Formulation of Lipid-Based Tableted Spray-Congealed Microparticles for Sustained Release of Vildagliptin: In Vitro and In Vivo Studies

 and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Preparation of VG Microparticles Using Spray Congealing Technology and Tablets Preparation
2.2. Yield of Production, Drug Recovery, and Particle Size Analysis
2.3. Differential Scanning Calorimetry (DSC)
2.4. X-ray Diffraction (XRD)
2.5. Scanning Electron Microscopy (SEM)
2.6. Dissolution Studies, Release Kinetics, and Aging Effect
2.7. Bioavailability Studies
2.8. Pharmacokinetic and Statistical Analysis
2.9. In Vitro–In Vivo Correlation (IVIVC) Studies
3. Results and Discussion
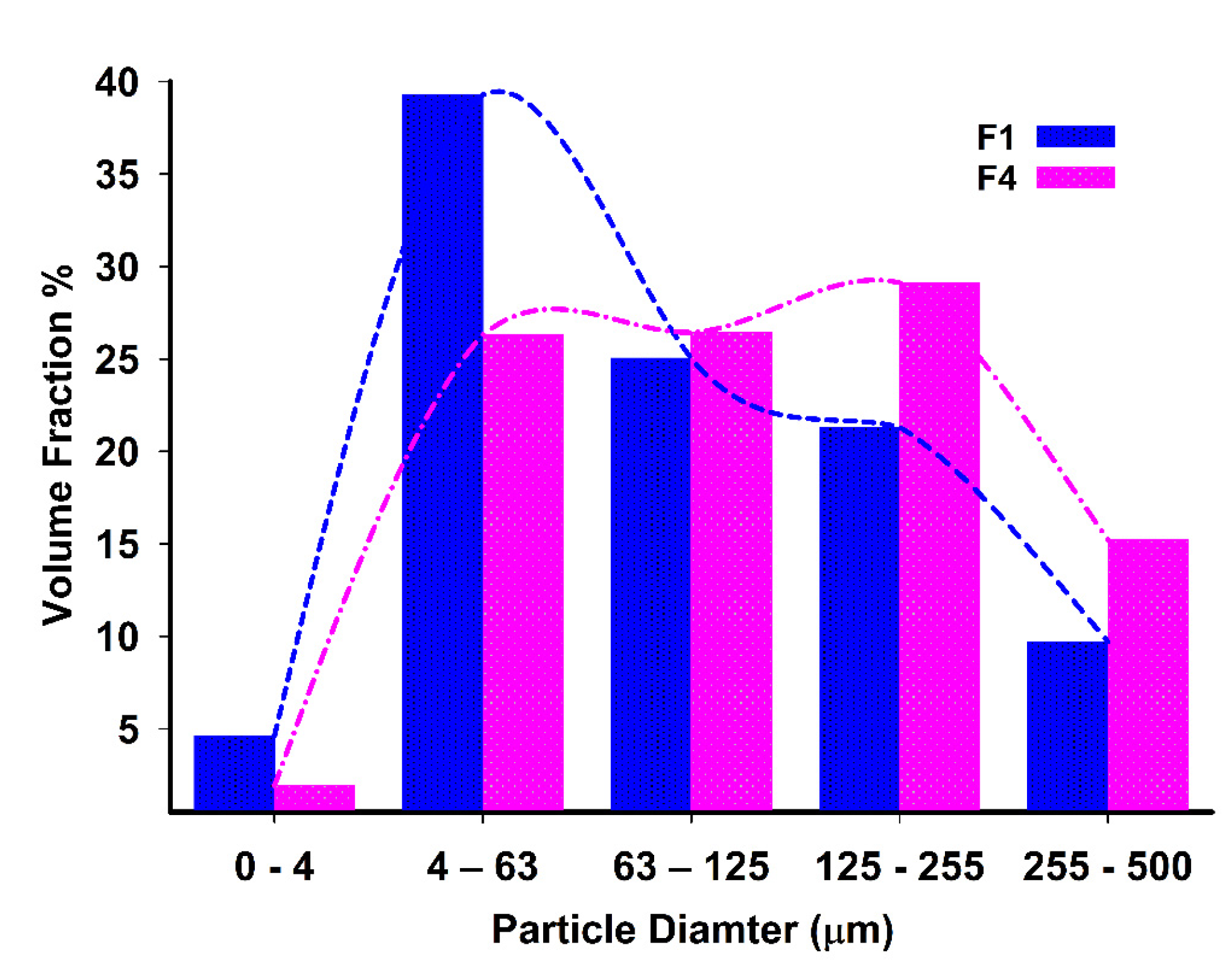
3.1. Yield, Content, and Particle Size Analysis
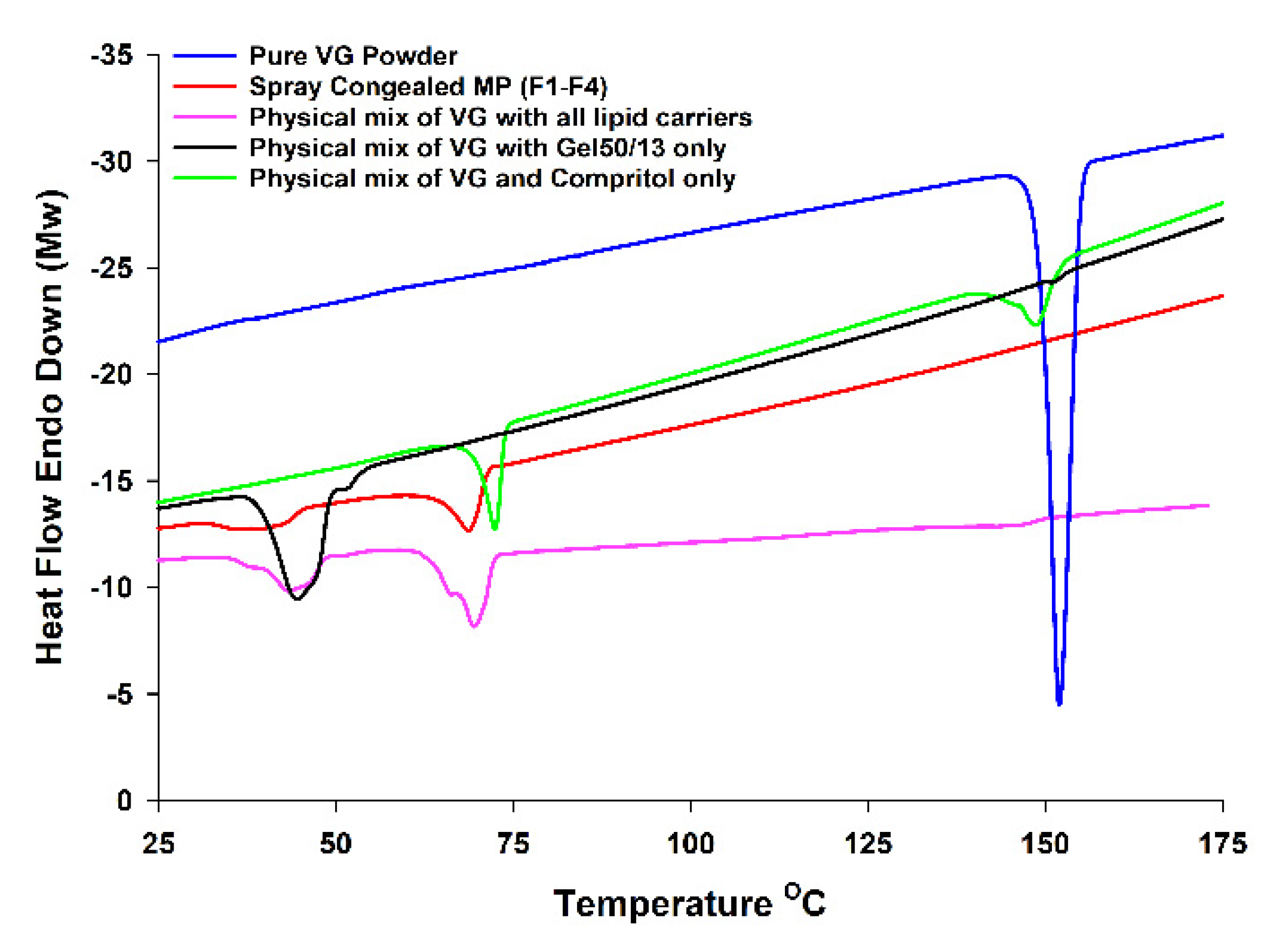
3.2. DSC and XRD Analysis
3.3. Morphological Analysis
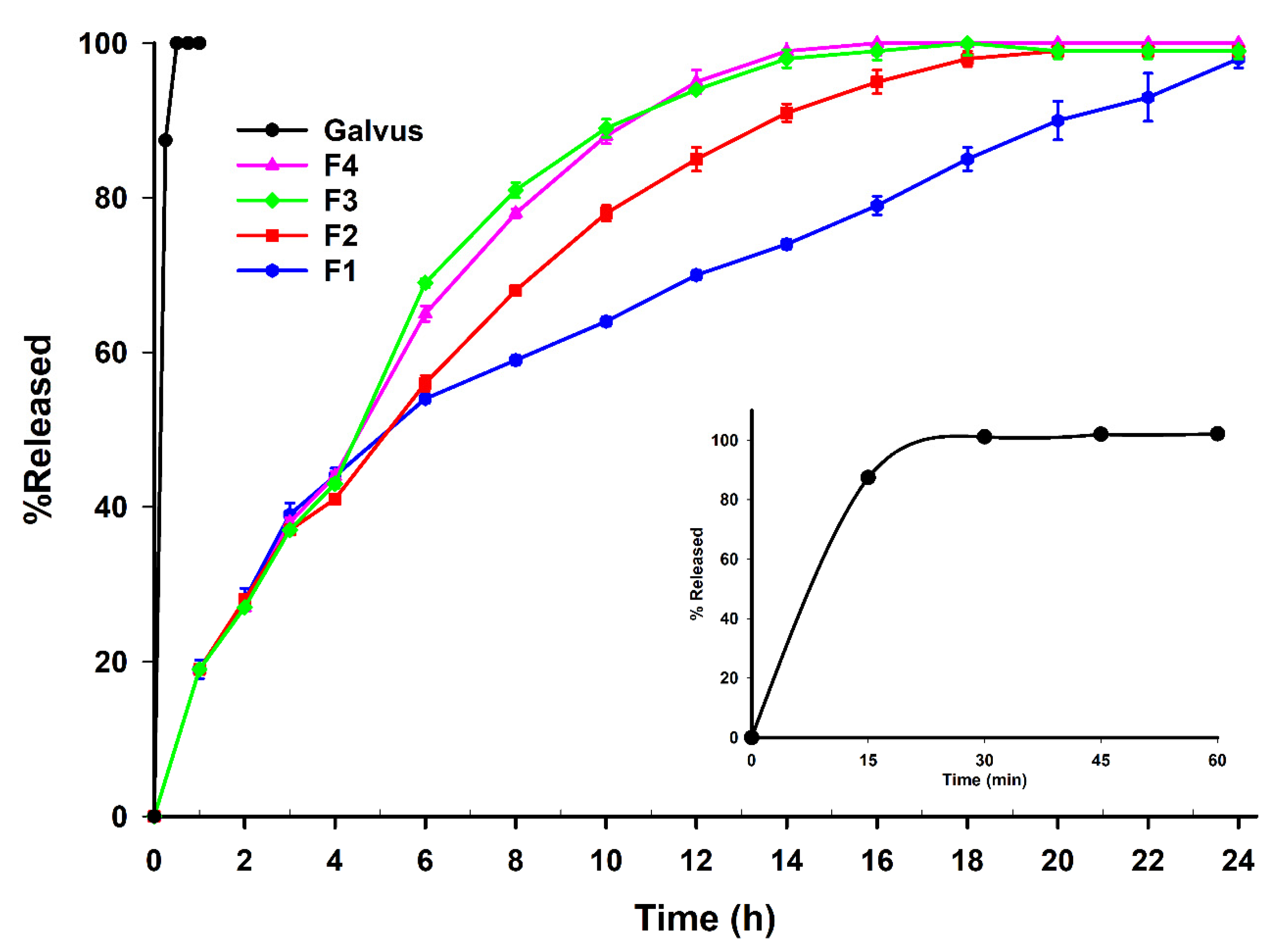
3.4. Dissolution Studies, Aging and Release Kinetics
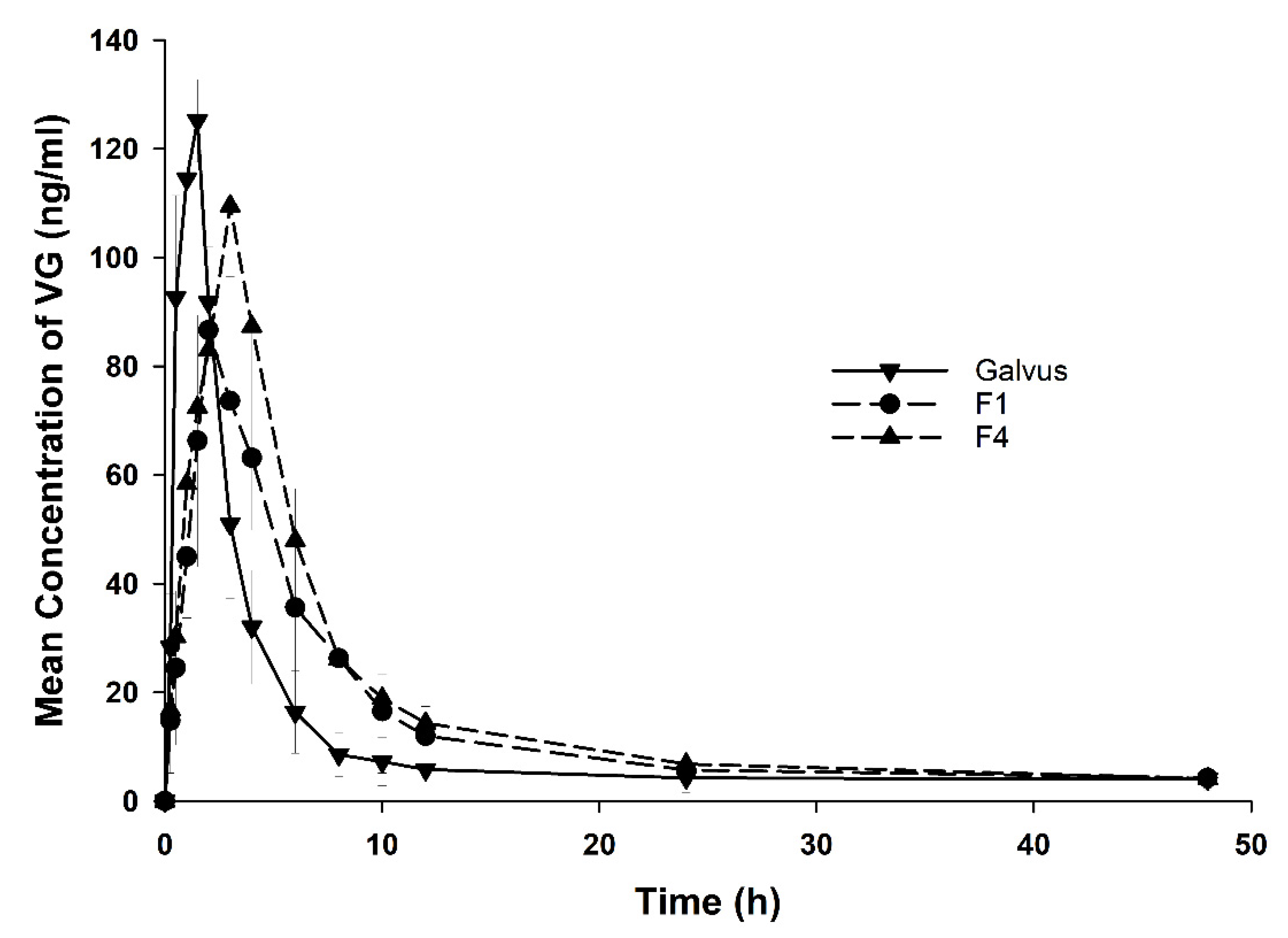
3.5. In Vivo Bioavailability Studies
3.6. In Vivo–In Vitro Correlation (IVIVC)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Particle Size Volume Percentage of Size Range (μm) | SPAN | ||||
|---|---|---|---|---|---|---|
| 0–4 | 4–63 | 63–125 | 125–255 | 255–500 | ||
| F1 | 4.63 | 39.29 | 25.04 | 21.32 | 9.70 | 2.516 * |
| F4 | 1.93 | 26.30 | 26.44 | 29.12 | 15.21 | 3.205 * |
| Equation # | Model | Equation * | Plot |
|---|---|---|---|
| 1s | Zero-Order | The amount of released drug versus time | |
| 2s | First-Order | The decimal logarithm of released bioactive versus time would yield a straight line with a slope of −K/2.303 | |
| 3s | Higuchi | Percentage of released drug versus square root of time | |
| 4s | Hixson-Crowell | The cubic root of remaining drug in matrix/tablet versus time | |
| 5s | Korsmeyer-Peppas | Log percentage of released bioactive versus log time |
| Model | Formulations | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | Galvus® | |||||||||||
| R2 | K | n | R2 | K | n | R2 | K | n | R2 | K | n | R2 | K | n | |
| Zero order (0–6 h) | 0.912 | 3.448 | - | 0.87 | 3.9 | - | 0.782 | 3.867 | - | 0.851 | 4.29 | - | 0.104 | 0.91 | - |
| Zero-order (post 6 h) | 0.997 | 2.445 | - | - | - | - | - | - | - | - | - | - | - | - | |
| First order | 0.964 | 0.117 | - | 0.921 | 0.15 | - | 0.944 | 0.18 | - | 0.967 | 0.19 | - | 0.986 | 8.53 | - |
| Higuchi model | 0.994 | 20.19 | - | 0.974 | 22.52 | - | 0.921 | 23.69 | - | 0.962 | 24.66 | - | - | - | - |
| Hixson Crowell | 0.929 | 0.03 | - | 0.920 | 0.04 | - | 0.945 | 0.05 | - | 0.960 | 0.05 | - | - | - | - |
| Korsmeyer Peppas | 0.990 | 22.08 | 0.47 | 0.987 | 23.76 | 0.48 | 0.965 | 28.16 | 0.43 | 0.981 | 25.97 | 0.48 | - | - | - |
References
- Foley, J.E.; Bhosekar, V.; Kawamori, R. Does the treatment of type 2 diabetes mellitus with the DPP-4 inhibitor vildagliptin reduce HbA1c to a greater extent in Japanese patients than in Caucasian patients? Vasc. Health Risk Manag. 2016, 12, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyenwe, E.A.; Jerkins, T.W.; Umpierrez, G.E.; Kitabchi, A.E. Management of type 2 diabetes: Evolving strategies for the treatment of patients with type 2 diabetes. Metabolism 2011, 60, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafeezuallah, K.; Abdulrauf, K.; Safirah, M.; Muhammad, H.; Syed, R.; Rai, S.; Asif, M.; Samar, A.; Mehwish, A. Preparation and In Vitro Evaluation of Sustained Release Microparticles of an Antidiabetic Drug. Lat. Am. J. Pharm. 2015, 34, 19–31. [Google Scholar]
- Mohan, V.; Zargar, A.; Chawla, M.; Joshi, A.; Ayyagari, U.; Sethi, B.; Gaurav, K.; Patted, U.R.H.; Bhagat, S.V.; Mane, A.I. Efficacy of a Combination of Metformin and Vildagliptin in Comparison to Metformin Alone in Type 2 Diabetes Mellitus: A Multicentre, Retrospective, Real-World Evidence Study. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 2925–2933. [Google Scholar] [CrossRef]
- Sunkara, G.; Sabo, R.; Wang, Y.; He, Y.L.; Campestrini, J.; Rosenberg, M.; Howard, D.; Dole, W.P. Dose proportionality and the effect of food on vildagliptin, a novel dipeptidyl peptidase IV inhibitor, in healthy volunteers. J. Clin. Pharmacol. 2007, 47, 1152–1158. [Google Scholar] [CrossRef]
- He, Y.-L.; Sadler, B.M.; Balez, R.S.; Wang, Y.; Campestrini, J.; Laurent, A.; Ligueros-Saylan, M.; Howard, D. The Absolute Oral Bioavailability and Population-Based Pharmacokinetic Modelling of a Novel Dipeptidyl peptidase-IV Inhibitor, Vildagliptin, in Healthy Volunteers. Clin. Pharmacokinet. 2007, 46, 787–802. [Google Scholar] [CrossRef]
- Dineshmohan, D.; Roja, G.; Harika, Y.; Harika, R.; Gupta, V.R.M. Effect Of Hydrophilic and Hydrophobic Polymer Combinations in Vildagliptin Sustained Release Tablets: Fabrication and In Vitro Characterization. Asian J. Pharm. 2015, 9, 298–301. [Google Scholar]
- Waghulde, M.; Rajput, R.; Mujumdar, A.; Naik, J. Production and evaluation of vildagliptin-loaded poly(dl-lactide) and poly(dl-lactide-glycolide) micro-/nanoparticles: Response surface methodology approach. Dry Technol. 2019, 37, 1265–1276. [Google Scholar] [CrossRef]
- Martins, R.M.; Siqueira, S.; Machado, M.O.; Freitas, L.A. The effect of homogenization method on the properties of carbamazepine microparticles prepared by spray congealing. J. Microencapsul. 2013, 30, 692–700. [Google Scholar] [CrossRef]
- Albertini, B.; Sabatino, M.D.; Melegari, C.; Passerini, N. Formulation of spray congealed microparticles with self-emulsifying ability for enhanced glibenclamide dissolution performance. J. Microencapsul. 2015, 32, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Albertini, B.; Di, S.M.; Melegari, C.; Passerini, N. Formulating SLMs as oral pulsatile system for potential delivery of melatonin to pediatric population. Int. J. Pharm. 2014, 469, 67–79. [Google Scholar] [CrossRef]
- Albertini, B.; Passerini, N.; Di Sabatino, M.; Vitali, B.; Brigidi, P.; Rodriguez, L. Polymer–lipid based mucoadhesive microspheres prepared by spray-congealing for the vaginal delivery of econazole nitrate. Eur. J. Pharm. Sci. 2009, 36, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Balducci, A.G.; Colombo, G.; Corace, G.; Cavallari, C.; Rodriguez, L.; Buttini, F.; Colombo, P.; Rossi, A. Layered lipid microcapsules for mesalazine delayed-release in children. Int. J. Pharm. 2011, 421, 293–300. [Google Scholar] [CrossRef]
- Passerini, N.; Qi, S.; Albertini, B.; Grassi, M.; Rodriguez, L.; Craig, D.Q. Solid lipid microparticles produced by spray congealing: Influence of the atomizer on microparticle characteristics and mathematical modeling of the drug release. J. Pharm. Sci. 2010, 99, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Scalia, S.; Traini, D.; Young, P.M.; Di Sabatino, M.; Passerini, N.; Albertini, B. Comparison of spray congealing and melt emulsification methods for the incorporation of the water-soluble salbutamol sulphate in lipid microparticles. Pharm. Dev. Technol. 2013, 18, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.H.; Heng, P.W.S.; Chan, L.W. Spray congealing as a microencapsulation technique to develop modified-release ibuprofen solid lipid microparticles: The effect of matrix type, polymeric additives and drug–matrix miscibility. J. Microencapsul. 2015, 32, 725–736. [Google Scholar] [CrossRef]
- Ilić, I.; Dreu, R.; Burjak, M.; Homar, M.; Kerč, J.; Srčič, S. Microparticle size control and glimepiride microencapsulation using spray congealing technology. Int. J. Pharm. 2009, 381, 176–183. [Google Scholar] [CrossRef]
- Aburahma, M.H.; Badr-Eldin, S.M. Compritol 888 ATO: A multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin. Drug Deliv. 2014, 11, 1865–1883. [Google Scholar] [CrossRef] [PubMed]
- Shilpa, N.P.; Neha, S.D.; Ankitkumar, S.J.; Prashant, P.K.; Urmila, M.T.; Nithya, G.; Chhanda, J.K.; Nabil, F.; Mangal, S.N. Compritol® 888 ATO a Lipid Excipient for Sustained Release of Highly Water Soluble Active: Formulation, Scale-up and IVIVC Study. Curr. Drug Deliv. 2013, 10, 548–556. [Google Scholar]
- Maniruzzaman, M.; Islam, M.T.; Halsey, S.; Amin, D.; Douroumis, D. Novel Controlled Release Polymer-Lipid Formulations Processed by Hot Melt Extrusion. AAPS PharmSciTech 2016, 17, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, K.C.; Patra, C.N.; Jena, G.K.; Ghose, D.; Jena, J.; Panda, S.K.; Sahu, M. Gelucire: A versatile polymer for modified release drug delivery system. Future J. Pharm. Sci. 2018, 4, 102–108. [Google Scholar] [CrossRef]
- European Medicines Agency: Guideline on Quality of Oral Modified Release Products. 2014. Available online: https://www.ema.europa.eu/en/quality-oral-modified-release-products#current-effective-version-section (accessed on 15 November 2021).
- Pontarolo, R.; Gimenez, A.C.; de Francisco, T.M.; Ribeiro, R.P.; Pontes, F.L.; Gasparetto, J.C. Simultaneous Determination of Metformin and Vildagliptin in Human Plasma by a HILIC–MS/MS Method. J. Chromatogr. B 2014, 965, 133–141. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Shah, V.P.; Tsong, Y.; Sathe, P.; Liu, J.-P. In Vitro Dissolution Profile Comparison—Statistics and Analysis of the Similarity Factor, f2. Pharm. Res. 1998, 15, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Duan, J.; Fisher, J. Application of Physiologically Based Absorption Modeling to Characterize the Pharmacokinetic Profiles of Oral Extended Release Methylphenidate Products in Adults. PLoS ONE 2016, 11, e0164641. [Google Scholar] [CrossRef]
- Maschke, A.; Becker, C.; Eyrich, D.; Kiermaier, J.; Blunk, T.; Gopferich, A. Development of a spray congealing process for the preparation of insulin-loaded lipid microparticles and characterization thereof. Eur. J. Pharm. Biopharm. 2007, 65, 175–187. [Google Scholar] [CrossRef]
- Gavory, C.; Abderrahmen, R.; Bordes, C.; Chaussy, D.; Belgacem, M.N.; Fessi, H.; Briancon, S. Encapsulation of a pressure sensitive adhesive by spray-cooling: Optimum formulation and processing conditions. Adv. Powder Technol. 2014, 25, 292–300. [Google Scholar] [CrossRef]
- Passerini, N.; Perissutti, B.; Albertini, B.; Franceschinis, E.; Lenaz, D.; Hasa, D.; Locatelli, I.; Voinovich, D. A new approach to enhance oral bioavailability of Silybum Marianum dry extract: Association of mechanochemical activation and spray congealing. Phytomedicine 2012, 19, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, C.; Rodriguez, L.; Albertini, B.; Passerini, N.; Rosetti, F.; Fini, A. Thermal and fractal analysis of diclofenac/Gelucire 50/13 microparticles obtained by ultrasound-assisted atomization. J. Pharm. Sci. 2005, 94, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cui, F.D.; Hayase, T.; Sunada, H. Preparation and Evaluation of Solid Dispersion for Nitrendipine—Carbopol and Nitrendipine—HPMCP Systems Using a Twin Screw Extruder. Chem. Pharm. Bull. 2005, 53, 1240–1245. [Google Scholar] [CrossRef] [Green Version]
- Suhail, M.; Wu, P.-C.; Minhas, M.U. Using Carbomer-Based Hydrogels for Control the Release Rate of Diclofenac Sodium: Preparation and In Vitro Evaluation. Pharmaceuticals 2020, 13, 399. [Google Scholar] [CrossRef]
- Saravani, K.; Prianka, D.; Sravani, R.; Reddy, S.; Kumar, V. Formulation and Evaluation of Vildagliptin Sustained Release Matrix Tablets. Int. J. Curr. Pharm. Res. 2014, 6, 69–75. [Google Scholar]
- Becker, K.; Salar-Behzadi, S.; Zimmer, A. Solvent-free melting techniques for the preparation of lipid-based solid oral formulations. Pharm. Res 2015, 32, 1519–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malekjani, N.; Jafari, S.M. Modeling the release of food bioactive ingredients from carriers/nanocarriers by the empirical, semiempirical, and mechanistic models. Compr. Rev. Food Sci. Food Saf. 2021, 20, 3–47. [Google Scholar] [CrossRef]
- Zhan, S.; Wang, J.; Wang, W.; Cui, L.; Zhao, Q. Preparation and in vitro release kinetics of nitrendipine-loaded PLLA–PEG–PLLA microparticles by supercritical solution impregnation process. RSC Adv. 2019, 9, 16167–16175. [Google Scholar] [CrossRef] [Green Version]
- Apu, A.S.; Pathan, A.H.; Shrestha, D.; Kibria, G.M.; Jalil, R.-u. Investigation of in vitro release kinetics of carbamazepine from Eudragit® RS PO and RL PO matrix tablets. Trop. J. Pharm. Res. 2009, 8, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, P.; Bhandari, S.K.; Islam, S.A.; Reza, M.S.; Adhikari, S. Design and Development of Immediate and Sustained Release Tablets of Vildagliptin. Res. J. Pharm. Biol. Chem. Sci. 2014, 5, 811. [Google Scholar]
- Hu, P.; Yin, Q.; Deckert, F.; Jiang, J.; Liu, D.; Kjems, L.; Dole, W.P.; He, Y.-L. Pharmacokinetics and Pharmacodynamics of Vildagliptin in Healthy Chinese Volunteers. J. Clin. Pharmacol. 2009, 49, 39–49. [Google Scholar] [CrossRef]
- Sharma, M.; Agrawal, S.S. Bioequivalence study of different brands of vildagliptin in healthy human subjects. Future J. Pharm. Sci. 2021, 7, 155. [Google Scholar] [CrossRef]
- The Food and Drug Administration. U.S. Extended Release Solid Dosage Forms: Development, Evaluation and Application of In Vitro/In Vivo Correlations. 1997. Available online: https://www.fda.gov/media/70939/download (accessed on 15 November 2021).
- Volpato, N.M.; Silva, R.L.; Brito, A.P.P.; Gonçalves, J.C.S.; Vaisman, M.; Noël, F. Multiple level C in vitro/in vivo correlation of dissolution profiles of two l-thyroxine tablets with pharmacokinetics data obtained from patients treated for hypothyroidism. Eur. J. Pharm. Sci. 2004, 21, 655–660. [Google Scholar] [CrossRef]
- Barzegar-Jalali, M.; Dastmalchi, S. Kinetic Analysis of Chlorpropamide Dissolution from Solid Dispersions. Drug Dev. Ind. Pharm. 2007, 33, 63–70. [Google Scholar] [CrossRef] [PubMed]


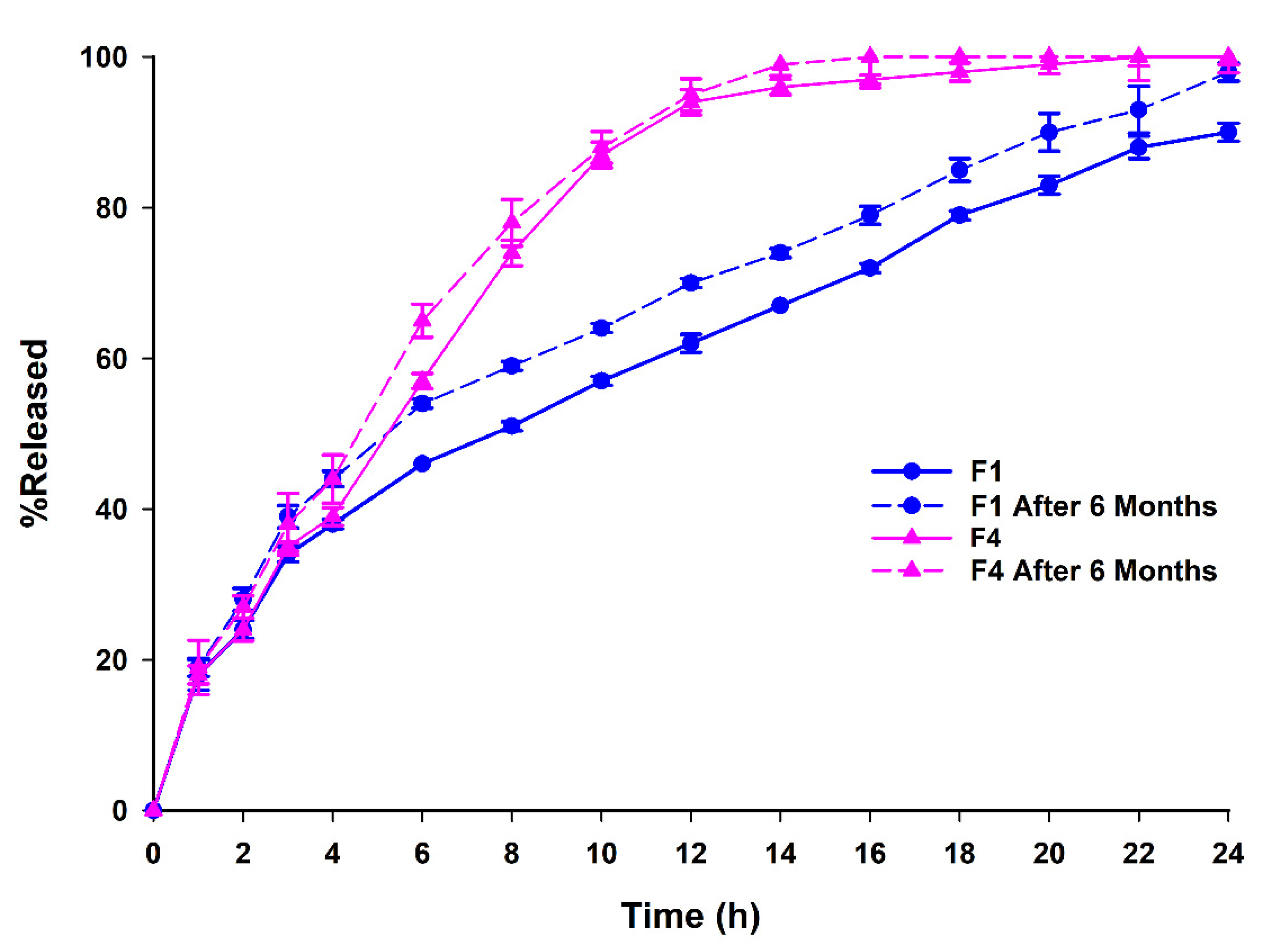
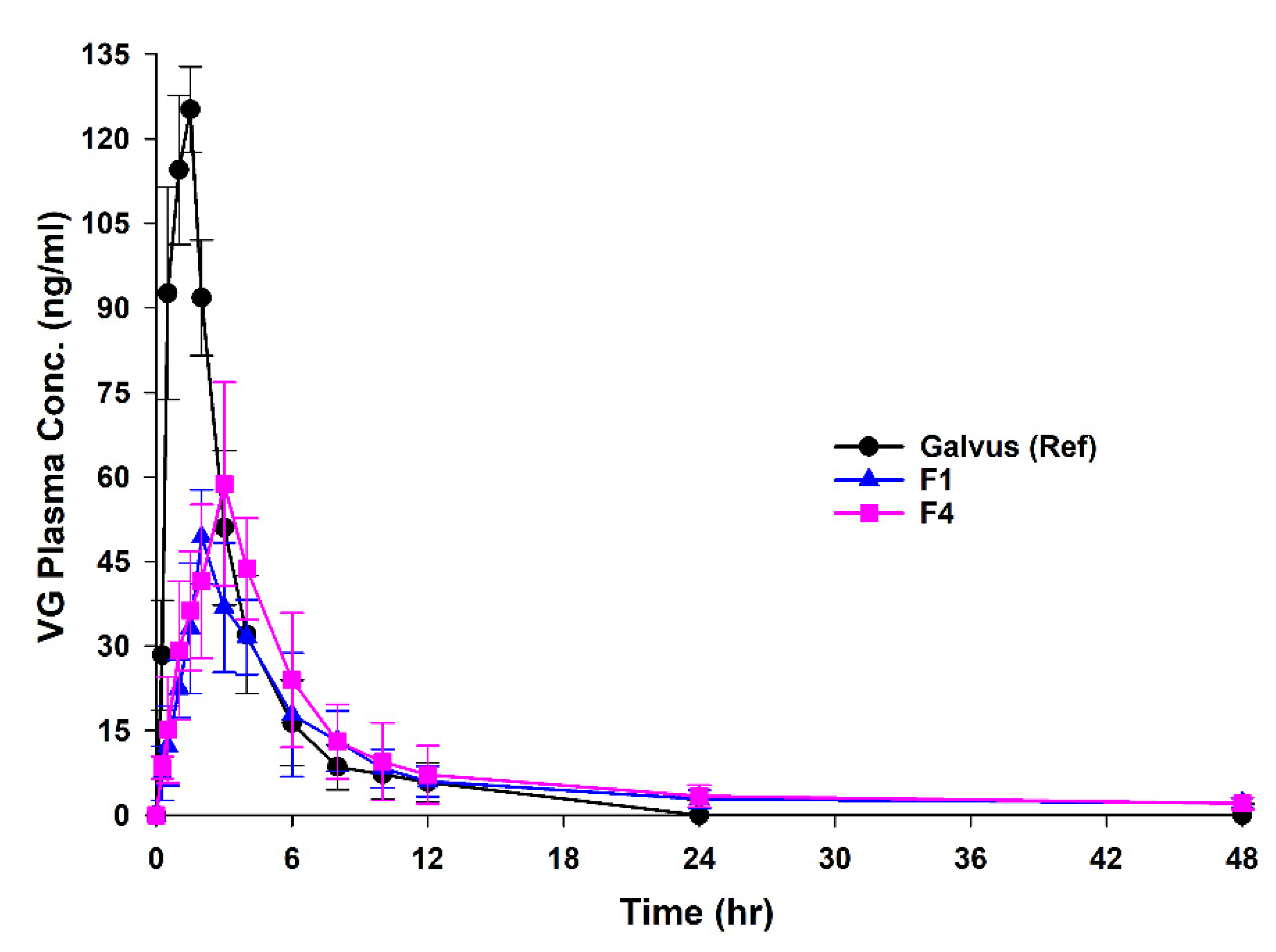

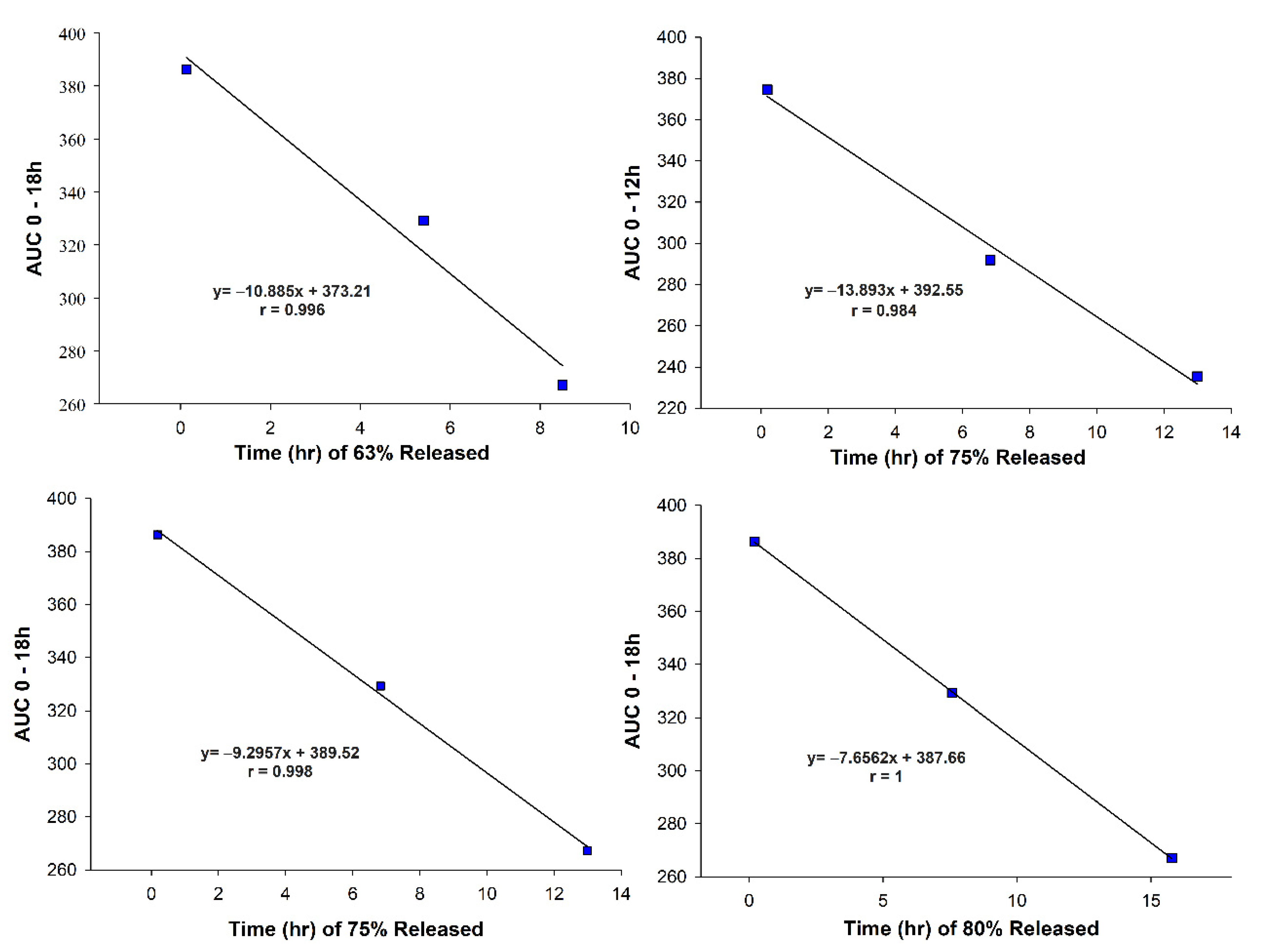
| Formula Code | VG (mg) * | Compritol® (mg) | Gelucire® 50/13 (mg) | Carbomer® (mg) |
|---|---|---|---|---|
| F1 | 100 | 500 | 300 | 0 |
| F2 | 100 | 500 | 280 | 20 |
| F3 | 100 | 500 | 260 | 40 |
| F4 | 100 | 500 | 210 | 90 |
| Formula | % Yield of MP | % VG Recovery |
|---|---|---|
| F1 | 79.00 | 98.80 |
| F2 | 71.50 | 95.40 |
| F3 | 73.00 | 90.10 |
| F4 | 76.00 | 94.40 |
| Formula | Cmax (ng/mL) | Tmax (h) | Ke (1/h) | T1/2 (h) | MRT (h) | AUC0–48 (h·ng/mL) | AUC0–∞ (h·ng/mL) | Rel. Bioav. |
|---|---|---|---|---|---|---|---|---|
| Galvus® | 147.3 ± 61.3 | 1.06 ± 0.58 | 0.34 ± 0.09 | 2.16 ± 0.56 | 2.87 ± 0.62 | 372.5 ± 50.6 | 388 ± 50.5 | 1.0 |
| F1 | 51.19 ± 11.9 * | 2.66 ± 0.86 * | 0.09 ± 0.03 * | 8.816 ± 2.9 * | 9.27 ± 3.66 * | 330 ± 99.1 | 359 ± 113 | 0.87 |
| F4 | 59 ± 14.7 *,# | 3.42 ± 1.1 *,# | 0.07 ± 0.0 *,# | 10.54 ± 2.32 * | 9.86 ± 21 *,# | 412.4 ± 108.1 | 443 ± 120.7 | 1.1 |
| Parameters of Weibull Model | Galvus® | F1 | F4 |
|---|---|---|---|
| R2 | 0.999 | 0.989 | 0.990 |
| Time for 25% release (T25%) (h) | 0.045 | 1.674 | 1.956 |
| Time for 50% release (T50%) (h) | 0.098 | 5.272 | 4.111 |
| Time for 63% release (Td) (h) | 0.135 | 8.495 | 5.406 |
| Time for 75% release (T75%) (h) | 0.180 | 12.993 | 6.827 |
| Time for 80% release (T80%) (h) | 0.205 | 15.776 | 7.575 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Zahabi, K.H.; Ben tkhayat, H.; Abu-Basha, E.; Sallam, A.S.; Younes, H.M. Formulation of Lipid-Based Tableted Spray-Congealed Microparticles for Sustained Release of Vildagliptin: In Vitro and In Vivo Studies. Pharmaceutics 2021, 13, 2158. https://doi.org/10.3390/pharmaceutics13122158
Al Zahabi KH, Ben tkhayat H, Abu-Basha E, Sallam AS, Younes HM. Formulation of Lipid-Based Tableted Spray-Congealed Microparticles for Sustained Release of Vildagliptin: In Vitro and In Vivo Studies. Pharmaceutics. 2021; 13(12):2158. https://doi.org/10.3390/pharmaceutics13122158
Chicago/Turabian StyleAl Zahabi, Khaled H., Hind Ben tkhayat, Ehab Abu-Basha, Al Sayed Sallam, and Husam M. Younes. 2021. "Formulation of Lipid-Based Tableted Spray-Congealed Microparticles for Sustained Release of Vildagliptin: In Vitro and In Vivo Studies" Pharmaceutics 13, no. 12: 2158. https://doi.org/10.3390/pharmaceutics13122158
APA StyleAl Zahabi, K. H., Ben tkhayat, H., Abu-Basha, E., Sallam, A. S., & Younes, H. M. (2021). Formulation of Lipid-Based Tableted Spray-Congealed Microparticles for Sustained Release of Vildagliptin: In Vitro and In Vivo Studies. Pharmaceutics, 13(12), 2158. https://doi.org/10.3390/pharmaceutics13122158