem Cell-Derived Neurons. Mol. Neurobiol. 2017, 54, 4507–4523. [Google Scholar] [CrossRef] [PubMed]
- Huguet, G.; Ey, E.; Bourgeron, T. The Genetic Landscapes of Autism Spectrum Disorders. Annu. Rev. Genom. Hum. Genet. 2013, 14, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Hyman, S.L.; Levy, S.E.; Myers, S.M.; Council on Children with Disabilities, Section on Developmental and Behavioral Pediatrics. Identification, Evaluation, and Management of Children with Autism Spectrum Disorder. Pediatrics 2019, 145, e20193447. [Google Scholar] [CrossRef]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors with Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.C.; Manaa, D.; Pawitan, Y.; Reichert, J.G.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Ziats, M.N.; Rennert, O.M. The Evolving Diagnostic and Genetic Landscapes of Autism Spectrum Disorder. Front. Genet. 2016, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; Napolioni, V. Autism genetics. Behav. Brain Res. 2013, 251, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Bury, L.A.; Wynshaw-Boris, A. Modeling Non-Syndromic Autism with Human-Induced Pluripotent Stem Cells. Neuropsychopharmacology 2017, 43, 219–220. [Google Scholar] [CrossRef] [PubMed]
- De La Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nat. Cell Biol. 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, X.; Wang, M.; Hu, Y.; Xue, K.; Gu, S.; Lv, L.; Huang, S.; Xie, W. Potential role of genomic imprinted genes and brain developmental related genes in autism. BMC Med Genom. 2020, 13, 54. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.H.; Greenberg, M.E. Activity-dependent neuronal signalling and autism spectrum disorder. Nat. Cell Biol. 2013, 493, 327–337. [Google Scholar] [CrossRef]
- Gilbert, J.; Man, H.-Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell. Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef]
- Tordjman, S.; Somogyi, E.; Coulon, N.; Kermarrec, S.; Cohen, D.; Bronsard, G.; Bonnot, O.; Weismann-Arcache, C.; Botbol, M.; Lauth, B.; et al. Gene × Environment Interactions in Autism Spectrum Disorders: Role of Epigenetic Mechanisms. Front. Psychiatry 2014, 5, 53. [Google Scholar] [CrossRef]
- Cheroni, C.; Caporale, N.; Testa, G. Autism spectrum disorder at the crossroad between genes and environment: Contributions, convergences, and interactions in ASD developmental pathophysiology. Mol. Autism 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Karahmadi, M.; Karimi, P.; Kamali, E.; Mousavi, S.M. Environmental factors influencing the risk of autism. J. Res. Med Sci. 2017, 22, 27. [Google Scholar] [CrossRef]
- Hallmayer, J. Genetic Heritability and Shared Environmental Factors Among Twin Pairs with Autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Bastaki, K.N.; Alwan, S.; Zahir, F.R. Maternal Prenatal Exposures in Pregnancy and Autism Spectrum Disorder: An Insight into the Epigenetics of Drugs and Diet as Key Environmental Influences. Adv. Neurobiol. 2020, 24, 143–162. [Google Scholar] [CrossRef]
- Brito, A.; Russo, F.B.; Muotri, A.R.; Beltrão-Braga, P.C.B. Autism spectrum disorders and disease modeling using stem cells. Cell Tissue Res. 2018, 371, 153–160. [Google Scholar] [CrossRef]
- Russo, F.B.; Freitas, B.C.; Pignatari, G.C.; Fernandes, I.R.; Sebat, J.; Muotri, A.R.; Beltrão-Braga, P.C.B. Modeling the Interplay Between Neurons and Astrocytes in Autism Using Human Induced Pluripotent Stem Cells. Biol. Psychiatry 2018, 83, 569–578. [Google Scholar] [CrossRef]
- Suzuki, I.K.; Vanderhaeghen, P. Is this a brain which I see before me? Modeling human neural development with pluripotent stem cells. Development 2015, 142, 3138–3150. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.-M.; Topol, A.; Brennand, K.J. From “Directed Differentiation” to “Neuronal Induction”: Modeling Neuropsychiatric Disease. Biomark. Insights 2015, 10, BMI.S20066–41. [Google Scholar] [CrossRef]
- Sacco, R.; Cacci, E.; Novarino, G. Neural stem cells in neuropsychiatric disorders. Curr. Opin. Neurobiol. 2018, 48, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Belinson, H.; Tian, Y.; Freitas, B.C.; Fu, C.; Vadodaria, K.; Beltrao-Braga, P.; Trujillo, C.A.; Mendes, A.P.; Padmanabhan, K.; et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol. Psychiatry 2017, 22, 820–835. [Google Scholar] [CrossRef]
- Wang, M.; Wei, P.-C.; Lim, C.K.; Gallina, I.S.; Marshall, S.; Marchetto, M.C.; Alt, F.W.; Gage, F.H. Increased Neural Progenitor Proliferation in a hiPSC Model of Autism Induces Replication Stress-Associated Genome Instability. Cell Stem Cell 2020, 26, 221–233.e6. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, S.M.; Magdalon, J.; Griesi-Oliveira, K.; Yamamoto, G.L.; Santacruz-Perez, C.; Fogo, M.; Passos-Bueno, M.R.; Sertié, A.L. Rare RELN variants affect Reelin-DAB1 signal transduction in autism spectrum disorder. Hum. Mutat. 2018, 39, 1372–1383. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Acab, A.; Gupta, A.R.; Sunaga, D.Y.; Chailangkarn, T.; Nicol, X.; Nunez, Y.; Walker, M.F.; Murdoch, J.D.; Sanders, S.J.; et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry 2015, 20, 1350–1365. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef]
- Muotri, A.R.; Marchetto, M.C.N.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nat. Cell Biol. 2010, 468, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Nageshappa, S.; Carromeu, C.; Trujillo, C.A.; Mesci, P.; Espuny-Camacho, I.; Pasciuto, E.; Vanderhaeghen, P.; Verfaillie, C.M.; Raitano, S.; Kumar, A.; et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 2016, 21, 178–188. [Google Scholar] [CrossRef]
- Rodrigues, D.C.; Mufteev, M.; Weatheritt, R.J.; Djuric, U.; Ha, K.C.; Ross, P.J.; Wei, W.; Piekna, A.; Sartori, M.A.; Byres, L.; et al. Shifts in Ribosome Engagement Impact Key Gene Sets in Neurodevelopment and Ubiquitination in Rett Syndrome. Cell Rep. 2020, 30, 4179–4196.e11. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Segal, M.; Ben-Yosef, D. Neural differentiation of fragile X human embryonic stem cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 2013, 374, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Achuta, V.S.; Möykkynen, T.; Peteri, U.-K.; Turconi, G.; Rivera, C.; Keinänen, K.; Castrén, M.L. Functional changes of AMPA responses in human induced pluripotent stem cell–derived neural progenitors in fragile X syndrome. Sci. Signal. 2018, 11, eaan8784. [Google Scholar] [CrossRef] [PubMed]
- Zucco, A.J.; Pozzo, V.D.; Afinogenova, A.; Hart, R.P.; Devinsky, O.; D’Arcangelo, G. Neural progenitors derived from Tuberous Sclerosis Complex patients exhibit attenuated PI3K/AKT signaling and delayed neuronal differentiation. Mol. Cell. Neurosci. 2018, 92, 149–163. [Google Scholar] [CrossRef]
- Martin, P.; Wagh, V.; Reis, S.A.; Erdin, S.; Beauchamp, R.L.; Shaikh, G.; Talkowski, M.; Thiele, E.; Sheridan, S.D.; Haggarty, S.J.; et al. TSC patient-derived isogenic neural progenitor cells reveal altered early neurodevelopmental phenotypes and rapamycin-induced MNK-eIF4E signaling. Mol. Autism 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Breen, M.S.; Browne, A.; Hoffman, G.E.; Stathopoulos, S.; Brennand, K.; Buxbaum, J.D.; Drapeau, E. Transcriptional signatures of participant-derived neural progenitor cells and neurons implicate altered Wnt signaling in Phelan-McDermid syndrome and autism. Mol. Autism 2020, 11, 1–23. [Google Scholar] [CrossRef]
- Paşca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Paşca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Voineagu, I.; Paşca, S.P.; Won, H.; Chandran, V.; Horvath, S.; E Dolmetsch, R.; Geschwind, D.H. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome Med. 2014, 6, 75. [Google Scholar] [CrossRef]
- Moon, U.Y.; Park, J.Y.; Park, R.; Cho, J.Y.; Hughes, L.J.; McKenna, J.T.; Goetzl, L.; Cho, S.-H.; Crino, P.B.; Gambello, M.J.; et al. Impaired Reelin-Dab1 Signaling Contributes to Neuronal Migration Deficits of Tuberous Sclerosis Complex. Cell Rep. 2015, 12, 965–978. [Google Scholar] [CrossRef]
- Vidal, S.; Xiol, C.; Pascual-Alonso, A.; O’Callaghan, M.; Pineda, M.; Armstrong, J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int. J. Mol. Sci. 2019, 20, 3925. [Google Scholar] [CrossRef] [PubMed]
- Kyle, S.M.; Vashi, N.; Justice, M.J. Rett syndrome: A neurological disorder with metabolic components. Open Biol. 2018, 8, 170216. [Google Scholar] [CrossRef]
- Banerjee, A.; Miller, M.T.; Li, K.; Sur, M.; E Kaufmann, W. Towards a better diagnosis and treatment of Rett syndrome: A model synaptic disorder. Brain 2019, 142, 239–248. [Google Scholar] [CrossRef]
- Rajaratnam, A.; Potter, L.A.; Biag, H.M.B.; Schneider, A.; Petrasic, I.C.; Hagerman, R.J. Review of Autism Profiles and Response to Sertraline in Fragile X Syndrome-Associated Autism vs. Non-syndromic Autism; Next Steps for Targeted Treatment. Front. Neurol. 2020, 11, 581429. [Google Scholar] [CrossRef]
- Sztainberg, Y.; Zoghbi, H.Y. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Volpi, A.; Sala, G.; Lesma, E.; Labriola, F.; Righetti, M.; Alfano, R.M.; Cozzolino, M. Tuberous sclerosis complex: New insights into clinical and therapeutic approach. J. Nephrol. 2018, 32, 355–363. [Google Scholar] [CrossRef]
- Vogels, A.; Droogmans, G.; Vergaelen, E.; Van Buggenhout, G.; Swillen, A. Recent developments in Phelan–McDermid syndrome research: An update on cognitive development, communication and psychiatric disorders. Curr. Opin. Psychiatry 2021, 34, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Lv, H.; Yang, T.; Du, X.; Sun, Y.; Xiao, B.; Fan, Y.; Luo, X.; Zhan, Y.; Wang, L.; et al. A 29 Mainland Chinese cohort of patients with Phelan–McDermid syndrome: Genotype–phenotype correlations and the role of SHANK3 haploinsufficiency in the important phenotypes. Orphanet J. Rare Dis. 2020, 15, 1–12. [Google Scholar] [CrossRef]
- Bekdash, R.; Klein, A.D.; Yazawa, M. Timothy syndrome iPSC modeling. Mol. Cell. Neurosci. 2020, 107, 103529. [Google Scholar] [CrossRef]
- Arai, Y.; Taverna, E. Neural Progenitor Cell Polarity and Cortical Development. Front. Cell. Neurosci. 2017, 11, 384. [Google Scholar] [CrossRef]
- Sajdel-Sulkowska, E.M.; Xu, M.; McGinnis, W.; Koibuchi, N. Brain Region-Specific Changes in Oxidative Stress and Neurotrophin Levels in Autism Spectrum Disorders (ASD). Cerebellum 2010, 10, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Ameis, S.H.; Catani, M. Altered white matter connectivity as a neural substrate for social impairment in Autism Spectrum Disorder. Cortex 2015, 62, 158–181. [Google Scholar] [CrossRef]
- Armstrong, D.D.; Dunn, K.; Antalffy, B. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J. Neuropathol. Exp. Neurol. 1998, 57, 1013–1017. [Google Scholar] [CrossRef]
- Belichenko, P.V.; Hagberg, B.; Dahlström, A. Morphological study of neocortical areas in Rett syndrome. Acta Neuropathol. 1997, 93, 50–61. [Google Scholar] [CrossRef]
- Hensch, T.K. Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 2005, 6, 877–888. [Google Scholar] [CrossRef]
- Rubenstein, J.L. Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr. Opin. Neurol. 2010, 23, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Uzunova, G.; Pallanti, S.; Hollander, E. Excitatory/inhibitory imbalance in autism spectrum disorders: Implications for interventions and therapeutics. World J. Biol. Psychiatry 2015, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Griesi-Oliveira, K.; Fogo, M.S.; Pinto, B.G.G.; Alves, A.Y.; Suzuki, A.M.; Morales, A.G.; Ezquina, S.; Sosa, O.J.; Sutton, G.J.; Sunaga-Franze, D.Y.; et al. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol. Psychiatry 2020, 1–17. [Google Scholar] [CrossRef]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef]
- Zaslavsky, K.; Zhang, W.-B.; McCready, F.P.; Rodrigues, D.C.; Deneault, E.; Loo, C.; Zhao, M.; Ross, P.J.; El Hajjar, J.; Romm, A.; et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat. Neurosci. 2019, 22, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Landucci, E.; Brindisi, M.; Bianciardi, L.; Catania, L.M.; Daga, S.; Croci, S.; Frullanti, E.; Fallerini, C.; Butini, S.; Brogi, S.; et al. iPSC-derived neurons profiling reveals GABAergic circuit disruption and acetylated α-tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res. 2018, 368, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Amenduni, M.; De Filippis, R.; Cheung, A.Y.L.; Disciglio, V.; Epistolato, M.C.; Ariani, F.; Mari, F.; Mencarelli, M.A.; Hayek, Y.; Renieri, A.; et al. iPS cells to model CDKL5-related disorders. Eur. J. Hum. Genet. 2011, 19, 1246–1255. [Google Scholar] [CrossRef]
- Yennawar, M.; White, R.S.; Jensen, F.E. AMPA Receptor Dysregulation and Therapeutic Interventions in a Mouse Model of CDKL5 Deficiency Disorder. J. Neurosci. 2019, 39, 4814–4828. [Google Scholar] [CrossRef]
- Mackay, C.I.; Wong, K.; Demarest, S.T.; Benke, T.A.; Downs, J.; Leonard, H. Exploring genotype-phenotype relationships in the CDKL5 deficiency disorder using an international dataset. Clin. Genet. 2021, 99, 157–165. [Google Scholar] [CrossRef]
- Ricciardi, S.; Ungaro, F.; Hambrock, M.; Rademacher, N.; Stefanelli, G.; Brambilla, D.; Sessa, A.; Magagnotti, C.; Bachi, A.; Giarda, E.; et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1–PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 2012, 14, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic Characterization of the FMR1 Gene and Aberrant Neurodevelopment in Human Induced Pluripotent Stem Cell Models of Fragile X Syndrome. PLoS ONE 2011, 6, e26203. [Google Scholar] [CrossRef]
- Halevy, T.; Czech, C.; Benvenisty, N. Molecular Mechanisms Regulating the Defects in Fragile X Syndrome Neurons Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Chen, X.; Feng, Y.; Zeng, Q.; Jiang, C.; Zhu, X.; Fan, G.; Xue, Z. Integrated transcriptome analysis of human iPS cells derived from a fragile X syndrome patient during neuronal differentiation. Sci. China Life Sci. 2016, 59, 1093–1105. [Google Scholar] [CrossRef]
- Boland, M.J.; Nazor, K.L.; Tran, H.T.; Szücs, A.; Lynch, C.L.; Paredes, R.; Tassone, F.; Sanna, P.P.; Hagerman, R.J.; Loring, J.F. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain 2017, 140, 582–598. [Google Scholar] [CrossRef]
- Utami, K.H.; Skotte, N.H.; Colaço, A.R.; Yusof, N.A.B.M.; Sim, B.; Yeo, X.Y.; Bae, H.-G.; Garcia-Miralles, M.; Radulescu, C.I.; Chen, Q.; et al. Integrative Analysis Identifies Key Molecular Signatures Underlying Neurodevelopmental Deficits in Fragile X Syndrome. Biol. Psychiatry 2020, 88, 500–511. [Google Scholar] [CrossRef]
- Doers, M.E.; Musser, M.T.; Nichol, R.; Berndt, E.R.; Baker, M.; Gomez, T.M.; Zhang, S.-C.; Abbeduto, L.; Bhattacharyya, A. iPSC-Derived Forebrain Neurons from FXS Individuals Show Defects in Initial Neurite Outgrowth. Stem Cells Dev. 2014, 23, 1777–1787. [Google Scholar] [CrossRef]
- Liu, J.; Kościelska, K.A.; Cao, Z.; Hulsizer, S.; Grace, N.; Mitchell, G.; Nacey, C.; Githinji, J.; McGee, J.; Garcia-Arocena, D.; et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum. Mol. Genet. 2012, 21, 3795–3805. [Google Scholar] [CrossRef]
- Li, Y.; Cao, J.; Chen, M.; Li, J.; Sun, Y.; Zhang, Y.; Zhu, Y.; Wang, L.; Zhang, C. Abnormal Neural Progenitor Cells Differentiated from Induced Pluripotent Stem Cells Partially Mimicked Development of TSC2 Neurological Abnormalities. Stem Cell Rep. 2017, 8, 883–893. [Google Scholar] [CrossRef]
- Nadadhur, A.G.; Alsaqati, M.; Gasparotto, L.; Cornelissen-Steijger, P.; Van Hugte, E.; Dooves, S.; Harwood, A.J.; Heine, V.M. Neuron-Glia Interactions Increase Neuronal Phenotypes in Tuberous Sclerosis Complex Patient iPSC-Derived Models. Stem Cell Rep. 2019, 12, 42–56. [Google Scholar] [CrossRef]
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/Inhibitory Synaptic Imbalance Leads to Hippocampal Hyperexcitability in Mouse Models of Tuberous Sclerosis. Neuron 2013, 78, 510–522. [Google Scholar] [CrossRef]
- Winden, K.D.; Sundberg, M.; Yang, C.; Wafa, S.M.; Dwyer, S.; Chen, P.-F.; Buttermore, E.D.; Sahin, M. Biallelic Mutations in TSC2 Lead to Abnormalities Associated with Cortical Tubers in Human iPSC-Derived Neurons. J. Neurosci. 2019, 39, 9294–9305. [Google Scholar] [CrossRef]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nat. Cell Biol. 2013, 503, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Darville, H.; Poulet, A.; Rodet-Amsellem, F.; Chatrousse, L.; Pernelle, J.; Boissart, C.; Héron, D.; Nava, C.; Perrier, A.; Jarrige, M.; et al. Human Pluripotent Stem Cell-derived Cortical Neurons for High Throughput Medication Screening in Autism: A Proof of Concept Study in SHANK3 Haploinsufficiency Syndrome. EBioMedicine 2016, 9, 293–305. [Google Scholar] [CrossRef]
- Gouder, L.; Vitrac, A.; Goubran-Botros, H.; Danckaert, A.; Tinevez, J.-Y.; André-Leroux, G.; Atanasova, E.; Lemière, N.; Biton, A.; Leblond, C.S.; et al. Altered spinogenesis in iPSC-derived cortical neurons from patients with autism carrying de novo SHANK3 mutations. Sci. Rep. 2019, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Danko, T.; Botelho, S.C.; Patzke, C.; Pak, C.; Wernig, M.; Südhof, T.C. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 2016, 352, aaf2669. [Google Scholar] [CrossRef]
- Krey, J.F.; Pasca, S.P.; Shcheglovitov, A.; Yazawa, M.; Schwemberger, R.; Rasmusson, R.L.; Dolmetsch, R.E. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 2013, 16, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakos, G.; Haveles, C.; Arjun, A.; Petrova, R.; Rana, A.; Portmann, T.; Paşca, S.P.; Palmer, T.D.; E Dolmetsch, R. Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. eLife 2019, 8, 51037. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Chen, P.-F.; Ng, K.Y.; Bourgois-Rocha, F.; Lemtiri-Chlieh, F.; Levine, E.S.; Lalande, M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader–Willi syndromes. Proc. Natl. Acad. Sci. USA 2010, 107, 17668–17673. [Google Scholar] [CrossRef] [PubMed]
- Madaan, M.; Mendez, M.D. Angelman Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Khatri, N.; Man, H.-Y. The Autism and Angelman Syndrome Protein Ube3A/E6AP: The Gene, E3 Ligase Ubiquitination Targets and Neurobiological Functions. Front. Mol. Neurosci. 2019, 12, 109. [Google Scholar] [CrossRef]
- Stanurova, J.; Neureiter, A.; Hiber, M.; Kessler, H.D.O.; Stolp, K.; Goetzke, R.; Klein, D.; Bankfalvi, A.; Klump, H.; Steenpass, L. Angelman syndrome-derived neurons display late onset of paternal UBE3A silencing. Sci. Rep. 2016, 6, 30792. [Google Scholar] [CrossRef]
- Fink, J.J.; Robinson, T.M.; Germain, N.D.; Sirois, C.L.; Bolduc, K.A.; Ward, A.J.; Rigo, F.; Chamberlain, S.J.; Levine, E.S. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun. 2017, 8, 15038. [Google Scholar] [CrossRef]
- Siracusa, R.; Fusco, R.; Cuzzocrea, S. Astrocytes: Role and Functions in Brain Pathologies. Front. Pharmacol. 2019, 10, 1114. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, C.; Ziats, M.N.; Rennert, O.M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol. Autism 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cerdeño, V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev. Neurobiol. 2017, 77, 393–404. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2004, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Saghazadeh, A.; Ataeinia, B.; Keynejad, K.; Abdolalizadeh, A.; Hirbod-Mobarakeh, A.; Rezaei, N. A meta-analysis of pro-inflammatory cytokines in autism spectrum disorders: Effects of age, gender, and latitude. J. Psychiatr. Res. 2019, 115, 90–102. [Google Scholar] [CrossRef]
- Malik, M.; Sheikh, A.M.; Wen, G.G.; Spivack, W.; Brown, W.T.; Li, X. Expression of inflammatory cytokines, Bcl2 and cathepsin D are altered in lymphoblasts of autistic subjects. Immunobiology 2011, 216, 80–85. [Google Scholar] [CrossRef]
- Enstrom, A.M.; Onore, C.E.; Van De Water, J.A.; Ashwood, P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain, Behav. Immun. 2010, 24, 64–71. [Google Scholar] [CrossRef]
- Williams, E.C.; Zhong, X.; Mohamed, A.; Li, R.; Liu, Y.; Dong, Q.; Ananiev, G.E.; Mok, J.C.C.; Lin, B.R.; Lu, J.; et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum. Mol. Genet. 2014, 23, 2968–2980. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Savas, J.N.; Miller, M.T.; Hu, X.; Carromeu, C.; Lavallée-Adam, M.; Freitas, B.C.G.; Muotri, A.R.; Yates, J.R.; Ghosh, A. Proteomic analyses reveal misregulation of LIN28 expression and delayed timing of glial differentiation in human iPS cells with MECP2 loss-of-function. PLoS ONE 2019, 14, e0212553. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y. Oligodendrocyte Physiology Modulating Axonal Excitability and Nerve Conduction. Adv. Exp. Med. Biol. 2019, 1190, 123–144. [Google Scholar] [CrossRef]
- Zheng, W.; Li, Q.; Zhao, C.; Da, Y.; Zhang, H.-L.; Chen, Z. Differentiation of Glial Cells From hiPSCs: Potential Applications in Neurological Diseases and Cell Replacement Therapy. Front. Cell. Neurosci. 2018, 12, 239. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Ingrassia, D.; Sarvadio, M.; Schiavi, S.; Steardo, L.; Verkhratsky, A.; Trezza, V.; Scuderi, C. Neuroglia in the autistic brain: Evidence from a preclinical model. Mol. Autism 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Corrigan, N.M.; Shaw, D.W.W.; Estes, A.M.; Richards, T.L.; Munson, J.; Friedman, S.D.; Dawson, G.; Artru, A.A.; Dager, S.R. Atypical Developmental Patterns of Brain Chemistry in Children with Autism Spectrum Disorder. JAMA Psychiatry 2013, 70, 964–974. [Google Scholar] [CrossRef]
- Kleinhans, N.M.; Schweinsburg, B.C.; Cohen, D.N.; Müller, R.-A.; Courchesne, E. N-acetyl aspartate in autism spectrum disorders: Regional effects and relationship to fMRI activation. Brain Res. 2007, 1162, 85–97. [Google Scholar] [CrossRef]
- Cheli, V.T.; González, D.A.S.; Zamora, N.N.; Lama, T.N.; Spreuer, V.; Rasmusson, R.L.; Bett, G.C.; Panagiotakos, G.; Paez, P.M. Enhanced oligodendrocyte maturation and myelination in a mouse model of Timothy syndrome. Glia 2018, 66, 2324–2339. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement from the American Heart Association. Circ. Genom. Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Miyagi-Shiohira, C.; Nakashima, Y. Induced Tissue-Specific Stem Cells and Epigenetic Memory in Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 930. [Google Scholar] [CrossRef]
- Ilieva, M.; Fex-Svenningsen, A.; Thorsen, M.; Michel, T.M. Psychiatry in a Dish: Stem Cells and Brain Organoids Modeling Autism Spectrum Disorders. Biol. Psychiatry 2018, 83, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.-Y.; Weick, J.P.; Yu, J.; Ma, L.-X.; Zhang, X.-Q.; Thomson, J.A.; Zhang, S.-C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef]
- Sasai, Y. Next-Generation Regenerative Medicine: Organogenesis from Stem Cells in 3D Culture. Cell Stem Cell 2013, 12, 520–530. [Google Scholar] [CrossRef]
- Paşca, A.M.; A Sloan, S.; E Clarke, L.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; A O’Rourke, N.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Paşca, S.P. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 2017, 95, 779–790.e6. [Google Scholar] [CrossRef] [PubMed]
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Levy, R.J.; Huguenard, J.R.; Pașca, S.P. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.T.; Paquola, A.C.M.; Stern, S.; Gosselin, D.; Ku, M.; Pena, M.; Kuret, T.J.M.; Liyanage, M.; Mansour, A.A.; Jaeger, B.N.; et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci. 2019, 22, 243–255. [Google Scholar] [CrossRef]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 2018, 23, 1051–1065. [Google Scholar] [CrossRef]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Hwang, S.-M.; Hysolli, E.; Cakir, B.; Kim, K.-Y.; Wang, W.; Kang, Y.-J.; Clement, E.M.; et al. Dysregulation of BRD4 Function Underlies the Functional Abnormalities of MeCP2 Mutant Neurons. Mol. Cell 2020, 79, 84–98.e9. [Google Scholar] [CrossRef]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef]
- Niu, W.; Parent, J.M. Modeling genetic epilepsies in a dish. Dev. Dyn. 2020, 249, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nat. Cell Biol. 2017, 545, 54–59. [Google Scholar] [CrossRef]
- Sun, A.X.; Yuan, Q.; Fukuda, M.; Yu, W.; Yan, H.; Lim, G.G.Y.; Nai, M.H.; D’Agostino, G.A.; Tran, H.-D.; Itahana, Y.; et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 2019, 366, 1486–1492. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; A Lancaster, M.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; A Knoblich, J. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Griffiths, R.; Price, D.J.; Mason, J.O. Cerebral organoids as tools to identify the developmental roots of autism. Mol. Autism 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A Neuroligin-3 Mutation Implicated in Autism Increases Inhibitory Synaptic Transmission in Mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef]
- Basu, R.; Duan, X.; Taylor, M.R.; Martin, E.A.; Muralidhar, S.; Wang, Y.; Gangi-Wellman, L.; Das, S.C.; Yamagata, M.; West, P.J.; et al. Heterophilic Type II Cadherins Are Required for High-Magnitude Synaptic Potentiation in the Hippocampus. Neuron 2017, 96, 160–176.e8. [Google Scholar] [CrossRef] [PubMed]
- Kolevzon, A.; Bush, L.; Wang, A.T.; Halpern, D.; Frank, Y.; Grodberg, D.; Rapaport, R.; Tavassoli, T.; Chaplin, W.; Soorya, L.; et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol. Autism 2014, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, O.S.; Ho, E.; Barnes, K.V.; O’Leary, H.M.; Pereira, L.M.; Finkelstein, Y.; Nelson, C.A.; Vogel-Farley, V.; DeGregorio, G.; Holm, I.A.; et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 4596–4601. [Google Scholar] [CrossRef]
- Sadowski, K.; Kotulska, K.; Schwartz, R.A.; Jóźwiak, S. Systemic effects of treatment with mTOR inhibitors in tuberous sclerosis complex: A comprehensive review. J. Eur. Acad. Dermatol. Venereol. 2015, 30, 586–594. [Google Scholar] [CrossRef]
- Russo, F.B.; Brito, A.; de Freitas, A.M.; Castanha, A.; de Freitas, B.C.; Beltrão-Braga, P.C.B. The use of iPSC technology for modeling Autism Spectrum Disorders. Neurobiol. Dis. 2019, 130, 104483. [Google Scholar] [CrossRef] [PubMed]
- Brick, D.J.; Nethercott, H.E.; Montesano, S.; Banuelos, M.G.; Stover, A.E.; Schutte, S.S.; O’Dowd, D.K.; Hagerman, R.J.; Ono, M.Y.; Hessl, D.R.; et al. The Autism Spectrum Disorders Stem Cell Resource at Children’s Hospital of Orange County: Implications for Disease Modeling and Drug Discovery. Stem Cells Transl. Med. 2014, 3, 1275–1286. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; A Barlow, K.; Wang, J.; Xiangdong, M.; Meng, X.; E Paschon, D.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2010, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; Dicarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Sci. 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- De La Vega, L.; Lee, C.; Sharma, R.; Amereh, M.; Willerth, S.M. 3D bioprinting models of neural tissues: The current state of the field and future directions. Brain Res. Bull. 2019, 150, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Salaris, F.; Rosa, A. Construction of 3D in vitro models by bioprinting human pluripotent stem cells: Challenges and opportunities. Brain Res. 2019, 1723, 146393. [Google Scholar] [CrossRef]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Enomura, M.; Yoshizawa, E.; Kimura, M.; Koike, H.; Ueno, Y.; Matsuzaki, T.; Yamazaki, T.; Toyohara, T.; Osafune, K.; et al. Vascularized and Complex Organ Buds from Diverse Tissues via Mesenchymal Cell-Driven Condensation. Cell Stem Cell 2015, 16, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Ham, O.; Jin, Y.B.; Kim, J.; Lee, M.-O. Blood vessel formation in cerebral organoids formed from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2020, 521, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Types | Advantages | Disadvantages |
|---|---|---|
| ESCs | Low cost Established protocols for maintenance in culture Any cell type differentiation (pluripotency) Efficient differentiation | Mutation rate Embryo destruction Ethical/political concerns Difficulty to obtain Lack of genetic/immunohistocompatibility match |
| MSCs | Availability Ease to isolate and expand No ethical concerns Trans-differentiation capacities Success in various clinical applications | Limited number of cell type differentiation (multipotency) Loss of proliferative and differentiation capacities over continuous passages Standardization difficulty Genetic heterogeneity |
| iPSCs | No ethical concerns Ease to obtain Use of abundant somatic cells of donor Any cell type differentiation (pluripotency) Genetic/immunohistocompatibility match Utility for drug development and developmental studies | Cost of production Difficulty of standardization, reproducibility and maintenance Tumorigenesis Genomic instability |
| Disease | Genetic Mutations in Samples (n) | iPSCs-Based Models | Relevant Findings | Effective Drugs | Reference |
|---|---|---|---|---|---|
| Non-syndromic ASD | different no ASD-related variants (3) | neurons and astrocytes | decreased synapses and release of excitatory neurotransmitters, glial dysfunction, and high levels of IL-6 | anti-IL-6 | [31] |
| different no ASD-related variants (8) | NPCs and neurons | increased proliferation in NPCs, abnormal neurogenesis, reduced synaptogenesis, and decreased release of inhibitory/excitatory neurotransmitters | IGF-1 | [35] | |
| different no ASD-related variants (3) | NPCs | hyperproliferation of NPCs | [36] | ||
| rare compound heterozygous missense variants in RELN (1) | NPCs | impaired crosstalk between mTORC1 and Reelin-DAB1 pathways | rapamycin | [37] | |
| de novo balanced translocation in TRPC6 (1) | NPCs and neurons | abnormal neuronal development and morphology, fewer dendritic spines and synapses | IGF-1 and hyperforin | [38] | |
| loss-of-function mutations in FOXG1 (4) | neurons and brain organoids | overproduction of GABAergic neurons and GABA neurotransmitter | [69] | ||
| heterozygous loss-of-function mutations in SHANK2 (2) | neurons | increased dendrite length and complexity, synapse number, and frequency of spontaneous excitatory post-synaptic currents | agonist of mGluRs DHPG | [70] | |
| different no ASD-related variants (8) | brain organoids | neurodevelopmental abnormalities triggered by temporal dysregulation of specific gene networks | [121] | ||
| heterozygous knockout of CHD8 (1) | brain organoids | enrichment of genes involved in GABAergic interneuron development and Wnt/β-catenin signaling | [122] | ||
| RTT | missense, frameshift and nonsense mutations in MECP2 (4) | NPCs and neurons | reduced soma size, dendritic spine densities and synapses, altered Ca2+ signaling, and electrophysiological defects | IGF-1 and gentamicin | [39] |
| frameshift mutation in MECP2 (1) | NPCs and neurons | increased frequency of de novo LINE-1 retrotransposition | [40] | ||
| different duplications in MECP2 (3) | NPCs and neurons | altered expression of neuronal progenitor genes, increased synaptogenesis and dendritic complexity with altered network synchronization | histone deacetylase inhibitor NCH-51 | [41] | |
| large deletion in MECP2 (1) | NPCs and cortical neurons | repressed translation and decreased ribosome engagement of NEDD4-family ubiquitin ligases | [42] | ||
| missense mutations in MECP2 (2) | neurons | impaired microtubule network and decreased acetylated α-tubulin | selective inhibitors of HDAC6 | [71] | |
| missense and nonsense mutations in MECP2 (3) | neurons and astrocytes | neuronal morphological abnormalities mediated by mutant astrocytes | IGF-1 and GPE | [105] | |
| missense and nonsense mutations in MECP2 (2) | astrocytes | perturbed astrocyte differentiation | [106] | ||
| missense and frameshift mutations in MECP2 (2) | brain organoids | impaired neurogenesis, neuronal differentiation and migration | [123] | ||
| missense and nonsense mutations in MECP2 (3) | brain organoids | cell-type-specific impairments | BET inhibitor JQ1 | [124] | |
| CDKL5 disorder | missense and nonsense mutations in CDKL5 (2) | neurons | decreased density of dendritic spines and reduced number of excitatory synapse | [72] | |
| translocation t(7;X) inactivating CDKL5 (1) | neurons | decreased density of dendritic spines and loss of synaptic contacts | [75] | ||
| FXS | >200 CGG repeats in 5′UTR FMR1 (3) | NPCs | abnormal expression of key NPC genes (SOX1, NOTCH1, PAX6) | [43] | |
| >200 CGG repeats in 5′UTR FMR1 (4) | NPCs | impaired Ca2+ signaling affecting neuronal differentiation | [44] | ||
| >700 CGG repeats in 5′UTR FMR1 (3) | neurons | impaired neuronal differentiation and maturation | [76] | ||
| FMR1 knockout (2) | neurons | abnormal synaptic transmission, neuronal differentiation, and cell proliferation | [80] | ||
| >700 CGG repeats in 5′UTR FMR1 (3) | neurons | altered neurite outgrowth and branching defects | [81] | ||
| 94 CGG repeats in 5′UTR FMR1 (1) | neurons | dysregulated Ca2+ signals, reduced synaptic protein expression, and shorter neurites | [82] | ||
| TSC | de novo mutations in TSC2 (2) | NPCs and neurons | delayed neuronal differentiation | [45] | |
| nonsense mutation in TSC1 (1) | NPCs | enhanced proliferation, aberrant neurite outgrowth, and enlarged cell size | rapamycin | [46] | |
| splicing mutation in TSC1 (1) | neurons | enlarged soma, decreased neurite length, and abnormal connections | [83] | ||
| de novo mutation in TSC1 and frameshift mutation in TSC2 (2) | co-cultures of cortical neurons and oligodendrocytes | cellular hypertrophy and increased axonal density | rapamycin | [84] | |
| single or biallelic mutations in TSC2 (2) | neurons | morphological changes | rapamycin | [86] | |
| loss-of-function mutations in TSC1 and TSC2 (2) | brain organoids | impaired developmental suppression of mTORC1 signaling by loss of either TSC1 or TSC2 | rapamycin | [125] | |
| PMDS | small/large 22q13.3 deletions and frameshift mutation in SHANK3 (7) | NPCs | disrupted neurogenesis leading to altered excitatory/inhibitory balance | [47] | |
| 22q13 deletion (2) | neurons | impaired excitatory synaptic transmission | IGF-1 | [87] | |
| de novo truncating and frameshift mutations in SHANK3 (2) | neurons | impaired excitatory synaptic transmission | lithium and valproic acid | [88] | |
| de novo truncating mutations in SHANK3 (4) | pyramidal neurons | decreased dendritic spines and altered spinogenesis | [89] | ||
| heterozygous and homozygous SHANK3 deletions (2) | neurons | decreased neurite outgrowth, hyperexcitability, increased input resistance, and disrupted excitatory synaptic transmission | [90] | ||
| TS | G406R missense mutation in CACNA1C (5) | NPCs and neurons | dysregulated Ca2+ signaling, impaired neuronal differentiation, increased TH and catecholamine expression | roscovitine | [48] |
| G406R missense mutation in CACNA1C (3) | NPCs | dysregulated Ca2+ signaling affecting neuronal development and function | [49] | ||
| G406R missense mutation in CACNA1C (2) | neurons | Ca2+-dependent dendritic retraction and altered cellular structure | C3 transferase | [91] | |
| G406R missense mutation in CACNA1C (3) | neurons | altered differentiation in the developing cortex | [92] | ||
| G406R missense mutation in CACNA1C (3) | brain organoids | delayed migration of inhibitory neurons | nimodipine | [127] | |
| AS | maternal 15q11-q13 deletion including UBE3A (2) | neurons | no phenotypic alterations | [93] | |
| 3bp deletion in the maternally inherited UBE3A (1) | neurons | late paternal UBE3A silencing during neuronal differentiation | [96] | ||
| large 15q11–q13 deletion (3) | neurons | reduced synaptic activity and plasticity | [97] | ||
| 15q11.2-q13 microdeletion including UBE3A (1) | brain organoids | neuronal hyperexcitability | BK channels antagonist | [128] | |
| PWS | paternal 15q11-13 deletion (1) | neurons | no phenotypic alterations | [93] |
| Limitations of iPSCs | Potential Solutions/Approaches |
|---|---|
| Limited amount of patient-derived cell lines | Generation of biobanks of cells derived from patients and unaffected individuals |
| Lack of proper ASD control | Use of TALEN and CRISPR/Cas9 genome-editing techniques to create isogenic cell lines |
| Line-to-line variability | Use of TALEN and CRISPR/Cas9 genome-editing techniques to create isogenic cell lines More defined differentiation procedures for both 2D and 3D cultures |
| Lack of organoid-to-organoid reproducibility | Use of 3D bioprinting models |
| Lack of vascularization | In Vivo transplantation in animal models Use of combined progenitors (neural and mesenchymal stem cells) Promotion of blood vessel formation by VEGF supplementation in brain organoids |
| Limited long-term maturation of brain organoids | Optimization of growth conditions by spinning bioreactors In vivo transplantation in animal models |
| High cost of culturing organoids | Miniaturized bioreactors with reduced incubator space and decreased volume of media |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa, C.; Combi, R.; Conconi, D.; Lavitrano, M. Patient-Derived Induced Pluripotent Stem Cells (iPSCs) and Cerebral Organoids for Drug Screening and Development in Autism Spectrum Disorder: Opportunities and Challenges. Pharmaceutics 2021, 13, 280. https://doi.org/10.3390/pharmaceutics13020280
Villa C, Combi R, Conconi D, Lavitrano M. Patient-Derived Induced Pluripotent Stem Cells (iPSCs) and Cerebral Organoids for Drug Screening and Development in Autism Spectrum Disorder: Opportunities and Challenges. Pharmaceutics. 2021; 13(2):280. https://doi.org/10.3390/pharmaceutics13020280
Chicago/Turabian StyleVilla, Chiara, Romina Combi, Donatella Conconi, and Marialuisa Lavitrano. 2021. "Patient-Derived Induced Pluripotent Stem Cells (iPSCs) and Cerebral Organoids for Drug Screening and Development in Autism Spectrum Disorder: Opportunities and Challenges" Pharmaceutics 13, no. 2: 280. https://doi.org/10.3390/pharmaceutics13020280
APA StyleVilla, C., Combi, R., Conconi, D., & Lavitrano, M. (2021). Patient-Derived Induced Pluripotent Stem Cells (iPSCs) and Cerebral Organoids for Drug Screening and Development in Autism Spectrum Disorder: Opportunities and Challenges. Pharmaceutics, 13(2), 280. https://doi.org/10.3390/pharmaceutics13020280