2.3. Synthesis
2.3.1. Compound 10a
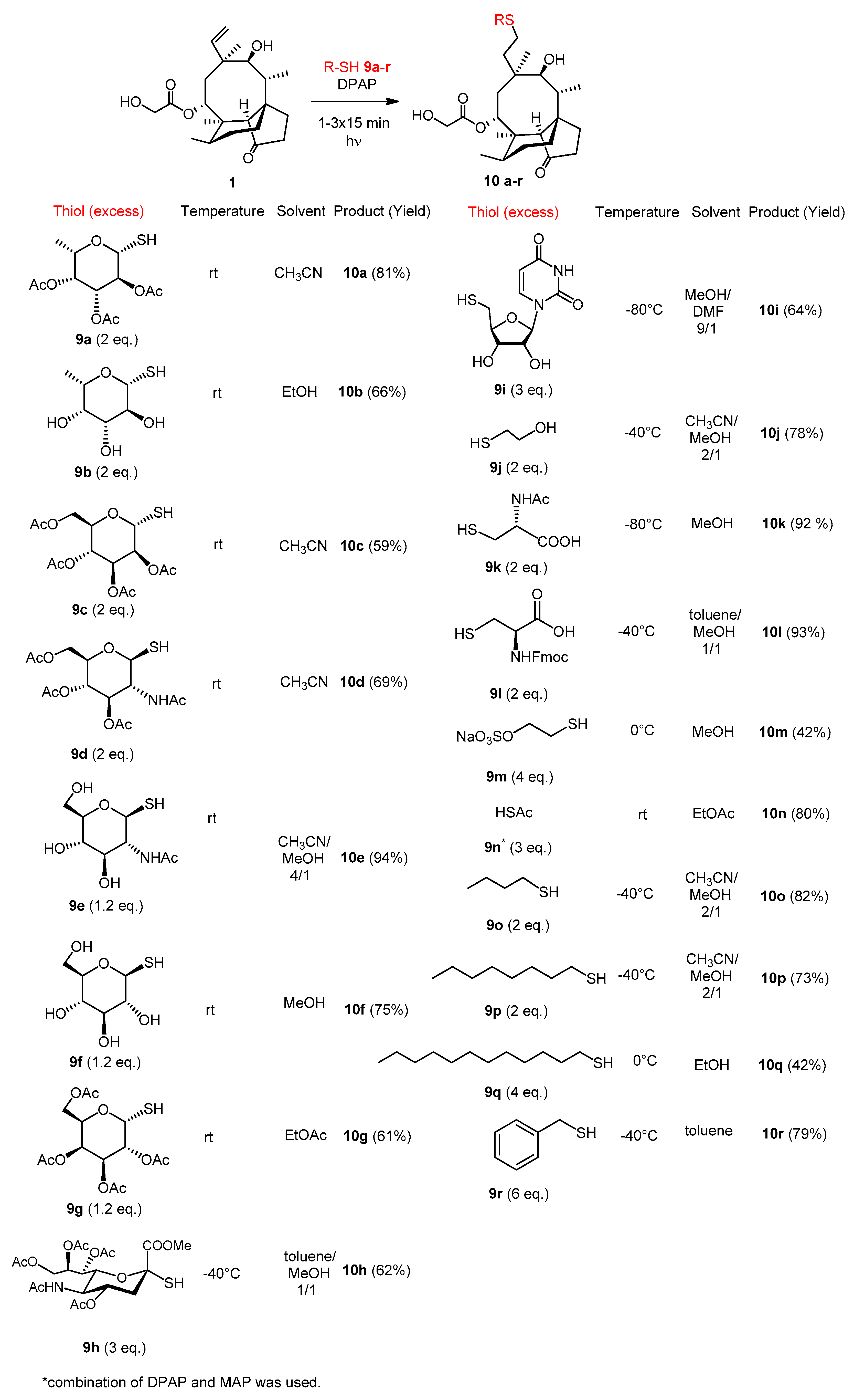
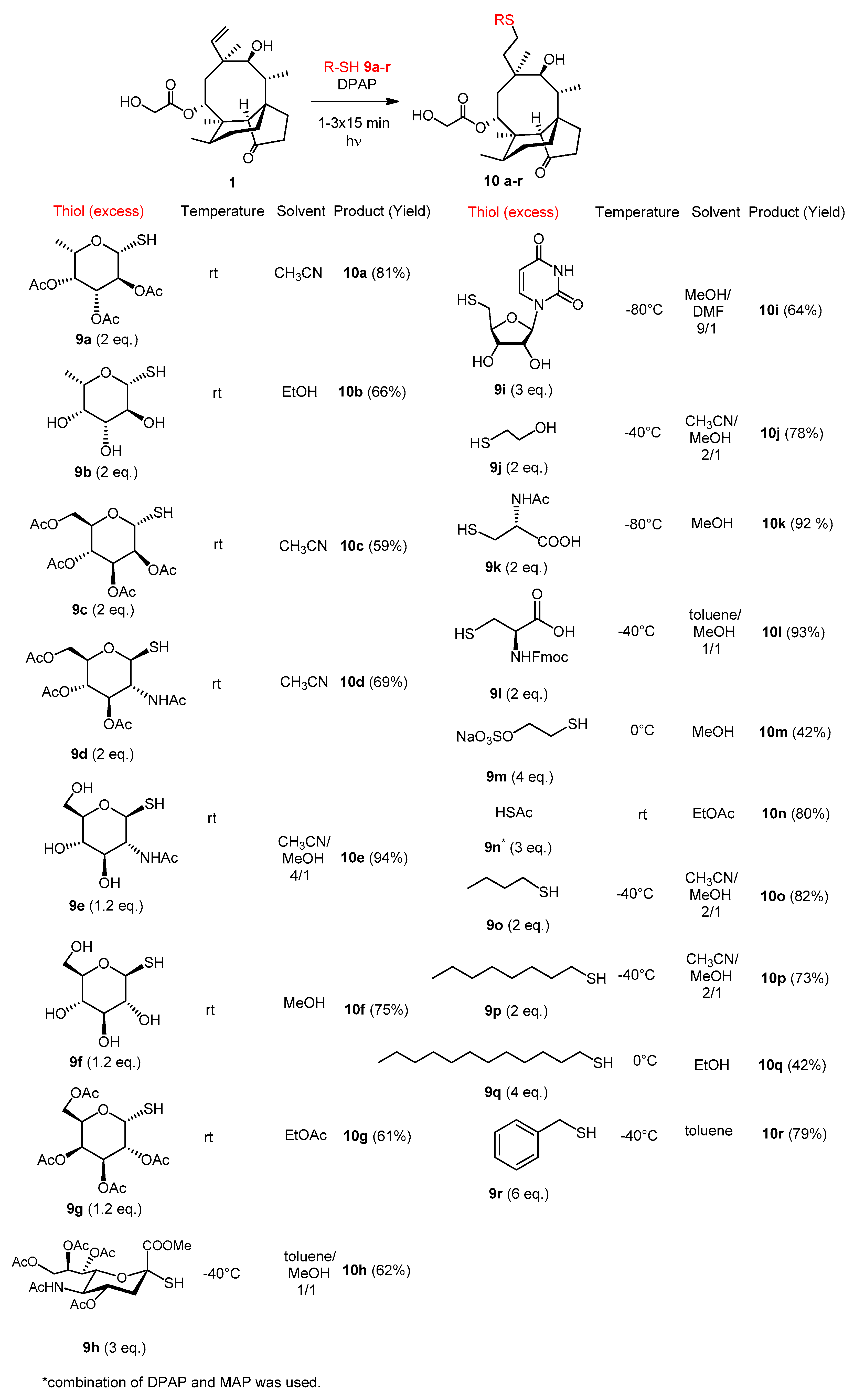
Pleuromutilin (95 mg, 0.25 mmol) and thiol 9a (153 mg, 0.5 mol, 2 × 1 equiv.) were reacted in CH3CN at room temperature according to the general method, using two irradiating cycles. The crude product was purified by flash column chromatography (CH2Cl2/acetone 99/1) to result in compound 10a as white powder (140 mg, 81%). Rf: 0.31 (CH2Cl2/acetone 9/1), [α]24D +43.0 (c 0.1, MeOH), m.p. 192–193 °C. 1H NMR (400 MHz, Chloroform-d) δ 5.62 (d, J = 8.2 Hz, 1H), 5.24 − 5.20 (m, 1H), 5.12 (t, J = 9.9 Hz, 1H), 5.03 (dd, J = 10.0, 3.3 Hz, 1H), 4.55 (d, J = 9.7 Hz, 1H), 4.12 − 3.97 (m, 2H), 3.96 − 3.86 (m, 1H), 3.38 (d, J = 5.7 Hz, 1H), 3.20 (s, 1H), 2.60 (dt, J = 11.9, 6.0 Hz, 1H), 2.45 (td, J = 12.0, 5.0 Hz, 1H), 2.30 (t, J = 6.7 Hz, 1H), 2.11 (d, J = 1.4 Hz, 9H), 2.03 (s, 4H), 1.92 (s, 3H), 1.86 − 1.67 (m, 2H), 1.65 − 1.39 (m, 3H), 1.35 (s, 4H), 1.21 (d, J = 5.2 Hz, 1H), 1.16 (d, J = 6.4 Hz, 3H), 1.14 − 1.01 (m, 1H), 0.99 (s, 3H), 0.90 (d, J = 7.0 Hz, 3H), 0.63 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 216.9, 172.2, 170.6, 170.2, 170.1, 82.9, 75.9, 72.8, 72.3, 70.6, 69.5, 67.7, 61.3, 58.2, 45.5, 42.0, 41.8, 41.2, 36.5, 34.5, 34.4, 30.9, 30.1, 29.6, 26.8, 26.6, 24.9, 24.8, 20.9, 20.6, 16.5, 16.4, 14.7, 11. HRMS (ESI): m/z calcd. for C34H52NaO12S: 707.3077 [M+Na]+; found: 707.3070.
2.3.2. Compound 10b
Pleuromutilin (125 mg, 0.33 mmol) and thiol 9b (119 mg, 0.66 mmol, 2 × 1 equiv.) were reacted in EtOH at room temperature according to the general method, using two irradiating cycles. The crude product was purified by flash column chromatography (CH2Cl2/MeOH 95/5) to result in compound 10b as white powder (121 mg, 66%). Rf: 0.31 (CH2Cl2/MeOH 9/1), [α]24D +12.22 (c 0.09, MeOH), m.p. 91–93 °C. 1H NMR (400 MHz, Methanol-d4) δ 5.78 (d, J = 8.2 Hz, 1H), 4.57 − 4.43 (m, 1H), 4.14 (d, J = 17.3 Hz, 1H), 4.02 (d, J = 17.1 Hz, 1H), 3.88 − 3.77 (m, 1H), 3.69 (d, J = 3.2 Hz, 1H), 3.62 − 3.51 (m, 2H), 3.47 (d, J = 5.8 Hz, 1H), 3.33 (t, J = 1.7 Hz, 1H), 2.65 (td, J = 12.3, 4.2 Hz, 1H), 2.52 (td, J = 12.6, 4.9 Hz, 1H), 2.38 (dd, J = 14.6, 4.6 Hz, 2H), 2.33 − 2.23 (m, 1H), 2.19 (t, J = 9.4 Hz, 2H), 2.10 (ddd, J = 17.7, 11.4, 5.3 Hz, 2H), 2.00 − 1.80 (m, 4H), 1.77 − 1.54 (m, 4H), 1.45 (s, 3H), 1.38 (d, J = 21.9 Hz, 1H), 1.30 (t, J = 5.9 Hz, 4H), 1.23 − 1.11 (m, 1H), 1.04 (d, J = 2.9 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.74 (d, J = 6.1 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 217.0 172.5, 85.3, 75.1, 74.9, 74.3, 71.9, 69.6, 69.0, 60.5, 57.9, 45.4, 41.7, 41.4, 41.0, 36.7, 34.8, 33.9, 30.1, 30.0, 26.7, 25.9, 24.7, 24.3, 15.8, 15.6, 13.9, 10.4. HRMS (ESI): m/z calcd for C28H46NaO9S: 581.2760 [M + Na]+; found: 581.2754.
2.3.3. Compound 10c
The reaction was carried out using the general method, starting from pleuromutilin (190 mg, 0.5 mmol) and thiol 9c (365 mg, 1.0 mmol, 2 × 1 equiv.) in CH3CN at room temperature, irradiating two times. The crude product was purified by flash column chromatography (CH2Cl2/acetone 95/5) to result in compound 10c as white powder (219 mg, 59%). Rf: 0.38 (CH2Cl2/acetone 9/1), [α]24D +107.0 (c 0.1, CHCl3), m.p. 127–130 °C. 1H NMR (400 MHz, Methanol-d4) δ 5.71 (d, J = 8.3 Hz, 1H), 5.48 (d, J = 1.5 Hz, 1H), 5.36 (dd, J = 3.2, 1.5 Hz, 1H), 5.31 (t, J = 9.9 Hz, 1H), 5.25 (dd, J = 10.1, 3.3 Hz, 1H), 4.46 (ddd, J = 9.6, 4.8, 2.4 Hz, 1H), 4.31 (dd, J = 12.3, 4.8 Hz, 1H), 4.15 (dd, J = 12.3, 2.5 Hz, 1H), 4.11 − 3.97 (m, 2H), 3.48 (d, J = 5.8 Hz, 1H), 2.72 − 2.48 (m, 2H), 2.36 (d, J = 7.1 Hz, 2H), 2.31 − 2.19 (m, 1H), 2.18 (d, J = 1.6 Hz, 5H), 2.08 (d, J = 5.8 Hz, 6H), 2.03 (dd, J = 9.9, 7.3 Hz, 2H), 1.99 (s, 1H), 1.97 − 1.79 (m, 4H), 1.76 − 1.54 (m, 4H), 1.45 (s, 3H), 1.30 (dd, J = 16.0, 6.9 Hz, 2H), 1.23 − 1.11 (m, 1H), 1.05 (s, 3H), 0.97 (d, J = 7.0 Hz, 3H), 0.75 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 217.0, 172.1, 171.0, 170.3, 170.2, 170.1, 81.5, 74.9, 70.9, 69.8, 69.0, 68.7, 66.1, 62.4, 60.6, 57.8, 45.4, 41.8, 41.7, 41.0, 36.6, 34.8, 33.9, 30.0, 29.7, 26.7, 26.0, 25.9, 24.3, 19.4, 19.4, 19.3, 19.2, 15.8, 13.9, 10.4. HRMS (ESI): m/z calcd. for C36H54NaO14S: 765.3132 [M + Na]+; found: 765.3125.
2.3.4. Compound 10d
The reaction was carried out using the general method, starting from pleuromutilin (190 mg, 0.5 mmol) and thiol 9d (364 mg, 1.0 mmol, 2 × 1 equiv.) in CH3CN at room temperature, irradiating two times. The crude product was purified by flash column chromatography (CH2Cl2/acetone 95/5) to result in compound 10d as white powder (255 mg, 69%). Rf: 0.11 (CH2Cl2/acetone 9/1), [α]24D +13.0 (c 0.1, CHCl3), m.p. 97–98 °C. 1H NMR (400 MHz, Chloroform-d) δ 6.79 (d, J = 9.3 Hz, 1H), 5.70 (d, J = 8.2 Hz, 1H), 5.41 − 5.24 (m, 1H), 5.13 (t, J = 9.7 Hz, 1H), 4.68 (d, J = 10.4 Hz, 1H), 4.25 (dd, J = 12.3, 4.7 Hz, 1H), 4.19 − 4.10 (m, 3H), 4.10 − 3.97 (m, 1H), 3.76 (ddd, J = 9.9, 4.6, 2.4 Hz, 1H), 3.42 (d, J = 5.7 Hz, 1H), 2.80 (td, J = 12.3, 4.3 Hz, 1H), 2.37 (tq, J = 12.8, 7.8, 6.2 Hz, 3H), 2.29 − 2.21 (m, 1H), 2.18 (s, 1H), 2.14 − 2.06 (m, 4H), 2.08 − 1.95 (m, 10H), 1.95 − 1.74 (m, 3H), 1.73 − 1.52 (m, 2H), 1.53 − 1.44 (m, 2H), 1.42 (s, 4H), 1.33 − 1.20 (m, 2H), 1.14 (td, J = 14.0, 4.5 Hz, 1H), 1.03 (s, 3H), 0.96 (d, J = 6.9 Hz, 3H), 0.70 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 217.0, 172.7, 171.1, 171.1, 170.9, 169.4, 86.1, 76.0, 75.8, 73.8, 69.6, 68.4, 62.5, 61.4, 58.2, 53.5, 45.5, 42.3, 41.8, 41.3, 36.5, 34.7, 34.4, 30.8, 30.2, 27.7, 26.9, 26.7, 24.8, 23.2, 20.8, 20.8, 20.7, 16.6, 14.7, 11.1. HRMS (ESI): m/z calcd. for C36H55NNaO13S: 764.3292 [M + Na]+; found: 764.3287.
2.3.5. Compound 10e
The reaction was carried out using the general method, starting from pleuromutilin (190 mg, 0.5 mmol) and thiol 9e (142 mg, 0.6 mmol, 1.2 equiv.) in CH3CN/MeOH 4/1 at room temperature, irradiating once. The crude product was purified by flash column chromatography (CH2Cl2/MeOH 8/2) to result in compound 10e as white amorphous solid (290 mg, 94%). Rf: 0.42 (CH2Cl2/MeOH 8/2), [α]24D +3.85 (c 0.13, CHCl3). 1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 9.4 Hz, 1H), 5.55 (d, J = 8.2 Hz, 1H), 5.39 (t, J = 6.6 Hz, 1H), 5.12 − 4.86 (m, 2H), 4.56 (d, J = 6.0 Hz, 1H), 4.50 (t, J = 6.0 Hz, 1H), 4.37 (d, J = 10.3 Hz, 1H), 4.21 − 4.11 (m, 1H), 4.03 − 3.79 (m, 2H), 3.75 − 3.63 (m, 1H), 3.22 − 3.05 (m, 3H), 2.64 − 2.48 (m, 2H), 2.46 − 2.30 (m, 2H), 2.18 (td, J = 12.7, 11.2, 6.8 Hz, 2H), 2.06 (dt, J = 18.7, 9.2 Hz, 1H), 1.94 − 1.77 (m, 5H), 1.67 (ttd, J = 22.0, 11.7, 7.8 Hz, 3H), 1.55 − 1.39 (m, 2H), 1.38 − 1.18 (m, 6H), 1.18 − 0.95 (m, 2H), 0.92 (s, 3H), 0.84 (d, J = 6.9 Hz, 3H), 0.62 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 217.7, 172.1, 169.6, 86.0, 81.2, 75.9, 74.1, 70.9, 68.3, 61.8, 60.8, 57.7, 55.2, 49.0, 45.4, 42.3, 41.8, 41.2, 36.8, 35.1, 34.4, 31.1, 30.6, 27.3, 26.7, 24.8, 23.5, 16.7, 15.0, 12.0. HRMS (ESI): m/z calcd for C30H49NNaO10S: 638.2975 [M + Na]+; found: 638.2969.
2.3.6. Compound 10f
The reaction was carried out using the general method, starting from pleuromutilin (190 mg, 0.5 mmol) and thiol 9f (118 mg, 0.6 mmol, 1.2 equiv.) in MeOH at room temperature, irradiating once. The crude product was purified by flash column chromatography (CH2Cl2/MeOH 8/2) and resulted in compound 10f as white amorphous solid (216 mg, 75%). Rf: 0.71 (CH2Cl2/MeOH 7/3), [α]24D= +17.8 (c 0.09, CHCl3). 1H NMR (400 MHz, DMSO-d6) δ 5.75 (d, J = 2.7 Hz, 1H), 5.55 (d, J = 8.1 Hz, 1H), 5.33 (s, 1H), 4.98 (d, J = 40.6 Hz, 2H), 4.59 (d, J = 5.7 Hz, 1H), 4.46 (s, 1H), 4.27 (dd, J = 9.6, 2.7 Hz, 1H), 3.90 (q, J = 17.0 Hz, 2H), 3.67 (d, J = 11.8 Hz, 1H), 3.15 (dd, J = 10.1, 6.6 Hz, 2H), 3.08 (d, J = 9.2 Hz, 1H), 2.96 (t, J = 9.3 Hz, 1H), 2.63 − 2.39 (m, 5H), 2.35 (s, 1H), 2.18 (t, J = 8.4 Hz, 2H), 2.15 − 2.00 (m, 1H), 2.00 − 1.79 (m, 2H), 1.80 − 1.56 (m, 2H), 1.58 − 1.40 (m, 2H), 1.38 − 1.20 (m, 6H), 1.17 − 0.97 (m, 1H), 0.94 (d, J = 2.7 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H), 0.63 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 217.6, 172.0, 86.3, 81.1, 78.6, 74.0, 73.8, 70.5, 68.3, 61.8, 60.9, 57.7, 45.4, 42.6, 41.8, 41.2, 36.8, 35.1, 34.4, 31.6, 30.6, 27.3, 27.1, 25.9, 24.8, 16.7, 15.0, 12.0. HRMS (ESI): m/z calcd. for C28H46NaO10S: 597.2709 [M + Na]+; found: 597.2704.
2.3.7. Compound 10g
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9g (109 mg, 0.3 mmol, 1.2 equiv.) in EtOAc at room temperature, irradiating once. The crude product was purified by flash column chromatography (CH2Cl2/acetone 95/5) to result in compound 10g as white amorphous solid (114 mg, 61%). Rf: 0.33 (CH2Cl2/acetone 9/1), [α]24D +19.3 (c 0.14, CHCl3). 1H NMR (360 MHz, Chloroform-d) δ 5.66 (d, J = 8.3 Hz, 1H), 5.43 (d, J = 3.3 Hz, 1H), 5.36 − 5.18 (m, 3H), 5.12 (dd, J = 10.0, 3.3 Hz, 1H), 4.66 (d, J = 9.9 Hz, 1H), 4.16 − 3.97 (m, 4H), 3.42 (d, J = 5.6 Hz, 1H), 2.81 (t, J = 5.7 Hz, 1H), 2.57 (t, J = 8.5 Hz, 2H), 2.40 − 2.11 (m, 6H), 2.05 (s, 4H), 1.99 (s, 3H), 1.86 (ddd, J = 32.2, 16.3, 8.6 Hz, 5H), 1.70 − 1.43 (m, 6H), 1.42 (s, 3H), 1.32 − 1.22 (m, 1H), 1.21 − 1.02 (m, 5H), 0.97 (d, J = 6.9 Hz, 3H), 0.70 (d, J = 6.8 Hz, 3H). 13C NMR (91 MHz, Chloroform-d) δ 216.6, 172.3, 170.6, 170.2, 169.8, 84.1, 76.1, 74.0, 71.9, 69.7, 67.4, 67.3, 61.4, 61.3, 58.1, 45.4, 41.9, 41.8, 41.3, 36.4, 34.4, 34.3, 30.5, 30.0, 26.8, 25.5, 24.8, 20.9, 20.6, 16.6, 14.7, 10.9. HRMS (ESI): m/z calcd. for C36H54NaO14S: 765.3132 [M + Na]+; found: 765.3126.
2.3.8. Compound 10h
The reaction was carried out using the general method, starting from pleuromutilin (48 mg, 0.125 mmol) and thiol 9h (191 mg, 0.375 mmol, 3 × 1 equiv.) in toluene/MeOH 1/1 at −40 °C, irradiating three times. The crude product was purified by flash column chromatography (CH2Cl2/acetone 9/1) to result in compound 10h as white amorphous solid (69 mg, 62%). Rf: 0.20 (CH2Cl2/acetone 8/2), [α]24D +46.7 (c 0.12, MeOH). 1H NMR (400 MHz, Chloroform-d) δ 5.59 (d, J = 8.1 Hz, 1H), 5.46 (d, J = 10.1 Hz, 1H), 5.31 (q, J = 4.9, 3.6 Hz, 2H), 4.95 − 4.76 (m, 1H), 4.37 − 4.22 (m, 1H), 4.16 − 3.94 (m, 4H), 3.94 − 3.66 (m, 4H), 3.38 (d, J = 5.5 Hz, 1H), 2.71 (dd, J = 12.7, 4.6 Hz, 1H), 2.62 (s, 1H), 2.57 (dd, J = 12.2, 4.1 Hz, 1H), 2.43 (td, J = 11.7, 5.2 Hz, 1H), 2.30 (dt, J = 10.0, 4.8 Hz, 1H), 2.22 − 2.06 (m, 6H), 2.02 (d, J = 1.8 Hz, 8H), 1.86 (s, 4H), 1.75 (dt, J = 9.8, 5.8 Hz, 2H), 1.68 − 1.50 (m, 2H), 1.50 − 1.42 (m, 1H), 1.41 (s, 4H), 1.38 − 1.29 (m, 1H), 1.24 (s, 5H), 1.10 (td, J = 14.1, 3.0 Hz, 1H), 1.03 (s, 3H), 0.91 (d, J = 6.8 Hz, 3H), 0.67 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 218.0, 172.4, 170.9, 170.8, 170.3, 170.2, 168.4, 83.3, 76.0, 73.9, 69.8, 69.7, 68.3, 67.2, 62.4, 61.3, 58.3, 53.8, 52.9, 49.3, 45.4, 41.7, 41.7, 41.4, 38.0, 36.5, 34.4, 30.1, 29.6, 29.3, 28.0, 26.8, 26.1, 24.8, 24.5, 23.1, 21.2, 20.9, 20.8, 16.6, 14.8, 11.0. HRMS (ESI): m/z calcd. for C42H63NNaO17S: 908.3714 [M+Na]+; found: 908.3709.
2.3.9. Compound 10i
The reaction was carried out using the general method, starting from pleuromutilin (190 mg, 0.5 mmol) and thiol 9i (390 mg, 1.5 mmol, 3 × 1 equiv.) in MeOH/DMF 9/1 at −80 °C, irradiating three times. The crude product was purified by flash column chromatography (CH2Cl2/MeOH 95/5 to 9:1) to result in compound 10i as white amorphous solid (204 mg, 64%). Rf: 0.51 (CH2Cl2/MeOH 8/2), [α]24D +14.0 (c 0.1, MeOH). 1H NMR (400 MHz, Methanol-d4) δ 7.80 (d, J = 8.0 Hz, 1H), 5.88 (d, J = 4.8 Hz, 1H), 5.74 (t, J = 8.1 Hz, 2H), 4.23 (t, J = 4.7 Hz, 1H), 4.13 (q, J = 5.3, 4.6 Hz, 3H), 4.07 (s, 1H), 4.02 (s, 1H), 3.44 (d, J = 5.8 Hz, 1H), 3.35 (s, 1H), 3.31 (p, J = 1.6 Hz, 1H), 3.01 − 2.90 (m, 2H), 2.59 (td, J = 11.9, 4.9 Hz, 1H), 2.46 (td, J = 12.1, 5.4 Hz, 1H), 2.34 (qd, J = 11.6, 9.9, 4.3 Hz, 3H), 2.25 (dd, J = 10.3, 7.9 Hz, 1H), 2.14 (dt, J = 19.1, 9.3 Hz, 1H), 2.02 − 1.78 (m, 4H), 1.72 − 1.52 (m, 3H), 1.48 − 1.32 (m, 5H), 1.26 (d, J = 16.3 Hz, 2H), 1.20 − 1.09 (m, 1H), 1.01 (s, 3H), 0.95 (d, J = 6.9 Hz, 3H), 0.72 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 173.5, 166.1, 152.3, 142.7, 103.0, 90.9, 85.0, 76.3, 75.0, 73.4, 70.3, 61.9, 59.3, 48.9, 46.8, 43.3, 43.1, 42.3, 38.0, 36.2, 35.3, 35.1, 31.8, 31.4, 29.4, 28.1, 27.4, 25.7, 17.0, 15.3, 11.8. HRMS (ESI): m/z calcd. for C31H46N2NaO10S: 661.2873 [M + Na]+; found: 661.2765.
2.3.10. Compound 10j
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9j (39 mg, 35µL, 0.5 mmol, 2 equiv.) in CH3CN/MeOH 2/1 at −40 °C, irradiating once. The crude product was purified by flash column chromatography (n-hexane/acetone 8/2) to result in compound 10j as white powder (89 mg, 78%). Rf: 0.10 (n-hexane/acetone 7/3), [α]24D +10.3 (c 0.35, MeOH), m.p. 189 °C. 1H NMR (400 MHz, Methanol-d4) δ 5.75 (d, J = 8.3 Hz, 1H, H-14), 4.03 (q, J = 17.1 Hz, 2H, H-22ab), 3.67 (t, J = 6.9 Hz, 2H, H-24ab), 3.44 (d, J = 5.9 Hz, 1H, H-11), 2.74 (dt, J = 13.8, 6.9 Hz, 1H, H-23a), 2.66 (dt, J = 13.6, 6.9 Hz, 1H, H-23b), 2.48 (td, J = 12.2, 4.7 Hz, 1H, H-19a*), 2.44 − 2.34 (m, 2H, H-10, H-19b*), 2.33 (s, 1H, H-4), 2.26 (dd, J = 19.2, 10.7 Hz, 1H, H-2a), 2.20 − 2.08 (m, 1H, H-2b), 2.03 − 1.93 (m, 1H, H-20a*), 1.92 − 1.80 (m, 3H, H-8a, H-13a, H-20b*), 1.73 − 1.65 (m, 1H, H-1a), 1.64 − 1.53 (m, 2H, H-6, H-7a), 1.45 (dd, J = 8.3, 6.2 Hz, 1H, H-1b), 1.42 (s, 3H, H-15abc), 1.37 (d, J = 10.8 Hz, 1H, H-7b), 1.32 − 1.25 (m, 1H, H-13b), 1.20 − 1.10 (m, 1H, H-8b), 1.01 (s, 3H, H-18abc), 0.95 (d, J = 7.0 Hz, 3H, H-17abc), 0.72 (d, J = 6.1 Hz, 3H, H-16abc). * H-19a↔H-20a and H-19b↔H-20b are interchangeable. 13C NMR (101 MHz, Methanol-d4) δ 219.6 (1C, C-3), 173.5 (1C, C-21), 76.4 (1C, C-11), 70.3 (1C, C-14), 62.8, 61.9 (2C, C-22, C-24), 59.3 (1C, C-4), 46.8 (1C, C-9), 43.1 (1C, C-13), 43.1 (1C, C-12), 42.2 (1C, C-5), 38.1 (1C, C-6), 36.2 (1C, C-10), 35.3, 34.9 (2C, C-2, C-23), 31.6, 31.4, 28.2, 28.1 (4C, C-7, C-19, C-8, C-20), 27.4 (1C, C-18), 25.7 (1C, C-1), 17.0 (1C, C-16), 15.3 (1C, C-15), 11.8 (1C, C-17). HRMS (ESI): m/z calcd. for C20H40NaO6S: 479.2443 [M + Na]+; found: 479.2438.
2.3.11. Compound 10k
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9k (82 mg, 0.5 mmol, 2 × 1 equiv.) in MeOH at −80 °C, irradiating two times. The crude product was purified by flash column chromatography (CH2Cl2/MeOH 9/1) to result in compound 10k as white-yellow powder (125 mg, 92%). Rf: 0.31 (CH2Cl2/MeOH 7/3), [α]24D +34.3 (c 0.14, MeOH), m.p. 73–74 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.85 (d, J = 6.2 Hz, 1H, NH), 5.56 (d, J = 7.8 Hz, 1H, H-14), 4.22 (s, 1H, H-24), 4.01 (d, J = 17.1 Hz, 1H, H-22a), 3.82 (d, J = 17.1 Hz, 1H, H-22b), 3.34 (d, J = 5.1 Hz, 1H, H-11), 2.91 (d, J = 9.7 Hz, 1H, H-23a), 2.74 (dd, J = 12.4, 5.8 Hz, 1H, H-23b), 2.46 − 2.25 (m, 3H, H-4, H-19ab*), 2.18 (t, J = 14.3 Hz, 2H, H-10, H-2a), 2.11 − 1.99 (m, 1H, H-2b), 1.86 (s, 3H, AcCH3), 1.81 (d, J = 8.0 Hz, 1H, H-13a), 1.64 (dd, J = 29.7, 9.9 Hz, 4H, H-8a, H-7a, H-20ab*), 1.44 (dd, J = 23.1, 9.1 Hz, 2H, H-6, H-1a), 1.32 (s, 3H, H-15abc), 1.29 − 1.22 (m, 2H, H-1b, H-7b), 1.09 (d, J = 16.0 Hz, 1H, H-13b), 1.00 (d, J = 12.7 Hz, 1H, H-8b), 0.91 (s, 3H, H-18abc), 0.82 (d, J = 6.6 Hz, 3H, H-17abc), 0.61 (d, J = 5.9 Hz, 3H, H-16abc). *H-19a↔H-20a, H-19b↔H-20b and C19↔20 are interchangeable signals. 13C NMR (101 MHz, DMSO-d6) δ 217.2 (1C, C-3), 172.3, 168.9 (3C, AcCO, COOH, C-21), 73.6 (1C, C-11), 67.7 (1C, C-14), 60.4 (1C, C-22), 57.3 (1C, C-4), 53.3 (1C, C-24), 55.0, 45.0, 41.4, 40.7 (4C, C-5, C-12, C-13, C-9), 36.4 (1C, C-6), 34.7 (1C, C-10), 34.5, 34.0 (2C, C-23, C-2), 30.2, 29.7 (2C, C-8, C-20*), 27.6 (1C, C-19*), 26.9 (1C, C-18), 26.7 (1C, C-1), 24.4 (1C, C-7), 22.8 (1C, AcCH3), 16.2 (1C, C-16), 14.56 (1C, C-15), 11.6 (1C, C-17). HRMS (MALDI): m/z calcd. for C27H43NNaO8S: 564.2607 [M + Na]+; found: 564.2616.
2.3.12. Compound 10l
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9l (172 mg, 0.5 mmol, 2 × 1 equiv.) in toluene/MeOH 1/1 at −40 °C, irradiating two times. The crude product was purified by flash column chromatography (toluene/MeOH 95/5) to result in compound 10l as yellow powder (169 mg, 93%). Rf: 0.11 (toluene/MeOH 8/2), [α]24D +15.3 (c 0.15, MeOH), m.p. 143–147 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.89 (d, J = 7.6 Hz, 2H), 7.74 (t, J = 6.9 Hz, 2H), 7.42 (t, J = 7.5 Hz, 2H), 7.34 (t, J = 7.5 Hz, 2H), 5.60 (d, J = 8.0 Hz, 1H), 4.32 − 4.23 (m, 2H), 4.22 (q, J = 6.7, 6.1 Hz, 2H), 4.06 (d, J = 17.1 Hz, 1H), 3.82 (d, J = 17.1 Hz, 1H), 3.00 (dd, J = 13.9, 4.5 Hz, 1H), 2.79 (dd, J = 13.5, 6.8 Hz, 1H), 2.37 (d, J = 21.5 Hz, 3H), 2.24 − 2.14 (m, 2H), 2.12 − 1.96 (m, 1H), 1.83 (dd, J = 16.2, 8.0 Hz, 1H), 1.67 (ddt, J = 37.8, 21.7, 10.0 Hz, 4H), 1.49 (d, J = 7.9 Hz, 2H), 1.40 − 1.28 (m, 5H), 1.30 − 1.21 (m, 5H), 1.11 (d, J = 15.8 Hz, 2H), 0.92 (s, 3H), 0.89 − 0.80 (m, 3H), 0.63 (d, J = 5.7 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 217.2, 172.20, 155.5, 143.9, 140.7, 127.6, 127.1, 125.4, 125.3, 120.1, 73.6, 67.5, 65.6, 60.3, 57.3, 46.7, 45.0, 41.3, 40.6, 36.3, 34.6, 33.9, 31.1, 30.2, 29.8, 29.0, 27.6, 26.9, 26.6, 24.4, 16.1, 14.5, 11.5. HRMS (MALDI): m/z calcd. for C40H51NNaO9S: 744.3182 [M + Na]+; found: 744.3163.
2.3.13. Compound 10m
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9m (165 mg, 1.0 mmol, 2 × 2 equiv.) in MeOH at 0 °C, irradiating two times. The crude product was purified by flash column chromatography (CH2Cl2/MeOH 95/5) to result in compound 10m as white powder (58 mg, 42%). Rf: 0.69 (CH2Cl2/MeOH 7/3), [α]24D +25.0 (c 0.12, MeOH), m.p. 113–114 °C. 1H NMR (400 MHz, Methanol-d4) δ 5.77 (d, J = 8.3 Hz, 1H), 4.19 − 3.93 (m, 2H), 3.47 (d, J = 5.9 Hz, 1H), 3.33 (p, J = 1.6 Hz, 1H), 3.09 (ddd, J = 9.1, 7.0, 2.2 Hz, 2H), 3.02 − 2.84 (m, 2H), 2.58 (td, J = 12.0, 4.7 Hz, 1H), 2.50 − 2.32 (m, 3H), 2.33 − 2.23 (m, 1H), 2.18 (d, J = 2.5 Hz, 1H), 1.91 (tdd, J = 25.3, 12.9, 4.1 Hz, 4H), 1.78 − 1.54 (m, 4H), 1.44 (s, 4H), 1.33 (d, J = 16.5 Hz, 2H), 1.27 − 1.08 (m, 1H), 1.05 (s, 3H), 0.97 (d, J = 7.0 Hz, 3H), 0.74 (d, J = 6.1 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 217.0, 172.0, 75.0, 69.0, 60.5, 57.9, 51.9, 45.5, 42.3, 41.7, 40.9, 36.7, 34.8, 33.9, 30.1, 30.0, 27.2, 26.7, 26.5, 25.9, 24.3, 15.6, 13.9, 10.4. HRMS (ESI): m/z calcd. for C24H39Na2O8S2: 565.1882 [M + Na]+; found: 565.1876.
2.3.14. Compound 10n
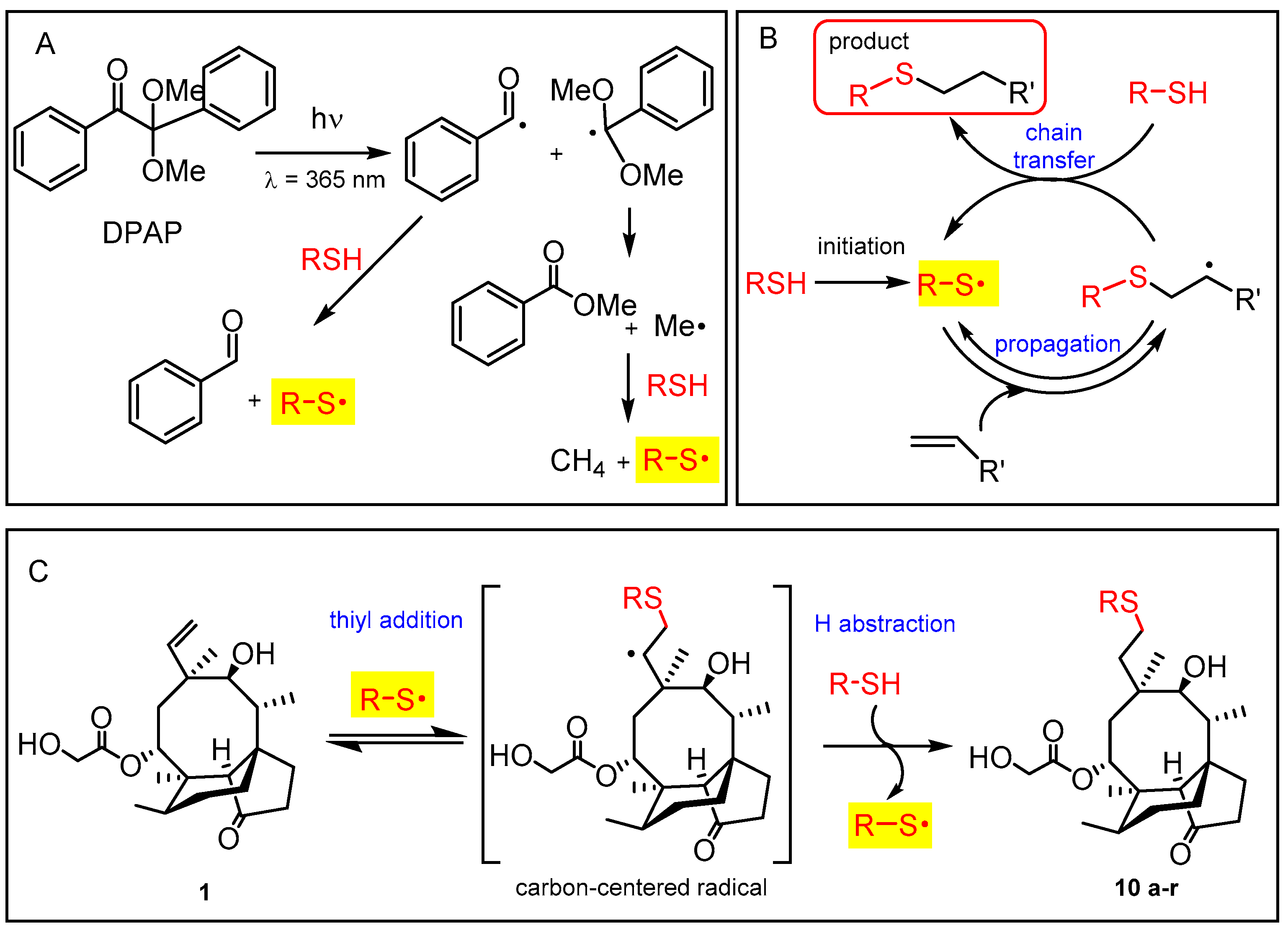
To the mixture of pleuromutilin (95 mg, 0.25 mmol) and thiol 9n (57 mg, 54 µL, 0.75 mmol, 3 equiv.) in EtOAc at room temperature, 0.1 equivalent of DPAP (2,2-dimethoxy-2-phenylacetophenone) (0.025 mmol, 6 mg) and 0.1 equivalent of MAP (4-methoxyacetophenone) (0.025mmol, 4 mg) were added. It was irradiated once for one hour. The crude product was purified by flash column chromatography (n-hexane/acetone 8/2) to result in compound 10n as white powder (91 mg, 80%). Rf: 0.36 (n-hexane/acetone 6/4), [α]24D +54.7 (c 0.15, MeOH), m.p. 188–189 °C. 1H NMR (400 MHz, Methanol-d4) δ 5.69 (d, J = 8.2 Hz, 1H), 4.14 − 3.87 (m, 2H), 3.44 (d, J = 6.1 Hz, 1H), 3.03 − 2.89 (m, 1H), 2.66 (td, J = 12.4, 5.0 Hz, 1H), 2.33 (d, J = 3.1 Hz, 2H), 2.30 (s, 4H), 2.28 − 2.17 (m, 1H), 2.18 − 2.05 (m, 1H), 1.97 − 1.77 (m, 5H), 1.73 − 1.51 (m, 3H), 1.42 (s, 4H), 1.31 (d, J = 16.4 Hz, 1H), 1.14 (td, J = 13.7, 4.4 Hz, 1H), 1.07 (s, 3H), 0.94 (d, J = 7.0 Hz, 3H), 0.71 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 217.0, 194.0, 168.0, 73.3, 67.3, 59.0, 56.4, 43.9, 40.2, 40.2, 39.6, 35.1, 33.4, 32.4, 28.6, 28.2, 27.6, 25.2, 24.2, 22.8, 14.1, 12.4, 8.9. HRMS (ESI): m/z calcd. for C24H38NaO6S: 477.2287 [M + Na]+; found: 477.2281.
2.3.15. Compound 10o
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9o (45 mg, 60 µL, 0.5 mmol, 2 × 1 equiv.) in CH3CN/MeOH 2/1 at −40 °C, irradiating two times. The crude product was purified by flash column chromatography (CH2Cl2/acetone 95/5) to result in compound 10o as white powder (97 mg, 82%). Rf: 0.56 (CH2Cl2/acetone 9/1), [α]24D +52.2 (c 0.18, CHCl3), m.p. 177–180 °C. 1H NMR (400 MHz, Chloroform-d) δ 5.65 (d, J = 8.3 Hz, 1H), 4.01 (q, J = 17.1 Hz, 2H), 3.38 (d, J = 5.8 Hz, 1H), 2.51 (dd, J = 7.9, 6.8 Hz, 2H), 2.41 (t, J = 8.3 Hz, 2H), 2.37 − 2.27 (m, 2H), 2.24 − 2.15 (m, 1H), 2.13 (s, 2H), 1.91 (t, J = 8.4 Hz, 2H), 1.83 − 1.68 (m, 2H), 1.62 − 1.41 (m, 7H), 1.36 (s, 5H), 1.30 − 1.18 (m, 2H), 1.08 (td, J = 13.8, 4.3 Hz, 1H), 0.99 (s, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.88 (t, J = 7.3 Hz, 3H), 0.64 (d, J = 6.8 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 217.1, 172.3, 76.0, 69.9, 61.3, 58.3, 45.5, 42.4, 41.8, 41.2, 36.5, 34.5, 34.4, 31.9, 31.8, 30.1, 30.1, 27.4, 26.8, 26.8, 24.8, 22.0, 16.5, 14.7, 13.7, 11.1. HRMS (ESI): m/z calcd. for C26H44NaO5S: 491.2807 [M + Na]+; found: 491.2801.
2.3.16. Compound 10p
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9p (73 mg, 87 µL, 0.5 mmol, 2 equiv.) in CH3CN/MeOH 2/1 at −40 °C, irradiating once. The crude product was purified by flash column chromatography (CH2Cl2/acetone 95/5) to result in compound 10p as white amorphous solid (96 mg, 73%). Rf: 0.66 (CH2Cl2/acetone 9/1), [α]24D +20.0 (c 0.04, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 5.68 (d, J = 8.3 Hz, 1H), 4.04 (qd, J = 17.0, 4.1 Hz, 2H), 3.41 (d, J = 5.8 Hz, 1H), 2.54 (t, J = 7.4 Hz, 2H), 2.45 (dt, J = 9.3, 3.0 Hz, 2H), 2.41 − 2.28 (m, 2H), 2.28 − 2.14 (m, 2H), 2.09 (d, J = 2.7 Hz, 1H), 1.94 (ddd, J = 9.9, 6.2, 2.4 Hz, 2H), 1.81 − 1.74 (m, 2H), 1.68 − 1.43 (m, 7H), 1.40 (s, 5H), 1.28 (dt, J = 10.0, 5.1 Hz, 10H), 1.11 (td, J = 13.8, 4.4 Hz, 1H), 1.03 (s, 3H), 0.96 (d, J = 7.0 Hz, 3H), 0.91 − 0.82 (m, 3H), 0.67 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 216.7, 172.3, 76.1, 70.0, 61.3, 58.3, 45.5, 42.4, 41.8, 41.2, 36.5, 34.5, 34.4, 32.3, 31.8, 30.1, 30.1, 29.8, 29.3, 29.2, 29.0, 27.4, 26.8, 26.8, 24.9, 22.6, 16.6, 14.7, 14.1, 11.1. HRMS (ESI): m/z calcd. for C30H52NaO5S: 547.3433 [M + Na]+; found: 547.3427.
2.3.17. Compound 10q
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9q (202 mg, 240 µL, 1.0 mmol, 2 × 2 equiv.) in EtOH at 0 °C, irradiating two times. The crude product was purified by flash column chromatography (CH2Cl2/acetone 95/5) to result in compound 10q as white amorphous solid (61 mg, 42%). Rf: 0.66 (CH2Cl2/acetone 9/1), [α]24D +47.4 (c 0.19, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 5.72 (d, J = 8.3 Hz, 1H), 4.06 (q, J = 17.0 Hz, 2H), 3.43 (d, J = 6.0 Hz, 1H), 2.57 (t, J = 7.4 Hz, 2H), 2.48 (td, J = 10.6, 9.7, 6.1 Hz, 2H), 2.40 (dd, J = 13.8, 6.1 Hz, 2H), 2.31 − 2.16 (m, 2H), 2.11 (s, 1H), 1.97 (dt, J = 10.8, 4.8 Hz, 2H), 1.82 (td, J = 15.5, 14.2, 5.8 Hz, 2H), 1.73 − 1.48 (m, 7H), 1.42 (d, J = 9.2 Hz, 5H), 1.28 (d, J = 5.3 Hz, 18H), 1.14 (td, J = 13.8, 4.3 Hz, 1H), 1.06 (s, 3H), 0.99 (d, J = 7.0 Hz, 3H), 0.89 (t, J = 6.6 Hz, 3H), 0.70 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 216.7, 172.3, 76.2, 70.1, 61.3, 58.3, 45.5, 42.5, 41.9, 41.2, 36.5, 34.5, 34.4, 32.4, 31.9, 30.1, 30.1, 29.8, 29.7, 29.6, 29.6, 29.3, 29.3, 29.0, 27.4, 26.8, 26.8, 24.9, 22.7, 16.5, 14.7, 14.1, 11.0. HRMS (ESI): m/z calcd. for C34H60NaO5S: 603.4059 [M + Na]+; found: 603.4052.
2.3.18. Compound 10r
The reaction was carried out using the general method, starting from pleuromutilin (95 mg, 0.25 mmol) and thiol 9r (186 mg, 177 µL, 1.5 mmol, 3 × 2 equiv.) in toluene at −40 °C, it was irradiated three times. The crude product was purified by flash column chromatography (CH2Cl2/acetone 97/3) to result in compound 10r as white powder (100 mg, 79%). Rf: 0.60 (CH2Cl2/acetone 9/1), [α]24D +67.1 (c 0.21, CHCl3), m.p. 150–151 °C. 1H NMR (400 MHz, Methanol-d4) δ 7.45 − 7.40 (m, 2H), 7.30 (td, J = 7.1, 6.1, 1.3 Hz, 2H), 7.25 − 7.19 (m, 1H), 5.74 (d, J = 8.3 Hz, 1H), 4.13 − 3.95 (m, 2H), 3.88 (d, J = 13.5 Hz, 1H), 3.74 (d, J = 13.5 Hz, 1H), 3.41 (d, J = 6.0 Hz, 1H), 2.37 (ddd, J = 9.7, 5.8, 2.5 Hz, 2H), 2.33 − 2.29 (m, 1H), 2.29 − 2.21 (m, 1H), 2.17 (s, 2H), 2.04 (ddd, J = 13.7, 10.3, 6.5 Hz, 1H), 1.95 − 1.77 (m, 4H), 1.64 (dt, J = 11.4, 6.7 Hz, 4H), 1.44 (s, 4H), 1.23 (d, J = 16.2 Hz, 1H), 1.15 (td, J = 13.4, 4.3 Hz, 1H), 0.98 (s, 3H), 0.83 (d, J = 6.9 Hz, 3H), 0.75 (d, J = 6.0 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 217.0, 171.9, 139.2, 128.7, 128.0, 126.4, 74.9, 68.9, 60.6, 57.9, 45.5, 42.5, 41.7, 40.8, 36.7, 35.1, 34.8, 33.9, 30.8, 30.0, 26.8, 26.3, 26.1, 24.4, 15.7, 14.0, 10.5. HRMS (ESI): m/z calcd. for C29H42NaO5S: 525.2651 [M + Na]+; found: 525.2643.
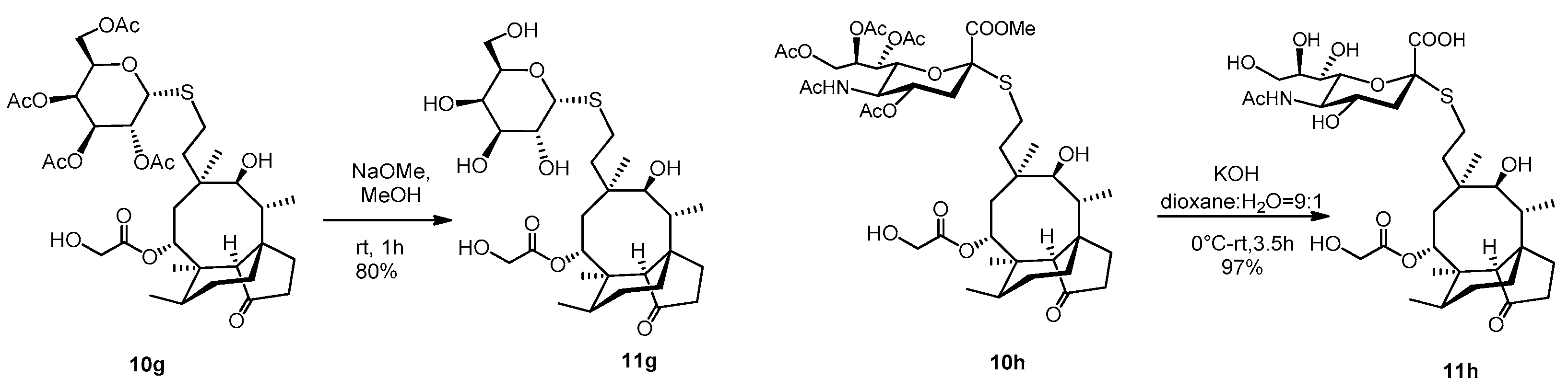
2.3.19. Compound 11g
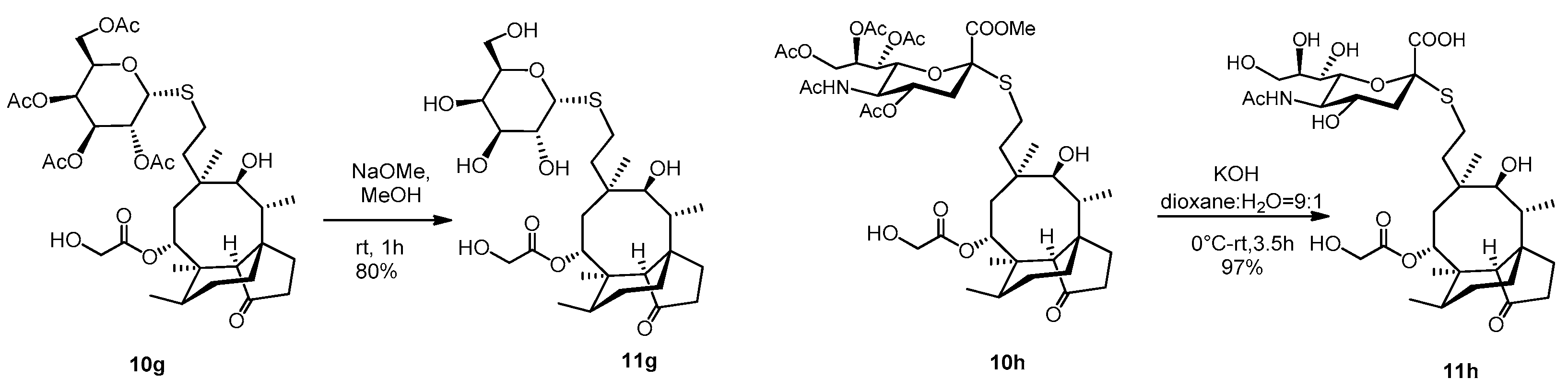
A catalytic amount of NaOMe (pH ~ 9) was added to a stirred solution of compound 10g (65 mg, 0.09 mmol) in dry MeOH (5 mL) and stirred for one hour at room temperature. The reaction mixture was neutralized with Amberlite IR-120 H+ ion-exchange resin, filtered and evaporated. Then, the crude product was purified by flash column chromatography (CH2Cl2/MeOH 8/2) to result in compound 11g as white amorphous solid (42 mg, 80%). Rf: 0.50 (CH2Cl2/MeOH 8/2), [α]24D= +26.4 (c 0.14, MeOH). 1H NMR (400 MHz, Methanol-d4) δ 5.74 (d, J = 8.3 Hz, 1H), 5.51 (s, 1H), 4.53 − 4.45 (m, 1H), 4.05 (m, 2H), 3.89 (d, J = 0.9 Hz, 1H), 3.85 − 3.76 (m, 1H), 3.75 − 3.66 (m, 2H), 3.56 − 3.51 (m, 2H), 3.46 (d, J = 5.9 Hz, 1H), 3.37 (s, 1H), 2.63 (tq, J = 17.8, 6.4, 5.4 Hz, 2H), 2.42 − 2.05 (m, 5H), 2.02 − 1.78 (m, 3H), 1.76 − 1.53 (m, 3H), 1.52 − 1.28 (m, 7H), 1.22 − 1.11 (m, 1H), 1.05 (s, 3H), 0.96 (d, J = 7.0 Hz, 3H), 0.74 (d, J = 6.5 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 211.6, 173.5, 88.2, 80.4, 76.5, 76.2, 71.7, 70.7, 70.4, 63.1, 61.9, 59.3, 46.8, 43.1, 42.8, 42.3, 38.0, 36.1, 35.3, 31.7, 31.4, 28.1, 27.4, 26.9, 25.7, 17.0, 15.3, 11.7. HRMS (ESI): m/z calcd. for C28H46NaO10S [M + Na]+ 597.2709, found: 597.2704.
2.3.20. Compound 11h
To a stirred solution of compound 10h (65 mg, 0.073 mmol) in dioxane:water = 9:1 (2 mL) 0.2 M aqueous solution of KOH (1.83 mL, 0.36 mmol, 5 equiv.) was added, and the reaction mixture was stirred for half an hour at 0 °C, then for 3 h at room temperature. The reaction mixture was neutralized with Amberlite IR-120 H+ ion-exchange resin, filtered and evaporated to obtain compound 11h as yellow amorphous solid (50 mg, 97%). Rf: 0.56 (CH3CN/H2O 9/1), [α]24D + 28.9 (c 0.09, MeOH). 1H NMR (400 MHz, DMSO-d6) δ 5.55 (d, J = 7.6 Hz, 1H, H-14), 4.09 (d, J = 5.7 Hz, 2H, H-22ab), 3.75 (dd, J = 19.5, 10.4 Hz, 4H), 3.63 − 3.56 (m, 2H), 3.51 (d, J = 9.7 Hz, 2H), 3.43 (d, J = 4.9 Hz, 1H, H-11), 3.27 (d, J = 1.2 Hz, 14H), 2.73 (dd, J = 12.4, 4.0 Hz, 1H), 2.65 − 2.56 (m, 1H), 2.47 (dd, J = 11.5, 3.7 Hz, 1H, H-19*a), 2.41 (s, 1H, H-19*b), 2.29 − 2.10 (m, 4H), 1.95 (s, 1H, H-20*a), 1.85 (s, 5H, H-20*b), 1.75 (d, J = 14.4 Hz, 1H, H-1a), 1.66 (d, J = 11.9 Hz, 2H), 1.59 − 1.50 (m, 1H), 1.49 − 1.39 (m, 4H, H-1b), 1.30 (s, 5H), 1.21 (dd, J = 29.0, 9.0 Hz, 6H), 1.09 − 0.97 (m, 2H), 0.91 (s, 3H, H18abc), 0.83 (d, J = 6.2 Hz, 3H, H-17abc), 0.60 (d, J = 5.3 Hz, 3H, H-16abc). * Interchangeable signals. 13C NMR (101 MHz, DMSO-d6) δ 174.3, 172.6, 171.1 (3C, C-1′, C-21, AcCO), 85.1 (1C, C-2′), 75.2, 73.9, 71.1, 69.2, 67.4, 66.9 53.6, 51.9 (8C, skeletal carbons, C-11, C-14), 63.4, 60.4, (2C, C-22, C-9′), 57.5 (1C, C-4), 45.0, 41.3, 40.8 (4C, C-12, C-13, C-5, C-9), 36.4 (1C, C-6), 34.0, 29.0, 27.4 (3C, C-19, C-20, C-8), 26.9 (1C, C-18), 25.5 (1C, C-1), 24.5 (1C, C-7), 22.3 (1C, AcCH3), 16.1 (1C, C-16), 14.6 (1C, C-15), 11.6 (1C, C-17). HRMS (MALDI): m/z calcd. for C33H53NNaO13S [M + Na]+ 726.3135, found: 726.3159.
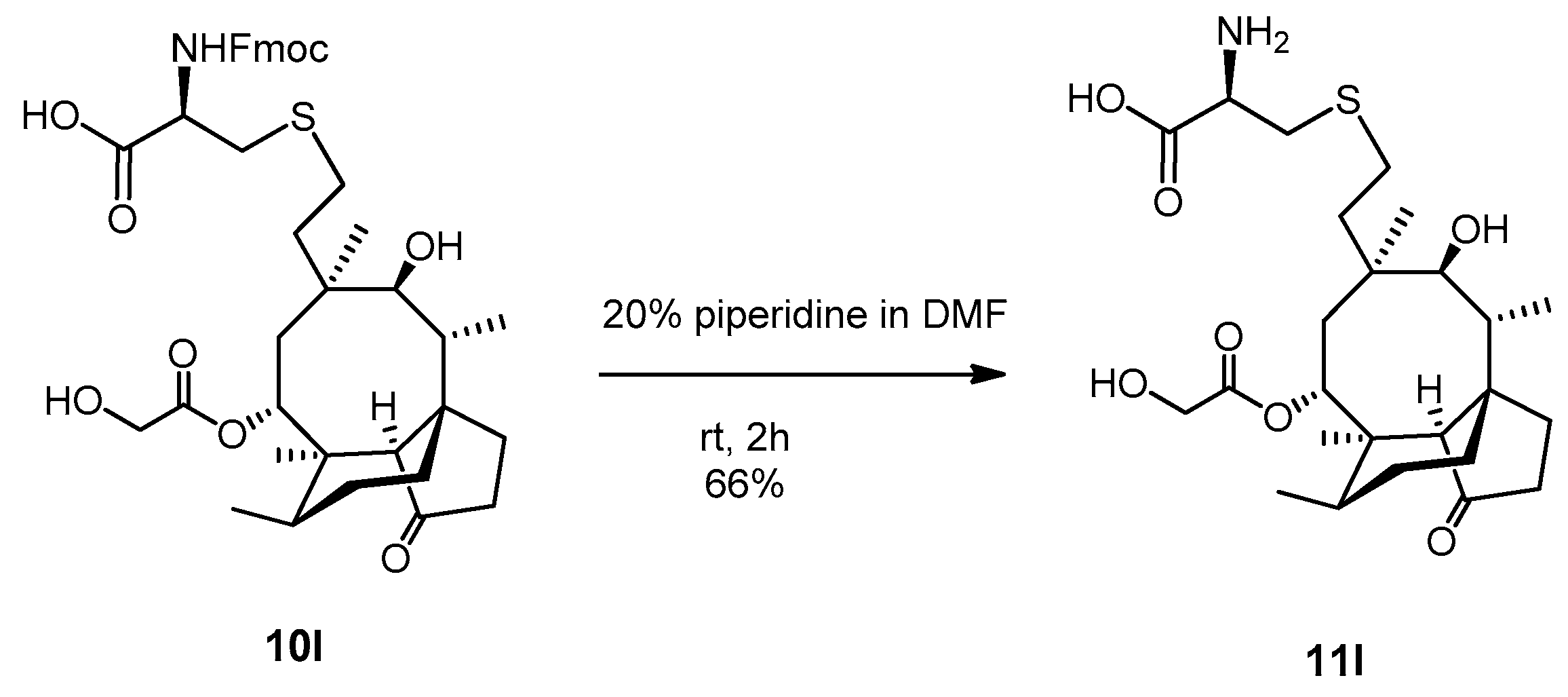
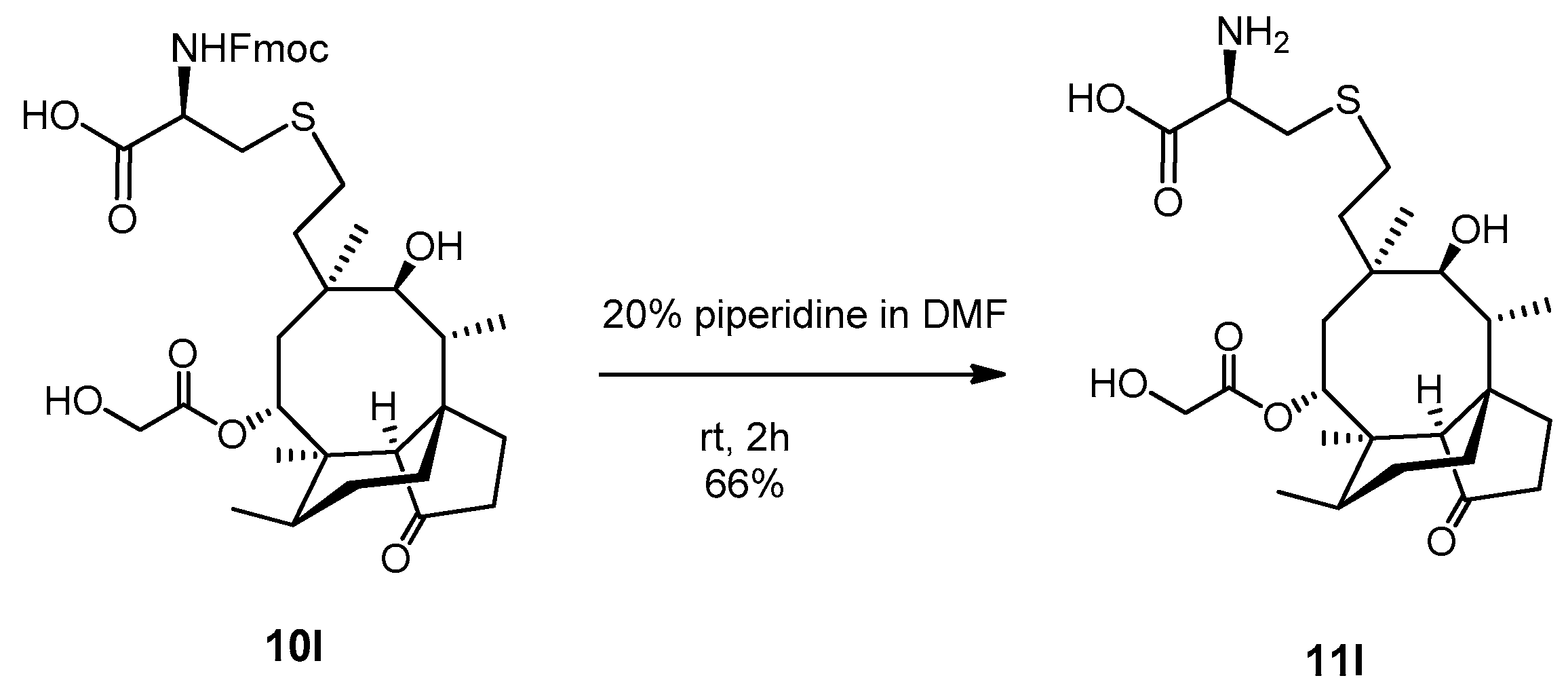
2.3.21. Compound 11l
Compound 10l (100 mg, 0.138 mmol) was dissolved in 20% piperidine solution in N,N-dimethylformamide (5 mL) and stirred for two hours at room temperature. The reaction mixture was evaporated, and the crude product was purified by flash column chromatography (CH3CN/H2O 9/1) to result in compound 11l as yellow powder (46 mg, 66%). Rf: 0.10 (CH3CN/H2O 9/1), [α]24D +41.7 (c 0.06, MeOH), m.p. 197–199 °C. 1H NMR (400 MHz, Methanol-d4) δ 5.69 (t, J = 8.6 Hz, 1H), 4.22 (dd, J = 25.1, 17.2 Hz, 1H), 4.04 (dd, J = 17.2, 13.4 Hz, 1H), 3.79 (dt, J = 10.9, 5.2 Hz, 1H), 3.47 (t, J = 5.0 Hz, 1H), 3.37 (s, 2H), 3.33 (p, J = 1.7 Hz, 1H), 3.22 (dd, J = 15.2, 4.8 Hz, 1H), 3.19 − 3.07 (m, 1H), 2.52 (tq, J = 10.8, 5.2, 4.8 Hz, 2H), 2.41 − 2.31 (m, 2H), 2.31 − 2.24 (m, 1H), 2.17 (q, J = 9.6 Hz, 1H), 2.13 − 1.98 (m, 1H), 1.98 − 1.48 (m, 5H), 1.45 (d, J = 3.7 Hz, 5H), 1.39 − 1.27 (m, 2H), 1.17 (td, J = 13.8, 4.3 Hz, 1H), 1.03 (d, J = 5.0 Hz, 3H), 0.95 (dd, J = 7.1, 1.8 Hz, 3H), 0.74 (dd, J = 7.0, 2.0 Hz, 3H). 13C NMR (101 MHz, Methanol-d4) δ 217.0, 190.0, 173.3, 74.9, 69.3, 60.5, 57.8, 53.8, 45.4, 41.6, 41.0, 36.6, 34.6, 33.9, 32.8, 30.0, 28.4, 28.1, 27.2, 26.8, 25.8, 24.2, 15.8, 13.9, 10.3. HRMS (ESI): m/z calcd. for C25H41NNaO7S [M + Na]+ 522.2501, found: 522.2496.
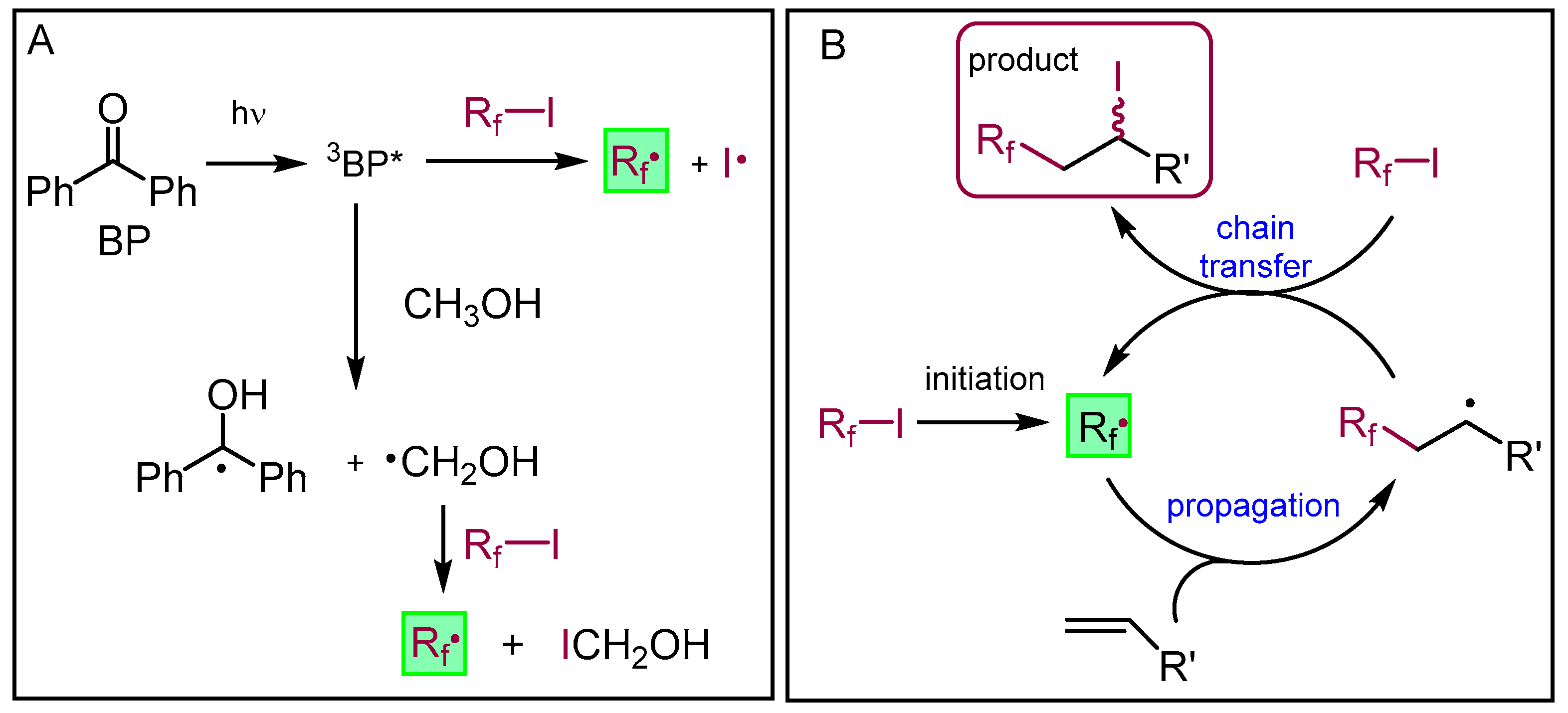
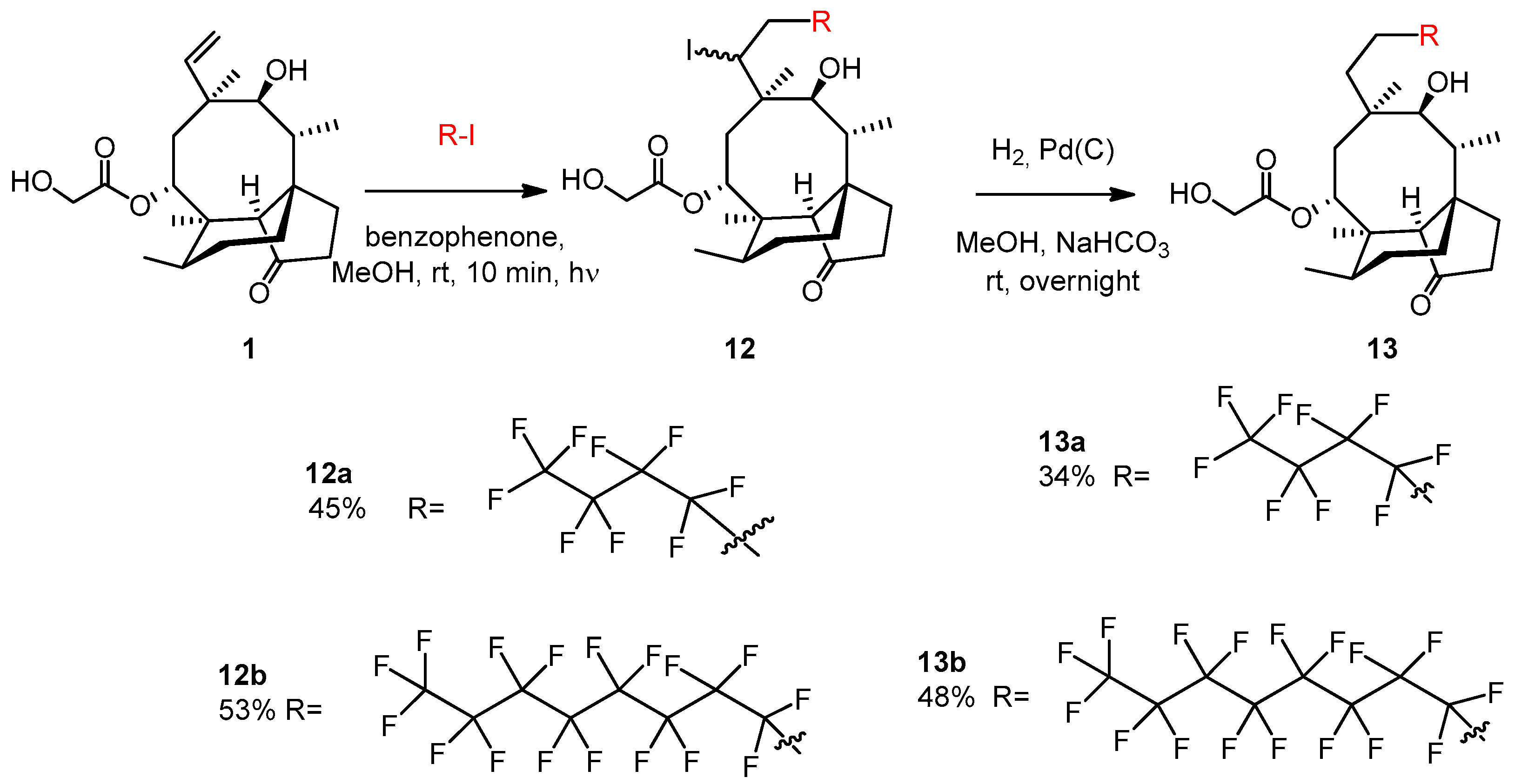
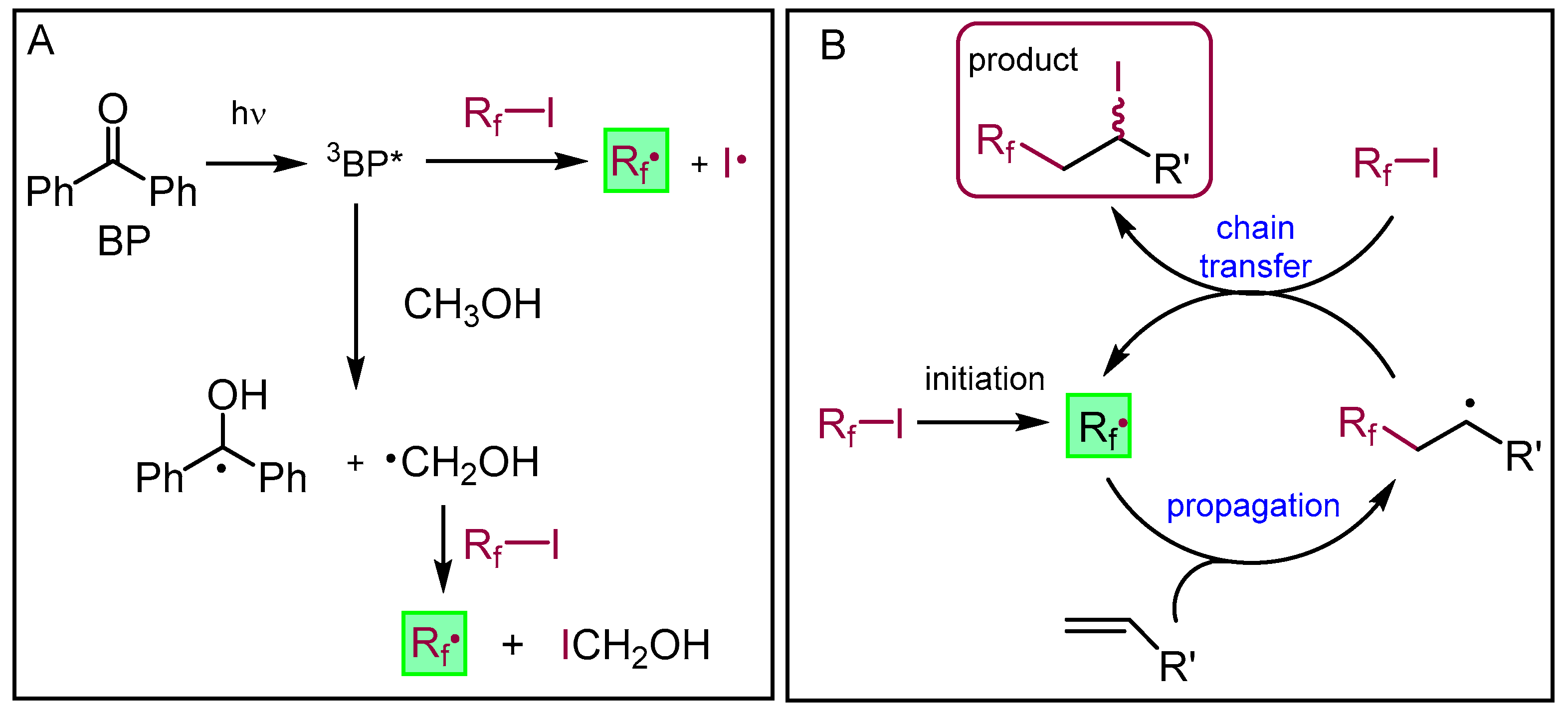
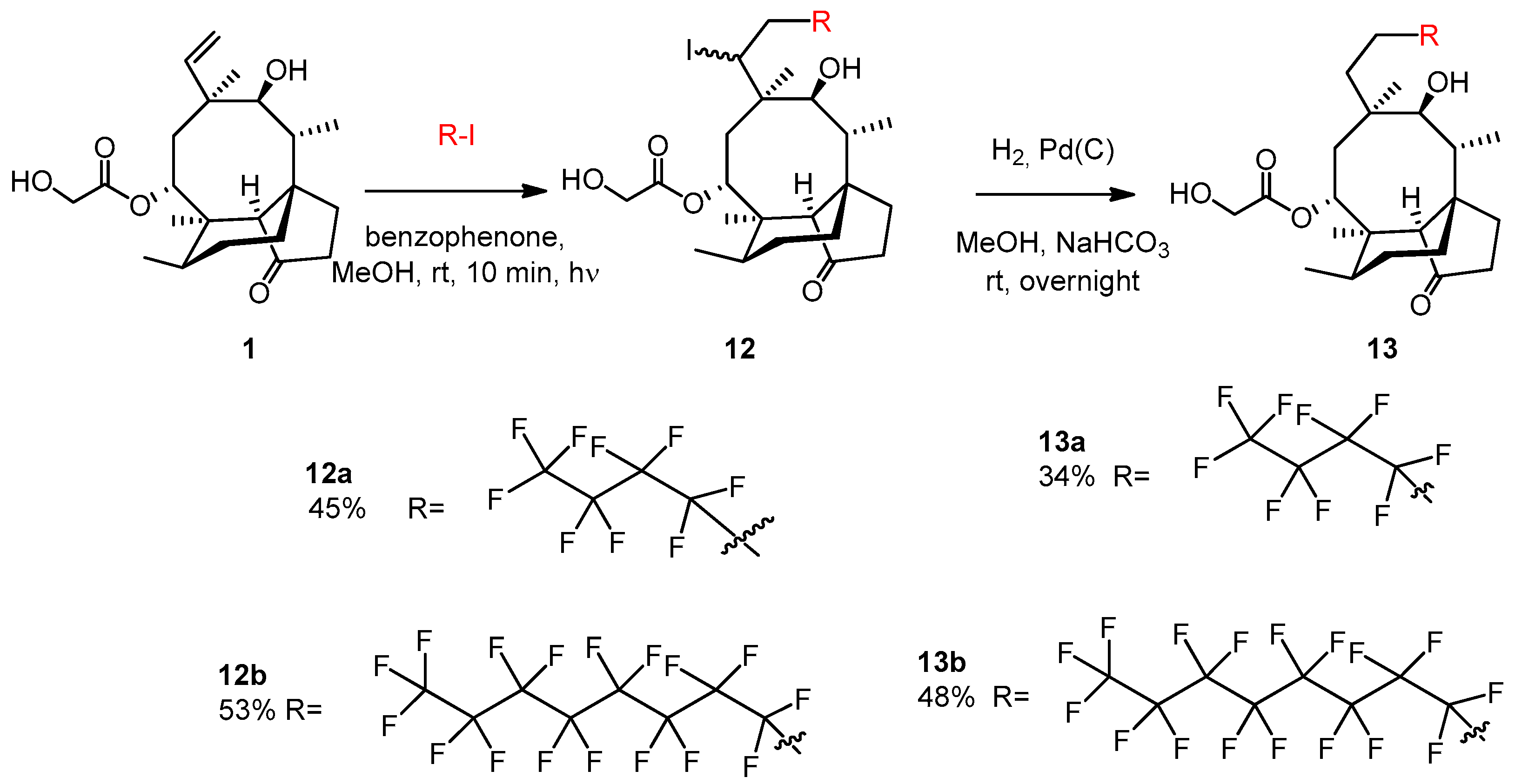
2.3.22. Compound 13a
Nonafluoro-1-iodobutane (103 μL, 0.6 mmol, 1.2 equiv.) and benzophenone (5 mg, 0.023 mmol) were added to the solution of pleuromutilin 1 (189 mg, 0.5 mmol) in methanol (5 mL). Argon gas was bubbled through the solution, and then irradiation occurred with UV light for 10 min. The solvent was evaporated, and the product was purified by flash column chromatography (CH2Cl2/acetone 95:5) to yield 12a (163 mg, 45%) as a colorless liquid. Rf = 0.51 (CH2Cl2/MeOH 95:5). To the solution of 12a (130 mg, 0.18 mmol), methanol (5 mL), 10% palladium on activated charcoal (25 mg) and NaHCO3 (53 mg, 2.5 equiv.) were added. The reaction mixture was stirred overnight under H2 atmosphere, then filtered through a pad of Celite, and the solvent was evaporated. The residue was dissolved in dichloromethane (50 mL) and the solution was washed with distilled water (10 mL) two times, and dried over anhydrous Na2SO4. The solvent was then evaporated in vacuum. The product was purified by flash column chromatography (CH2Cl2/acetone 97:3 then 95:5) to yield 13a (36 mg, 34%) as a colorless liquid. Rf = 0.25 (CH2Cl2/acetone 97:3), [α]24D +11.8 (c 0.22, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 5.63 (d, J = 8.2 Hz, 1H, H-14), 4.77 (dd, J = 9.3, 6.5 Hz, 1H), 4.20 − 4.12 (m, 1H, H-22a), 4.06 (dd, J = 18.3, 5.8 Hz, 1H, H-22b), 3.62 (s, 1H, H-11), 3.21 (d, J = 6.0 Hz, 1H), 2.62 (t, J = 5.3 Hz, 1H), 2.44 − 2.37 (m, 1H), 2.24 (dd, J = 17.1, 8.2 Hz, 3H), 2.16 (s, 1H, H-2b), 1.92 (s, 3H), 1.82 (d, J = 16.2 Hz, 2H), 1.77 − 1.70 (m, 2H), 1.64 (dd, J = 16.2, 8.3 Hz, 1H), 1.51 (t, J = 8.6 Hz, 2H), 1.44 (s, 3H, H-15abc), 1.18 (d, J = 6.8 Hz, 1H), 1.16 − 1.09 (m, 1H), 1.05 (s, 3H, H-18abc), 0.94 (d, J = 7.0 Hz, 3H, H-17abc), 0.69 (t, J = 6.0 Hz, 3H, H-16abc). 13C NMR (101 MHz, Chloroform-d) δ 174.9 (1C, C-21), 74.7, 71.1 (2C, C-11, C-14), 63.3, 61.5 (1C, C-22), 58.6, 46.4, 45.8, 42.0, 40.8 (4C, C-5, C-12, C-13, C-9), 36.3, 34.9 (2C, C-6, C-10), 34.4, 34.2 (2C, C-2, C-23), 29.8, 27.1 (2C, C-7, C-19), 24.7, 20.5, 17.0 (1C, C-16), 14.6 (1C, C-15), 10.5 (1C, C-17). HRMS (ESI): m/z calcd. for C26H35F9KO5 [M + K]+ 637.2188, found: 637.2182.
2.3.23. Compound 13b
Heptadecafluoro-1-iodooctane (0.160 mL, 327 mg, 0.6 mmol, 1.2 equiv.) and benzophenone (5 mg, 0.023 mmol) were added to the solution of pleuromutilin 1 (0.189 g, 0.5 mmol) in methanol (5 mL). Argon gas was bubbled through the solution and then irradiation occurred with UV light for 10 min. The solvent was evaporated, and the product was purified by flash column chromatography (CH2Cl2/acetone 97:3) to yield 12b (247 mg, 53%) as a colorless liquid. Rf = 0.55 (CH2Cl2/acetone 95:5). To the solution of 12b (0.120 g, 0.25 mmol) in methanol (5 mL) 10% palladium on activated charcoal (25 mg) and NaHCO3 (53 mg, 2.5 equiv.) were added. The reaction mixture was stirred overnight under H2 atmosphere, then filtered through a pad of Celite, and the solvent was evaporated. The residue was dissolved in dichloromethane (50 mL) and the solution was washed with distilled water (10 mL) two times, and dried with anhydrous Na2SO4. Then, the solvent was evaporated in vacuum. The product was purified by flash column chromatography (CH2Cl2/acetone 97:3) to yield 13b (49 mg, 48%) as a colorless liquid. Rf=0.22 (CH2Cl2/acetone 95:5), [α]24D +4.71 (c 0.34, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 5.64 (d, J = 8.2 Hz, 1H), 4.78 (dd, J = 9.0, 4.9 Hz, 1H), 4.20 − 4.00 (m, 2H), 3.62 (d, J = 4.0 Hz, 1H), 3.19 (s, 1H), 2.59 (s, 7H), 2.47 − 2.36 (m, 1H), 2.24 (dd, J = 17.1, 8.4 Hz, 3H), 1.88 (s, 1H), 1.82 (d, J = 16.2 Hz, 2H), 1.78 − 1.59 (m, 4H), 1.51 (dd, J = 11.5, 5.9 Hz, 2H), 1.44 (s, 3H, H-15abc), 1.17 (s, 1H), 1.16 − 1.09 (m, 1H), 1.05 (s, 3H, H-18abc), 0.94 (d, J = 7.0 Hz, 3H, H-17abc), 0.70 (d, J = 7.0 Hz, 3H, H-16abc). 13C NMR (101 MHz, Chloroform-d) δ 174.9 (1C, C-21), 74.8, 71.1 (2C, C-11, C-14), 63.4, 61.6 (1C, C-22), 58.6 (1C, C-4), 46.4, 45.9, 42.0, 40.9 (4C, C-5, C-12, C-13, C-9), 36.3, 34.4, 34.2, 29.9, 27.1, 24.7, 20.5, 16.9 (1C, C-16), 14.7 (1C, C-15), 10.5 (1C, C-17). HRMS (ESI): m/z calcd. for C30H35F17KO5 [M + K]+ 837.2060, found: 837.2054.
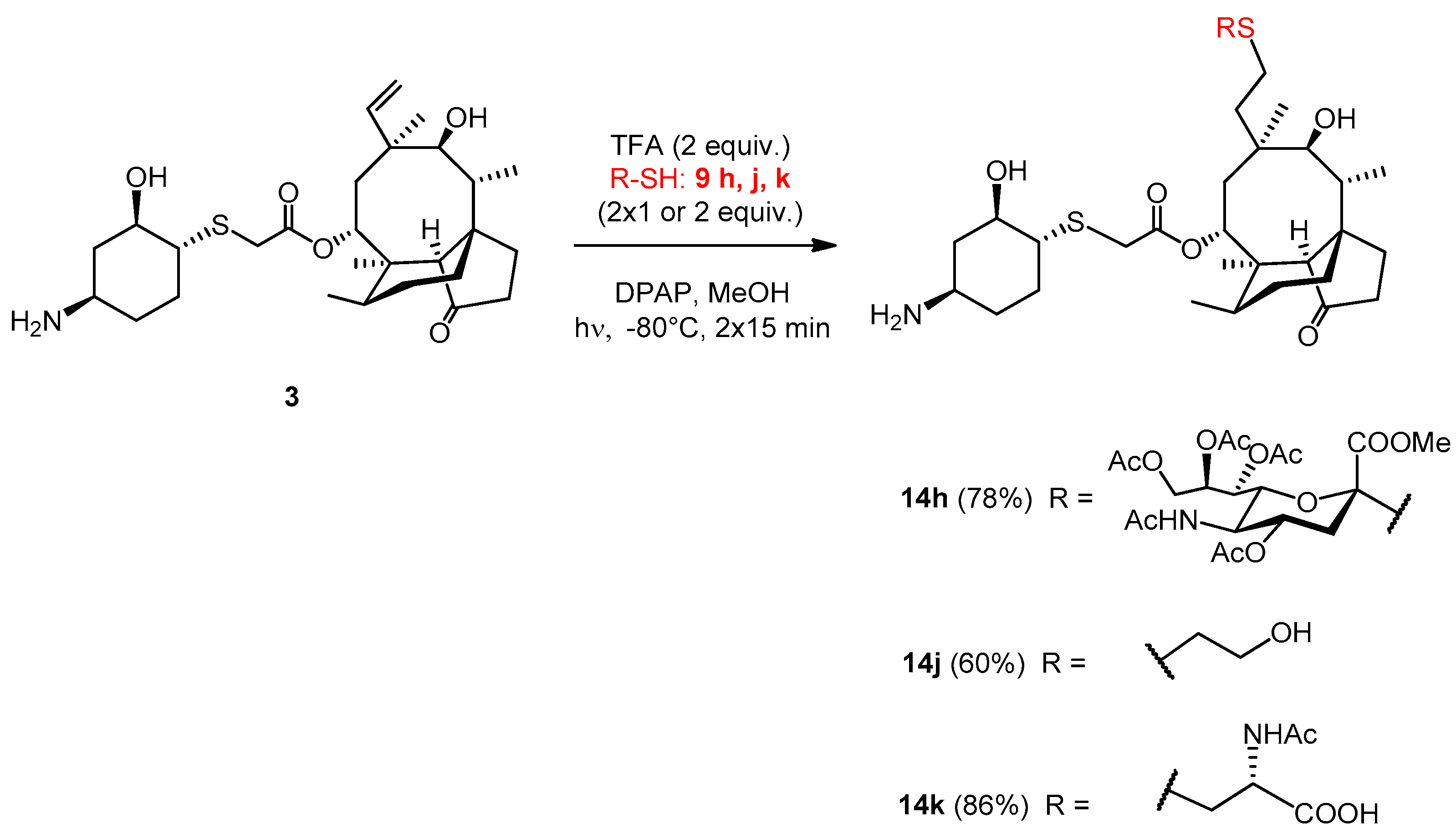
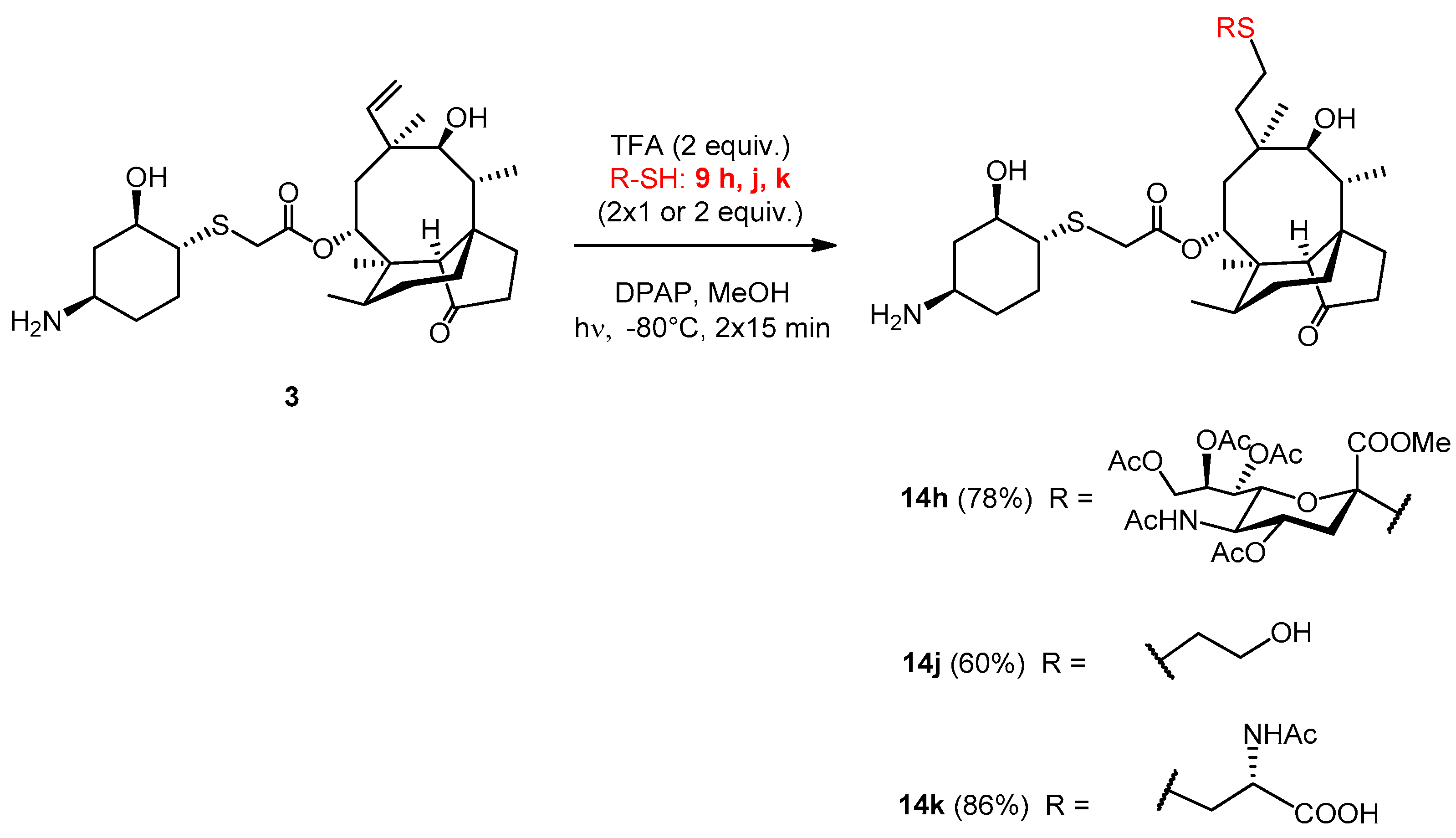
2.3.24. Compound 14h
The reaction was carried out using the general method, starting from lefamulin (51 mg, 0.1 mmol), trifluoroacetic acid (15 µL, 0.2 mmol, 2 equiv.) and thiol 9h (203 mg, 0.4 mmol, 2 × 2 equiv.) in methanol at −80 °C. It was irradiated two times. The reaction mixture was co-evaporated with toluene three times and the crude product was purified by flash column chromatography (CH2Cl2/MeOH 85/15) to result in compound 14h as white amorphous solid (79 mg, 78%). Rf: 0.56 (CH2Cl2/MeOH 7/3), [α]24D +24.4 (c 0.09, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 6.5 Hz, 1H, NH), 5.42 (d, J = 6.8 Hz, 1H, H-14), 5.20 (d, J = 2.3 Hz, 1H, H-8″), 5.16 (d, J = 7.1 Hz, 1H), 4.69 (s, 1H, H-4″), 4.65 (s, 1H, OH), 4.21 (d, J = 12.1 Hz, 1H, H-9″a), 4.04 (dd, J = 11.6, 4.5 Hz, 1H, H-9″b), 3.84 (s, 1H, H-5″), 3.80 (d, J = 1.6 Hz, 1H), 3.54 (d, J = 14.5 Hz, 1H), 3.42 (s, 5H), 3.33 (d, J = 12.8 Hz, 3H, H-11, H-2′), 3.02 (s, 1H, H-4′), 2.64 (d, J = 8.7 Hz, 1H, H-3″a), 2.56 (s, 1H, H-1′), 2.51 (d, J = 1.4 Hz, 2H), 2.35 (s, 1H, H-4), 2.16 (d, J = 10.9 Hz, 2H, H-5′a, H-10), 2.07 (d, J = 1.7 Hz, 3H, AcCH3), 2.01 (s, 3H, AcCH3), 1.96 (s, 3H, AcCH3), 1.92 (d, J = 1.7 Hz, 3H, AcCH3), 1.83 (d, J = 16.5 Hz, 1H, H-13a), 1.75 (d, J = 12.3 Hz, 1H, H-3″b), 1.65 (s, 5H, AcCH3, H-7a, H-8a), 1.49 (s, 2H, H-6), 1.37 (s, 3H, H-15abc), 1.23 (s, 6H, H-7b), 1.13 (d, J = 16.1 Hz, 2H, H-13b), 1.08 − 0.95 (m, 2H, H-8b), 0.90 (s, 3H, H-18abc), 0.80 (d, J = 5.4 Hz, 3H, H-17abc), 0.64 (d, J = 5.7 Hz, 3H, H-16abc). 13C NMR (101 MHz, DMSO-d6) δ 217.1 (1C, C-3), 170.1, 169.7, 169.4, 169.3, 169.3, 169.1, 168.4 (7C, 7×CO), 83.1 (1C, C-2″), 73.7, 73.3, 72.3, 67.3 (4C, C-11, C-2′, C-7″, C-6″), 69.6 (1C, C-4″), 68.9 (1C, C-14), 68.4 (1C, C-8″), 61.8 (1C, C-9″), 57.2 (1C, C-4), 54.9, 53.0 (1C, COOCH3), 49.7 (1C, C-1′), 47.8 (1C, C-5″), 47.2 (1C, C-4′), 44.9 (1C, C-9), 41.2, 40.6 (3C, C-13, C-12, C-5), 36.4 (1C, C-6), 34.4 (1C, C-10), 33.9 (1C, C-5′), 33.7 (1C, C-22), 30.1 (1C, C-8), 29.8, 29.6, 28.2, 26.8, 23.7 (6C, C-19, C-20, C-3′, C-6′, C-1, C-2), 26.2 (1C, C-18), 24.3 (1C, C-7), 22.6 (1C, NAcCH3), 20.9, 20.7, 20.6, 20.6 (4C, 4xOAcCH3), 16.8 (1C, C-16), 14.6 (1C, C-15), 11.5 (1C, C-17). HRMS (ESI): m/z calcd. for C48H75N2O17S2 [M + H]+ 1015.4507, found: 1015.4502.
2.3.25. Compound 14j
The reaction was carried out using the general method, starting from lefamulin (51 mg, 0.1 mmol), trifluoroacetic acid (15.3 µL, 0.2 mmol, 2 equiv.) and thiol 9j (31 mg, 28 µL, 0.4 mmol, 2 × 2 equiv.) in methanol at −80 °C. It was irradiated two times. The reaction mixture was co-evaporated with toluene three times and the crude product was purified by flash column chromatography (CH3CN/H2O 9/1) to result in compound 14j as white-yellow amorphous solid (35 mg, 60%). Rf: 0.19 (CH3CN/H2O 9/1), [α]24D +50.0 (c 0.07, H2O). 1H NMR (400 MHz, DMSO-d6) δ 5.51 (d, J = 7.6 Hz, 1H, H-14), 4.61 (s, 1H, OH), 3.63 − 3.46 (m, 3H, H-24ab), 3.40 − 3.29 (m, 3H, H-11, H-2′), 2.98 (s, 1H, H-4′), 2.57 (t, J = 7.0 Hz, 2H, H-23ab), 2.51 (s, 2H), 2.37 (t, J = 13.0 Hz, 3H, H-4), 2.23 − 2.02 (m, 3H, H-10, H-3′a), 1.96 (s, 3H), 1.91 − 1.78 (m, 2H, H-13a, H-5′a), 1.67 (t, J = 16.9 Hz, 3H, H-7a, H-8a), 1.49 (d, J = 5.9 Hz, 1H, H-6), 1.42 (d, J = 15.5 Hz, 1H), 1.37 (s, 3H, H-15abc), 1.33 − 1.18 (m, 7H, H-7b, H-3′a, H-5′b, H-6′ab), 1.12 (d, J = 16.0 Hz, 1H, H-13b), 1.07 − 0.98 (m, 1H, H-8b), 0.93 (s, 3H, H-18abc), 0.83 (d, J = 6.3 Hz, 3H, H-17abc), 0.64 (d, J = 6.2 Hz, 3H, H-16abc). 13C NMR (101 MHz, DMSO-d6) δ 217.1 (1C, C-3), 169.3 (1C, C-21), 73.5, 72.3 (2C, C-11, C-2′), 68.7 (1C, C-14), 61.0 (1C, C-24), 57.2 (1C, C-4), 49.5 (1C, C-1′), 47.3 (1C, C-4′), 45.0, 41.3, 40.6, 40.3 (4C, C-5, C-13, C-12, C-9), 36.4 (1C, C-6), 34.6 (1C, C-10), 33.9, 33.5, 30.5, 30.1, 28.3, 27.0, 26.7 (8C, C-3′, C-5′, C-6′, C-1, C-2, C-19, C-20, C-23), 26.9 (1C, C-18), 24.3 (1C, C-7), 16.5 (1C, C-16), 14.6 (1C, C-15), 11.5 (1C, C-17). HRMS (ESI): m/z calcd. for C30H52NO6S2 [M + H]+ 586.3236, found: 586.3230.
2.3.26. Compound 14k
The reaction was carried out using the general method, starting from lefamulin (50.8 mg, 0.1 mmol), trifluoroacetic acid (15.3 µL, 0.2 mmol, 2 equiv.) and thiol 9k (33 mg, 0.2 mmol, 2 × 1 equiv.) in methanol at −80 °C. It was irradiated two times. The reaction mixture was evaporated with added toluene three times and the crude product was purified by flash column chromatography (CH3CN/H2O 9/1) resulted in compound 14k as white-yellow amorphous solid (57.4 mg, 86%). Rf: 0.46 (CH3CN/H2O 85/15), [α]24D +36.0 (c 0.2, H2O). 1H NMR (400 MHz, DMSO-d6) δ 7.42 (d, J = 7.1 Hz, 1H, NH), 5.50 (d, J = 8.0 Hz, 1H, H-14), 4.57 (d, J = 4.4 Hz, 1H, OH), 4.08 (dd, J = 12.1, 5.4 Hz, 1H, H-24), 3.68 (d, J = 16.2 Hz, 2H, H-22a), 3.35 (dd, J = 21.8, 11.0 Hz, 2H, 10H, H-2′, H-11), 3.19 − 3.13 (m, 2H, H-22b), 2.95 (dt, J = 19.3, 9.7 Hz, 2H, H-23a, H-4′), 2.74 − 2.60 (m, 2H, H-23b, H-1′), 2.53 − 2.49 (m, 2H), 2.42 − 2.32 (m, 2H, H-4), 2.32 − 2.22 (m, 1H), 2.18 (dd, J = 12.1, 6.1 Hz, 2H, H-10, H-5′a), 2.03 (ddd, J = 21.3, 17.6, 5.7 Hz, 4H, H-5′b), 1.84 (s, 3H, AcCH3), 1.79 (dd, J = 13.9, 9.4 Hz, 3H, H-13a), 1.72 − 1.60 (m, 6H, H-7a, H-8a), 1.48 (dd, J = 11.9, 9.0 Hz, 1H, H-6), 1.41 (d, J = 13.3 Hz, 1H, H-6′a), 1.33 (s, 3H, H-15abc), 1.31 − 1.23 (m, 3H, H-7b), 1.18 (dd, J = 9.7, 6.4 Hz, 2H, H-13b), 1.07 − 0.97 (m, 1H, H-8b), 0.91 (s, 3H, H-18abc), 0.83 (d, J = 6.8 Hz, 3H, H-17abc), 0.60 (d, J = 6.6 Hz, 3H, H-16abc). 13C NMR (101 MHz, DMSO-d6) δ 217.3 (1C, C-3), 173.0 (1C, C-21), 169.0, 168.4 (2C, AcCO, COOH), 73.6 (2C, C-2′, C-11), 72.8, 68.5 (1C, C-14), 57.2 (1C, C-4), 53.9 (1C, C-24), 47.4, 47.2 (1C, C-4′, C-1′), 41.2 (1C, C-13), 45.0, 40.7 (3C, C-5, C-12, C-9), 36.4 (1C, C-6), 35.9 (1C, C-23), 34.5 (1C, C-10), 34.0 (1C, C-5′), 33.0 (1C, C-22), 30.1 (1C, C-8), 31.1, 29.2, 28.2, 27.7 (4C, C-19, C-20, C-3′, C-6′), 26.8 (1C, C-18), 24.3 (1C, C-7), 22.9 (1C, AcCH3), 16.4 (1C, C-16), 14.6 (1C, C-15), 11.5 (1C, C-17). HRMS (MALDI): m/z calcd. for C33H54N2NaO8S2 [M + Na]+ 693.3219, found: 693.3219.
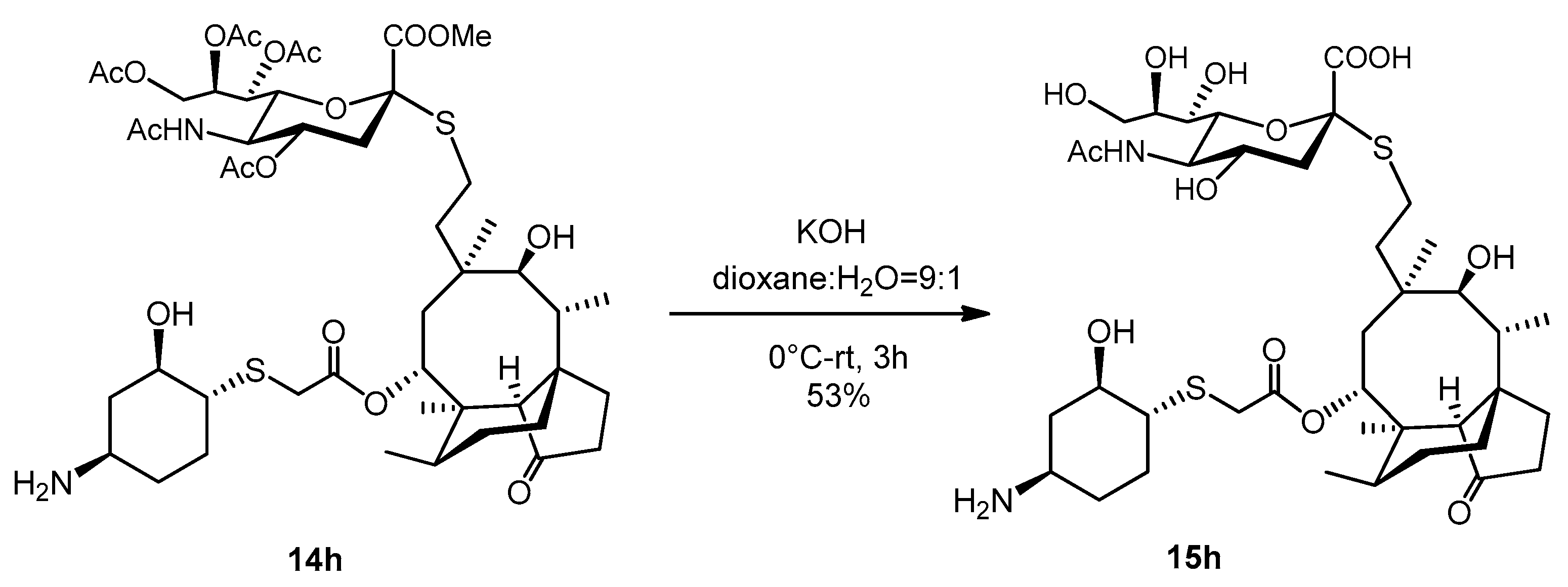
2.3.27. Compound 15h
To a stirred solution of compound 14h (58 mg, 0.06 mmol) in dioxane: water = 9:1 (2 mL) and 1.5mL of 0.2 M aqueous solution of KOH (1.5 mL, 0.3 mmol, 5 equiv.) was added (pH = 12). The reaction mixture was stirred for half an hour at 0 °C, then stirred for 3 h at room temperature. The reaction mixture was neutralized with Amberlite IR-120 H+ ion-exchange resin, filtered, evaporated, and purified by flash column chromatography (CH3CN/H2O 9/1) to obtain compound 15h as white amorphous solid (25 mg, 53%). Rf: 0.4 (CH3CN/H2O 8/2), [α]24D -3.33 (c 0.06, DMSO). 1H NMR (400 MHz, DMSO-d6) δ 8.05 (d, J = 7.9 Hz, 1H, NH), 5.49 (d, J = 7.8 Hz, 1H, H-14), 5.17 (s, 1H, H-8″), 4.87 (s, 1H), 4.69 (s, 1H, H-4″), 4.48 (d, J = 5.5 Hz, 1H), 4.08 (s, 1H), 3.60 (t, J = 14.9 Hz, 4H, H-5″, H-9″a, H-11, H-2′), 3.26 (s, 1H, H-9″b), 3.16 (s, 2H), 2.98 (s, 1H, H-4′), 2.89 (s, 1H), 2.74 (s, H), 2.69 (dd, J = 8.6, 3.6 Hz, 1H, H-22a), 2.63 (d, J = 12.8 Hz, 1H, H-1′), 2.56 (d, J = 6.7 Hz, 1H, H-22b), 2.35 (s, 1H, H-4), 2.21 − 2.08 (m, 3H, H-5′a, H-10), 2.03 (d, J = 9.2 Hz, 3H, H-5′b), 1.89 (s, 3H), 1.77 (t, J = 16.3 Hz, 4H, H-13a), 1.63 (dd, J = 21.1, 9.8 Hz, 3H, H-7a, H-8a), 1.48 (dd, J = 21.3, 10.7 Hz, 5H, H-6), 1.33 (s, 3H), 1.24 (s, 3H, H-7b), 1.19 (s, 3H, H-13b), 1.03 (dd, J = 16.5, 4.5 Hz, 2H, H-8b), 0.94 (s, 3H, H-18abc), 0.83 (d, J = 6.5 Hz, 3H, H-17abc), 0.61 (d, J = 6.3 Hz, 3H, H-16abc). 13C NMR (101 MHz, DMSO-d6) δ 217.2 (1C, C-3), 172.4, 172.2, 168.5 (3C, AcCO, C-1″, C-21), 84.8 (1C, C-2″), 75.2, 73.5, 72.0, 71.2 (3C, C-11, C-2′), 68.8, 68.6 (2C, C-8″, C-14), 67.7, 63.2 (1C, C-9″), 57.2 (1C, C-4), 54.9, 52.8, 49.8 (1C, C-1′), 47.3, 47.1 (1C, C-4′), 44.9 (1C, C-9), 42.3, 41.2, 40.9 (3C, C-12, C-13, C-5), 36.4 (1C, C-6), 35.8, 34.4 (1C, C-10), 34.0 (1C, C-5′), 33.0 (1C, C-22), 31.1, 28.9, 30.1 (1C, C-8), 26.8 (1C, C-18), 24.4 (1C, C-7), 24.2 (1C, C-22), 22.5 (1C, AcCH3), 16.4 (1C, C-, 6), 14.6 (1C, C-15), 11.4 (1C, C-17). HRMS (ESI): m/z calcd. for C39H64N2NaO13S2 [M + Na]+ 855.3748, found: 855.3746.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}