
Bacterial-Derived Outer Membrane Vesicles are Potent Adjuvants that Drive Humoral and Cellular Immune Responses



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
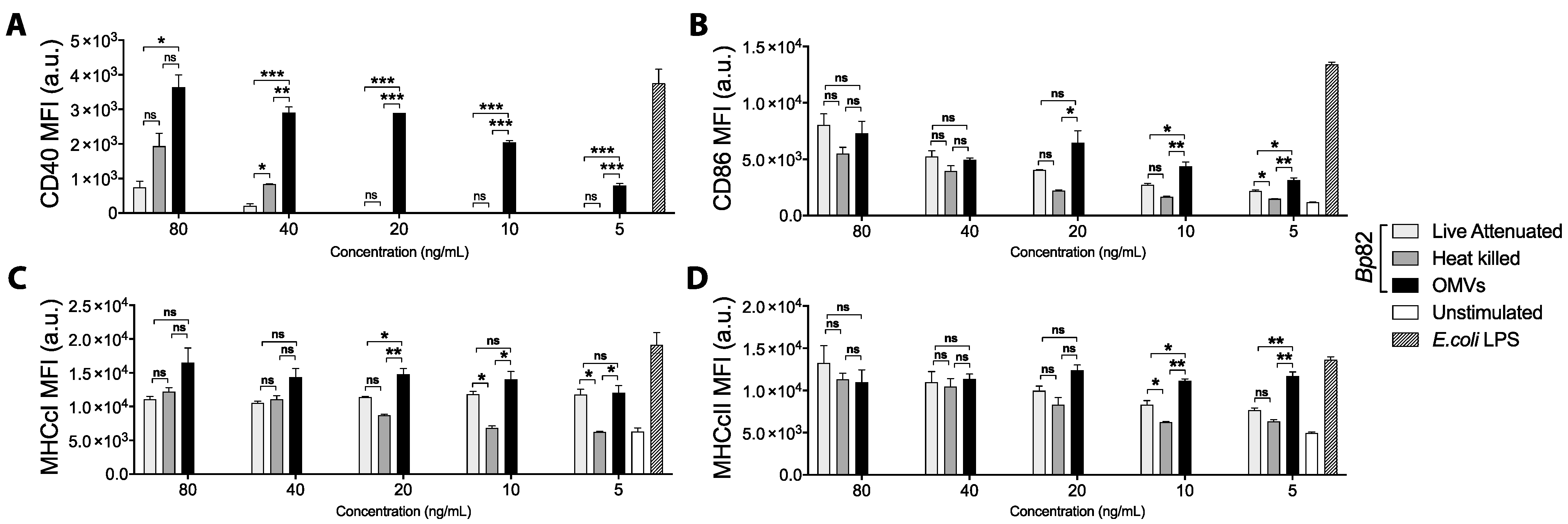
3.1. Outer Membrane Vesicles (OMVs) Activate Antigen-Presenting Cells Better than Heat-Inactivated or Live-Attenuated Bacteria
3.2. OMV Adjuvant Promotes Antigen-Specific CD4 and CD8 T Cells to Co-Delivered Peptides
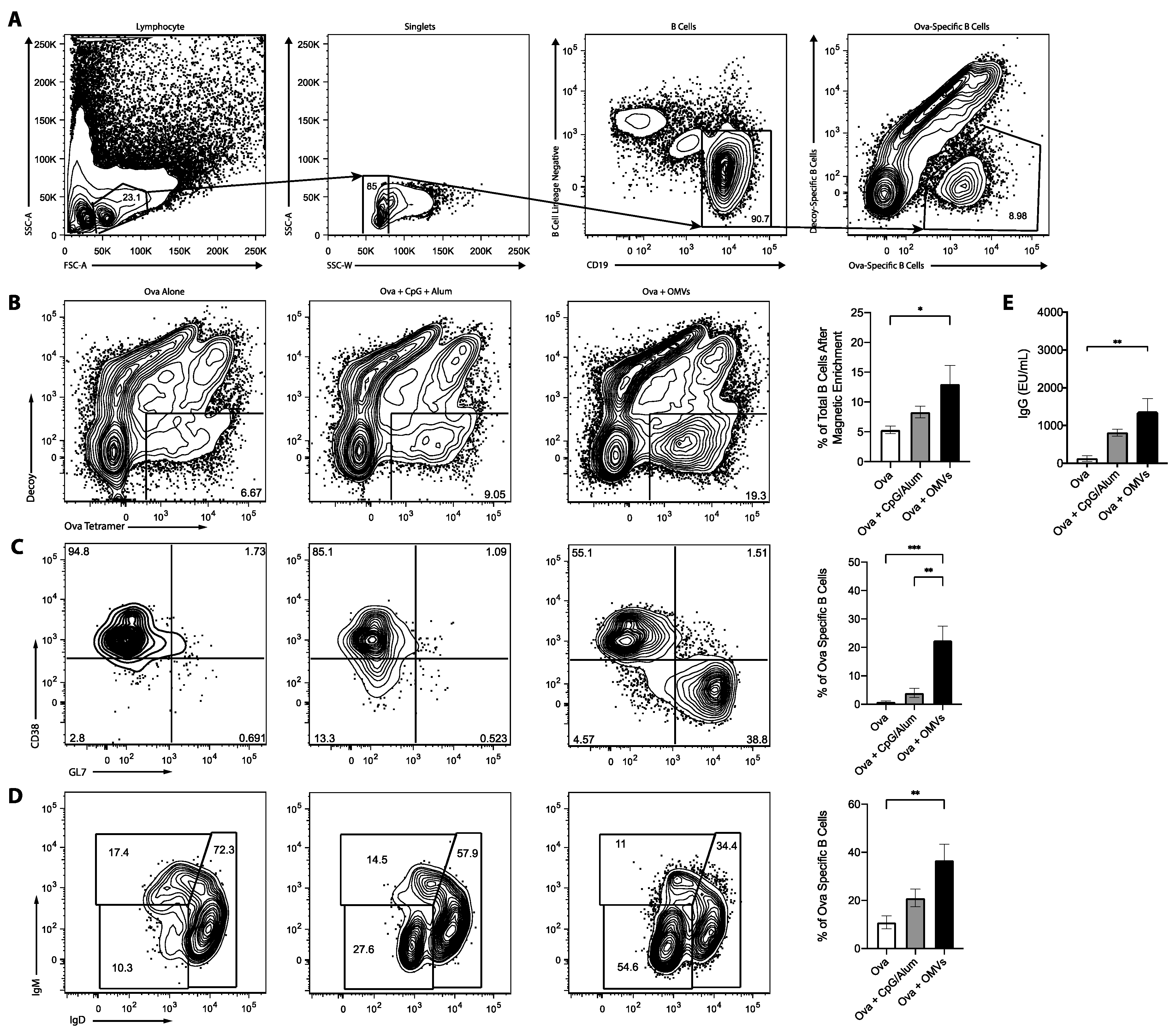
3.3. OMV Adjuvant Elicits B Cell and Antibody Responses to a Co-Delivered Protein Antigen
3.4. Pre-Existing Antibody Does Not Inhibit OMV Adjuvanticity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andreano, E.; D’Oro, U.; Rappuoli, R.; Finco, O. Vaccine Evolution and Its Application to Fight Modern Threats. Front. Immunol. 2019, 10, 1722. [Google Scholar] [CrossRef] [PubMed]
- López-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-Assembling Protein Nanoparticles in the Design of Vaccines. Comput. Struct. Biotechnol. J. 2016, 14, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Zhai, L.; Tumban, E. Virus-like Particle-Based L2 Vaccines against HPVs: Where Are We Today? Viruses 2019, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, Y.; Du, J. Human Papillomavirus Vaccines: An Updated Review. Vaccines 2020, 8, 391. [Google Scholar] [CrossRef]
- Borrow, R.; Taha, M.-K.; Giuliani, M.M.; Pizza, M.; Banzhoff, A.; Bekkat-Berkani, R. Methods to Evaluate Serogroup B Meningococcal Vaccines: From Predictions to Real-World Evidence. J. Infect. 2020, 81, 862–872. [Google Scholar] [CrossRef]
- Zurita, M.E.; Wilk, M.M.; Carriquiriborde, F.; Bartel, E.; Moreno, G.; Misiak, A.; Mills, K.H.G.; Hozbor, D. A Pertussis Outer Membrane Vesicle-Based Vaccine Induces Lung-Resident Memory CD4 T Cells and Protection Against Bordetella Pertussis, Including Pertactin Deficient Strains. Front. Cell Infect. Microbiol. 2019, 9, 125. [Google Scholar] [CrossRef]
- Guebre-Xabier, M.; Patel, N.; Tian, J.-H.; Zhou, B.; Maciejewski, S.; Lam, K.; Portnoff, A.D.; Massare, M.J.; Frieman, M.B.; Piedra, P.A.; et al. NVX-CoV2373 Vaccine Protects Cynomolgus Macaque Upper and Lower Airways against SARS-CoV-2 Challenge. Vaccine 2020, 38, 7892–7896. [Google Scholar] [CrossRef]
- Gerritzen, M.J.H.; Martens, D.E.; Wijffels, R.H.; van der Pol, L.; Stork, M. Bioengineering Bacterial Outer Membrane Vesicles as Vaccine Platform. Biotechnol. Adv. 2017, 35, 565–574. [Google Scholar] [CrossRef]
- Zepeda-Cervantes, J.; Ramírez-Jarquín, J.O.; Vaca, L. Interaction Between Virus-Like Particles (VLPs) and Pattern Recognition Receptors (PRRs) From Dendritic Cells (DCs): Toward Better Engineering of VLPs. Front. Immunol. 2020, 11, 1100. [Google Scholar] [CrossRef]
- Syed, F.M.; Khan, M.A.; Nasti, T.H.; Ahmad, N.; Mohammad, O. Antigen Entrapped in the Escheriosomes Leads to the Generation of CD4+ Helper and CD8+ Cytotoxic T Cell Response. Vaccine 2003, 21, 2383–2393. [Google Scholar] [CrossRef]
- Nieves, W.; Asakrah, S.; Qazi, O.; Brown, K.A.; Kurtz, J.; AuCoin, D.P.; McLachlan, J.B.; Roy, C.J.; Morici, L.A. A Naturally Derived Outer-Membrane Vesicle Vaccine Protects against Lethal Pulmonary Burkholderia pseudomallei Infection. Vaccine 2011, 29, 8381–8389. [Google Scholar] [CrossRef] [PubMed]
- Nieves, W.; Petersen, H.; Judy, B.M.; Blumentritt, C.A.; Russell-Lodrigue, K.; Roy, C.J.; Torres, A.G.; Morici, L.A. A Burkholderia pseudomallei Outer Membrane Vesicle Vaccine Provides Protection against Lethal Sepsis. Clin. Vaccine Immunol. 2014, 21, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H.; Nieves, W.; Russell-Lodrigue, K.; Roy, C.J.; Morici, L.A. Evaluation of a Burkholderia pseudomallei Outer Membrane Vesicle Vaccine in Nonhuman Primates. Procedia Vaccinol. 2014, 8, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.M.; Settles, E.W.; Davitt, C.; Gellings, P.; Kikendall, N.; Hoffmann, J.; Wang, Y.; Bitoun, J.; Lodrigue, K.-R.; Sahl, J.W.; et al. Burkholderia pseudomallei OMVs Derived from Infection Mimicking Conditions Elicit Similar Protection to a Live-Attenuated Vaccine. npjVaccines 2021. [Google Scholar] [CrossRef]
- Mancini, F.; Rossi, O.; Necchi, F.; Micoli, F. OMV Vaccines and the Role of TLR Agonists in Immune Response. Int. J. Mol. Sci 2020, 21, 4416. [Google Scholar] [CrossRef]
- Elizagaray, M.L.; Gomes, M.T.R.; Guimaraes, E.S.; Rumbo, M.; Hozbor, D.F.; Oliveira, S.C.; Moreno, G. Canonical and Non-Canonical Inflammasome Activation by Outer Membrane Vesicles Derived from Bordetella Pertussis. Front. Immunol. 2020, 11, 1879. [Google Scholar] [CrossRef]
- Finethy, R.; Luoma, S.; Orench-Rivera, N.; Feeley, E.M.; Haldar, A.K.; Yamamoto, M.; Kanneganti, T.-D.; Kuehn, M.J.; Coers, J. Inflammasome Activation by Bacterial Outer Membrane Vesicles Requires Guanylate Binding Proteins. MBio 2017, 8, e01188-17. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V.A.K. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef]
- Acevedo, R.; Zayas, C.; Norheim, G.; Fernández, S.; Cedré, B.; Aranguren, Y.; Cuello, M.; Rodriguez, Y.; González, H.; Mandiarote, A.; et al. Outer Membrane Vesicles Extracted from Neisseria Meningitidis Serogroup X for Prevention of Meningococcal Disease in Africa. Pharm. Res. 2017, 121, 194–201. [Google Scholar] [CrossRef]
- Dowling, D.J.; Sanders, H.; Cheng, W.K.; Joshi, S.; Brightman, S.; Bergelson, I.; Pietrasanta, C.; van Haren, S.D.; van Amsterdam, S.; Fernandez, J.; et al. A Meningococcal Outer Membrane Vesicle Vaccine Incorporating Genetically Attenuated Endotoxin Dissociates Inflammation from Immunogenicity. Front. Immunol. 2016, 7, 562. [Google Scholar] [CrossRef]
- Norris, M.H.; Schweizer, H.P.; Tuanyok, A. Structural Diversity of Burkholderia pseudomallei Lipopolysaccharides Affects Innate Immune Signaling. PLoS Negl. Trop. D 2017, 11, e0005571. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.M.; Davitt, C.J.H.; Motyka, N.; Kikendall, N.L.; Russell-Lodrigue, K.; Roy, C.J.; Morici, L.A. A Burkholderia pseudomallei Outer Membrane Vesicle Vaccine Provides Cross Protection against Inhalational Glanders in Mice and Non-Human Primates. Vaccines 2017, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Kukutsch, N.; Ogilvie, A.L.J.; Rößner, S.; Koch, F.; Romani, N.; Schuler, G. An Advanced Culture Method for Generating Large Quantities of Highly Pure Dendritic Cells from Mouse Bone Marrow. J. Immunol. Methods 1999, 223, 77–92. [Google Scholar] [CrossRef]
- Taylor, J.J.; Martinez, R.J.; Titcombe, P.J.; Barsness, L.O.; Thomas, S.R.; Zhang, N.; Katzman, S.D.; Jenkins, M.K.; Mueller, D.L. Deletion and Anergy of Polyclonal B Cells Specific for Ubiquitous Membrane-Bound Self-Antigen. J. Exp. Med. 2012, 209, 2065–2077. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Chen, Y.; Lim, Y.-C.; Tan, G.-Y.G.; Liu, Y.; Lim, Y.-T.; MacAry, P.; Gan, Y.-H. Suppression of Host Innate Immune Response by Burkholderia pseudomallei through the Virulence Factor TssM. J. Immunol. 2010, 184, 5160–5171. [Google Scholar] [CrossRef]
- Moon, J.J.; Chu, H.H.; Pepper, M.; McSorley, S.J.; Jameson, S.C.; Kedl, R.M.; Jenkins, M.K. Naive CD4(+) T Cell Frequency Varies for Different Epitopes and Predicts Repertoire Diversity and Response Magnitude. Immunity 2007, 27, 203–213. [Google Scholar] [CrossRef]
- Schott, E.; Bertho, N.; Ge, Q.; Maurice, M.M.; Ploegh, H.L. Class I Negative CD8 T Cells Reveal the Confounding Role of Peptide-Transfer onto CD8 T Cells Stimulated with Soluble H2-Kb Molecules. Proc. Natl. Acad. Sci. USA 2002, 99, 13735–13740. [Google Scholar] [CrossRef]
- Giordano, N.P.; Cian, M.B.; Dalebroux, Z.D. Outer Membrane Lipid Secretion and the Innate Immune Response to Gram-Negative Bacteria. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef]
- McKee, A.S.; Marrack, P. Old and New Adjuvants. Curr. Opin. Immunol. 2017, 47, 44–51. [Google Scholar] [CrossRef]
- Heineman, T.C.; Cunningham, A.; Levin, M. Understanding the Immunology of Shingrix, a Recombinant Glycoprotein E Adjuvanted Herpes Zoster Vaccine. Curr. Opin. Immunol. 2019, 59, 42–48. [Google Scholar] [CrossRef]
- Egan, K.; Hook, L.M.; Naughton, A.; Friedman, H.M.; Awasthi, S. Herpes Simplex Virus Type 2 Trivalent Protein Vaccine Containing Glycoproteins C, D and E Protects Guinea Pigs against HSV-1 Genital Infection. Hum. Vaccines Immunother. 2020, 16, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Hook, L.M.; Swaminathan, G.; Cairns, T.M.; Brooks, B.; Smith, J.S.; Ditto, N.T.; Gindy, M.E.; Bett, A.J.; Espeseth, A.S.; et al. Antibody Responses to Crucial Functional Epitopes as a Novel Approach to Assess Immunogenicity of Vaccine Adjuvants. Vaccine 2019, 37, 3770–3778. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, L.; Stork, M.; van der Ley, P. Outer Membrane Vesicles as Platform Vaccine Technology. Biotechnol. J. 2015, 10, 1689–1706. [Google Scholar] [CrossRef] [PubMed]
- Micoli, F.; Alfini, R.; Benedetto, R.D.; Necchi, F.; Schiavo, F.; Mancini, F.; Carducci, M.; Palmieri, E.; Balocchi, C.; Gasperini, G.; et al. GMMA Is a Versatile Platform to Design Effective Multivalent Combination Vaccines. Vaccines 2020, 8, 540. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prior, J.T.; Davitt, C.; Kurtz, J.; Gellings, P.; McLachlan, J.B.; Morici, L.A. Bacterial-Derived Outer Membrane Vesicles are Potent Adjuvants that Drive Humoral and Cellular Immune Responses. Pharmaceutics 2021, 13, 131. https://doi.org/10.3390/pharmaceutics13020131
Prior JT, Davitt C, Kurtz J, Gellings P, McLachlan JB, Morici LA. Bacterial-Derived Outer Membrane Vesicles are Potent Adjuvants that Drive Humoral and Cellular Immune Responses. Pharmaceutics. 2021; 13(2):131. https://doi.org/10.3390/pharmaceutics13020131
Chicago/Turabian StylePrior, J. Timothy, Christopher Davitt, Jonathan Kurtz, Patrick Gellings, James B. McLachlan, and Lisa A. Morici. 2021. "Bacterial-Derived Outer Membrane Vesicles are Potent Adjuvants that Drive Humoral and Cellular Immune Responses" Pharmaceutics 13, no. 2: 131. https://doi.org/10.3390/pharmaceutics13020131
APA StylePrior, J. T., Davitt, C., Kurtz, J., Gellings, P., McLachlan, J. B., & Morici, L. A. (2021). Bacterial-Derived Outer Membrane Vesicles are Potent Adjuvants that Drive Humoral and Cellular Immune Responses. Pharmaceutics, 13(2), 131. https://doi.org/10.3390/pharmaceutics13020131