Delivery of Protein Kinase A by CRISPRMAX and Its Effects on Breast Cancer Stem-Like Properties

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Enzyme Delivery by Lipofectamine CRISPRMAX Reagent
2.3. Characterization of PKAs- and BSAs-Loaded CRISPRMAX Complexes
2.4. Encapsulation Efficiency
2.5. Release Efficiency
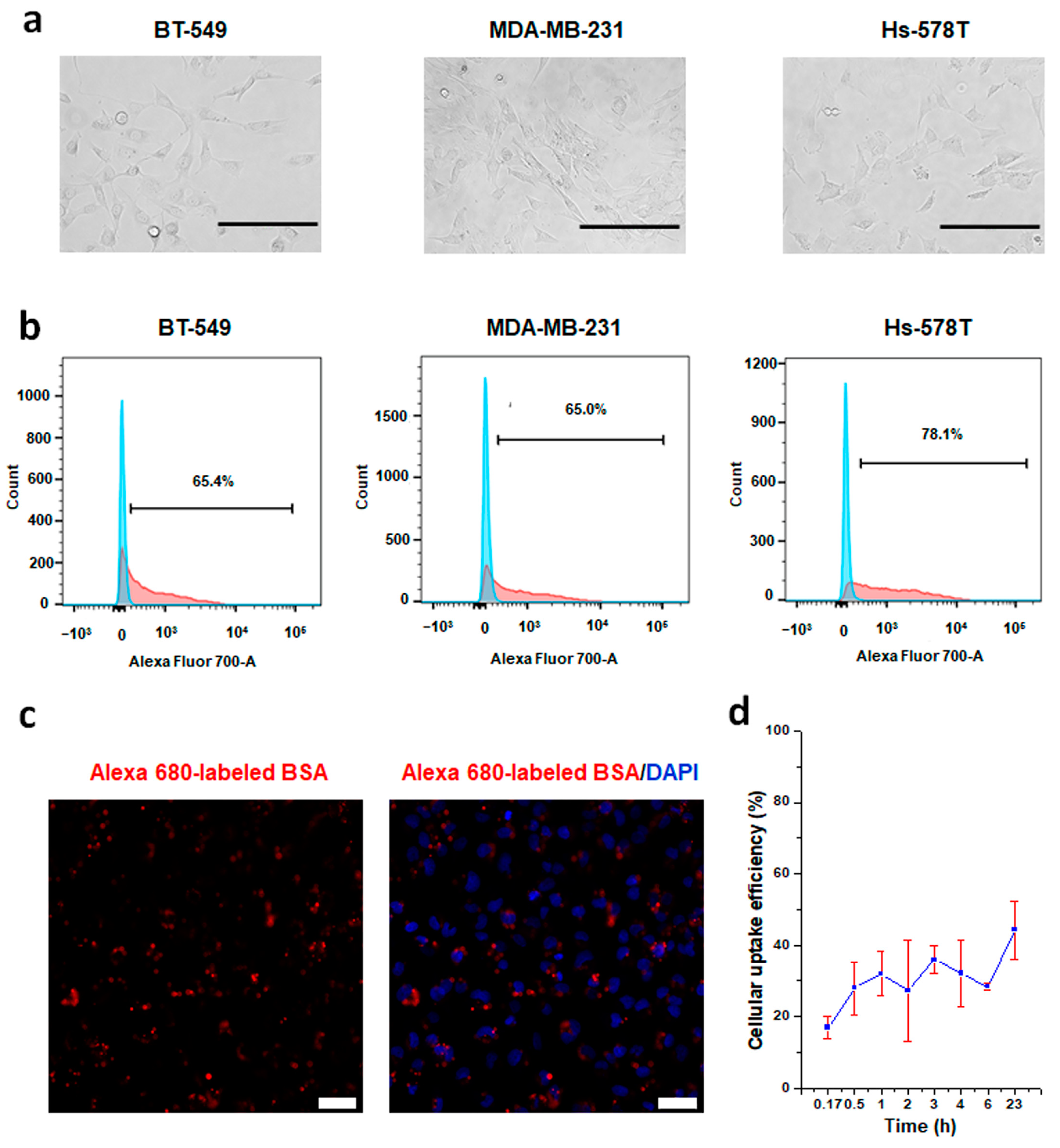
2.6. Cellular Uptake Efficiency Analysis of CRISPRMAX-BSA-Alexa Fluor 680
2.7. Immunofluorescence Analysis by Confocal Laser Scanning Microcopy
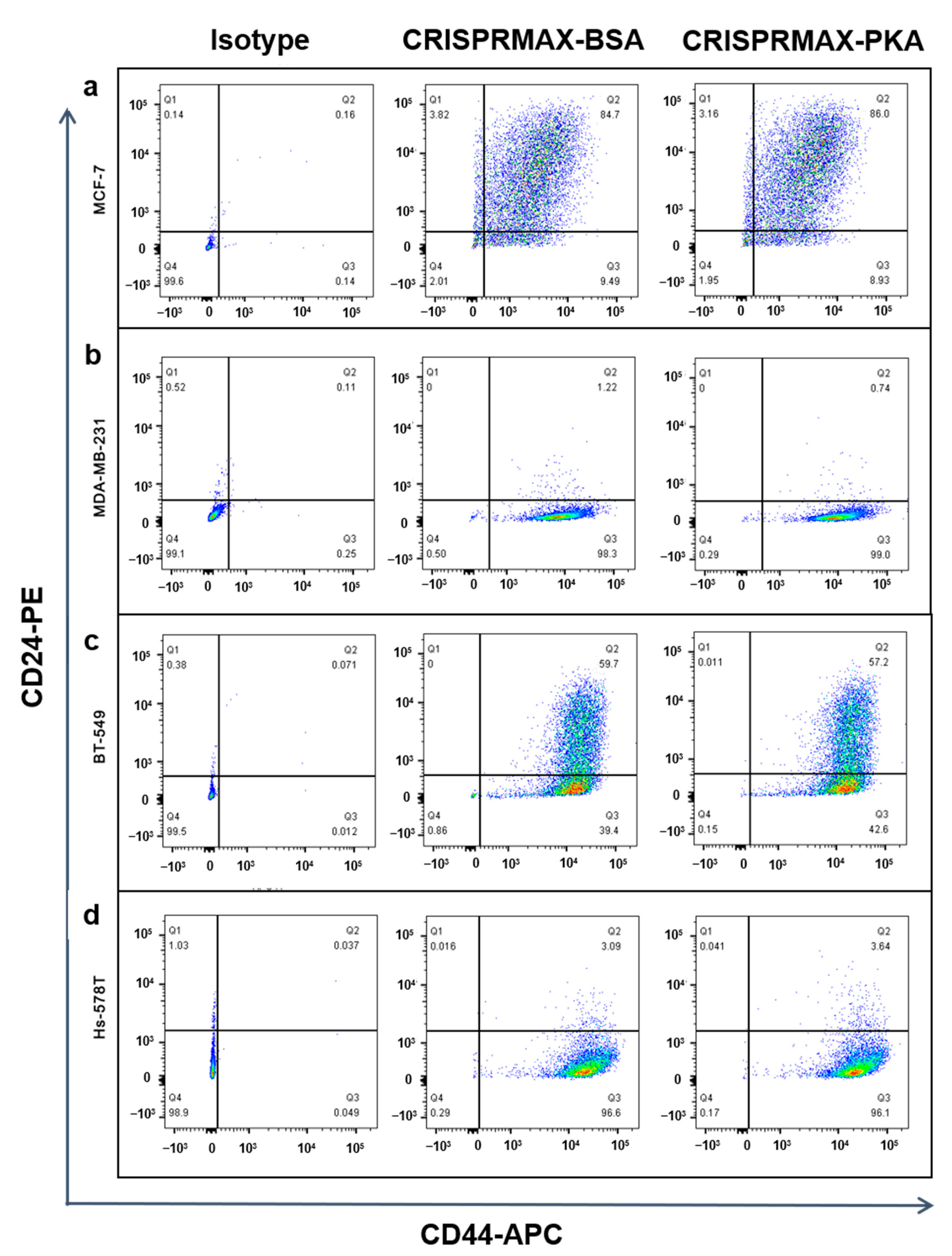
2.8. Flow Cytometry
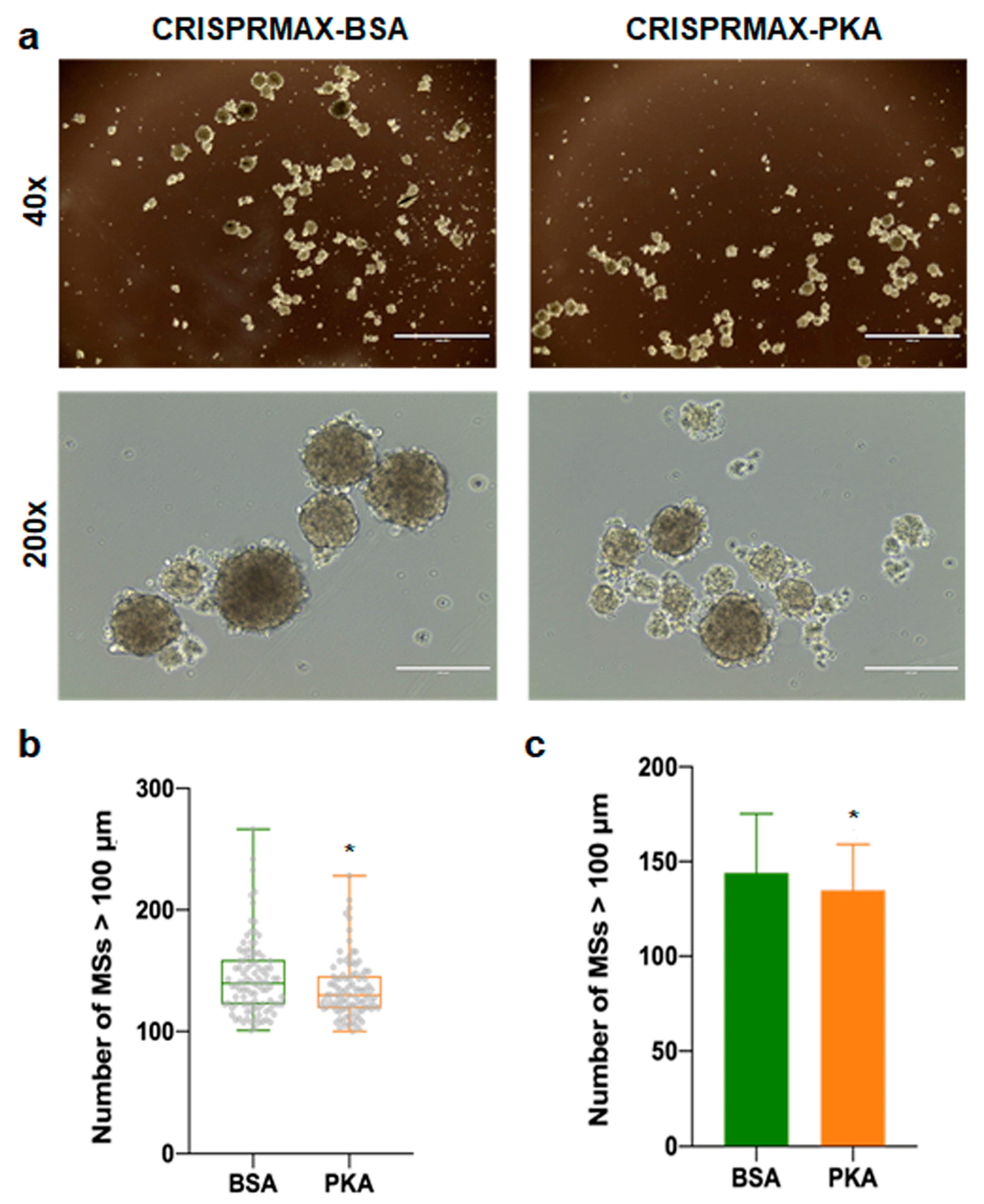
2.9. Mammosphere Formation Assay
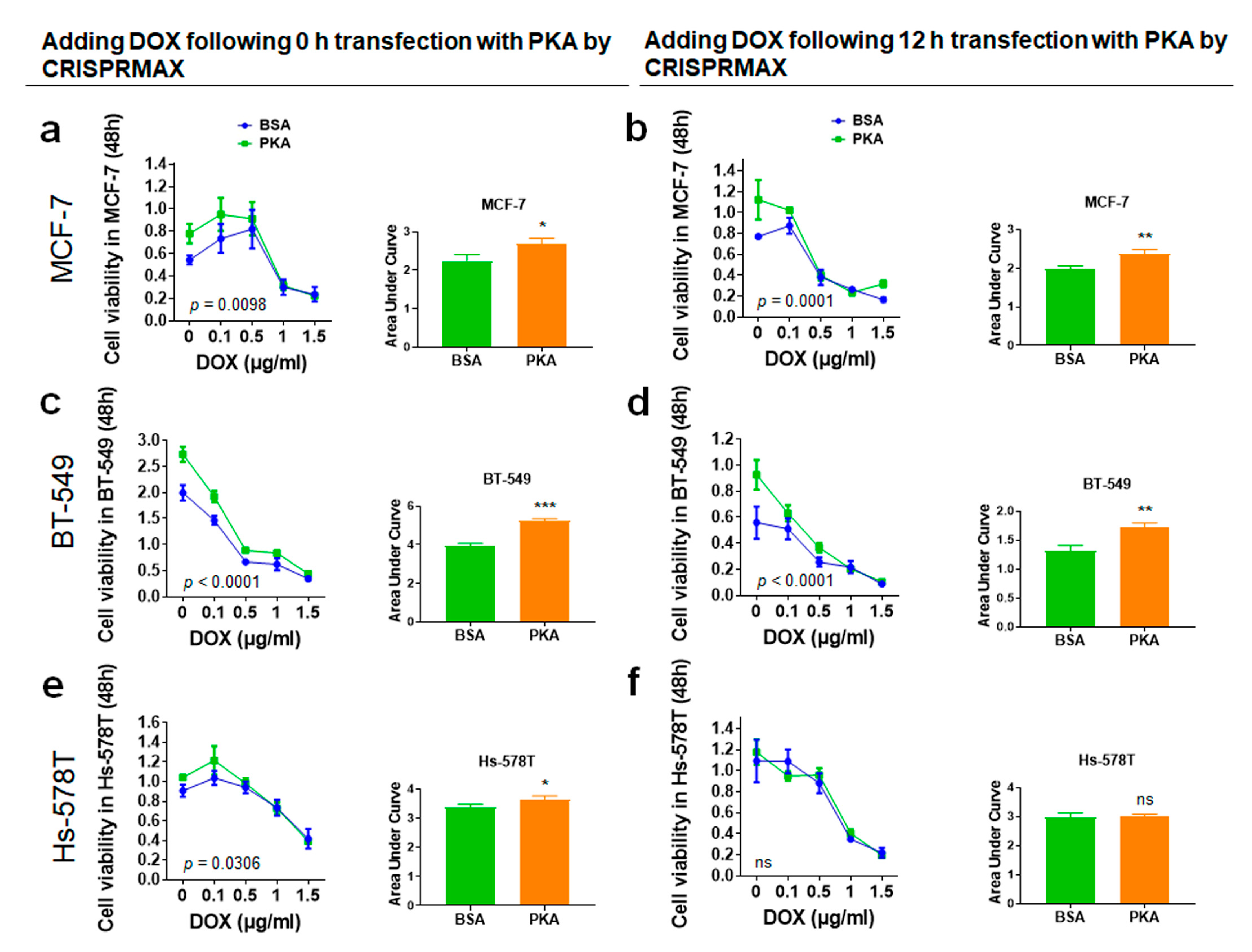
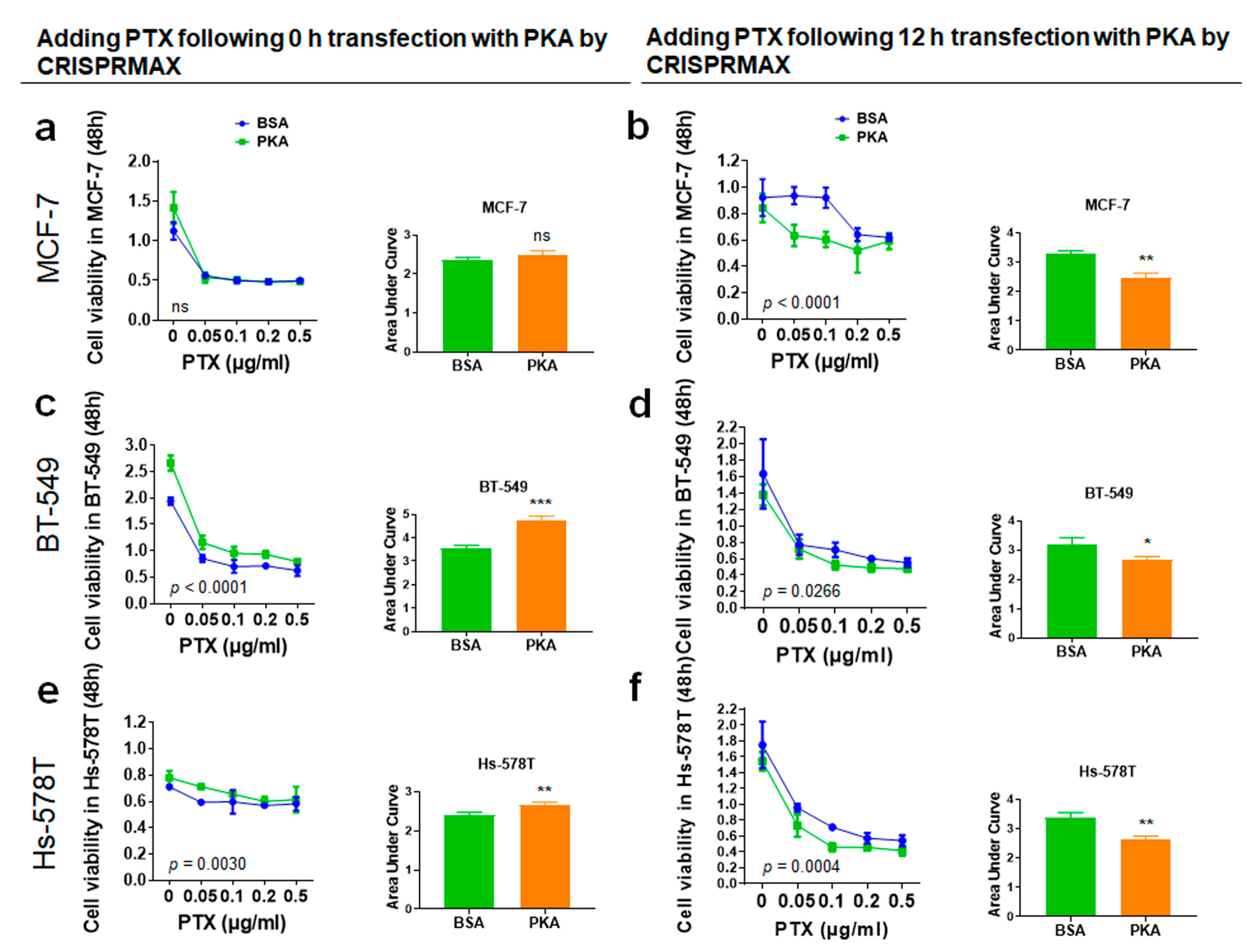
2.10. Chemoresistance Assay
2.11. Statistical Analyses
3. Results
3.1. Characterization of PKAs- and BSA-Loaded CRISPRMAX Complexes
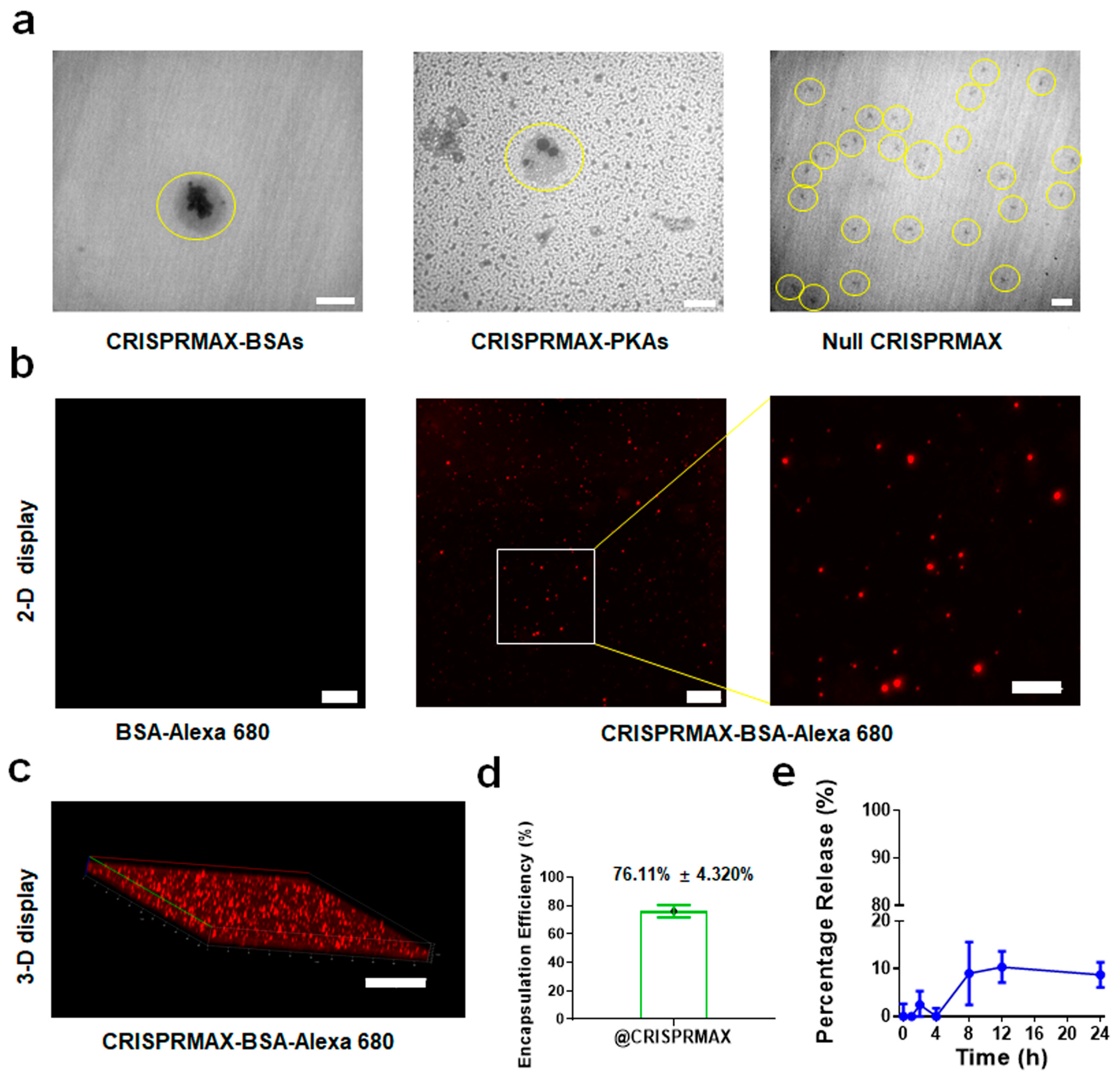
3.1.1. Morphology
3.1.2. Encapsulation Efficiency and Release Efficiency of CRISPRMAX
3.1.3. Determination of Zeta Potential and Particle Size of the Prepared Lipid Nanoparticles
3.2. Establishment of a CRISPRMAX-Based Enzyme Delivery System
3.2.1. Establishment of a CRISPRMAX-Based Protein Delivery System Using Alexa 680-Labeled BSA as a Model Protein
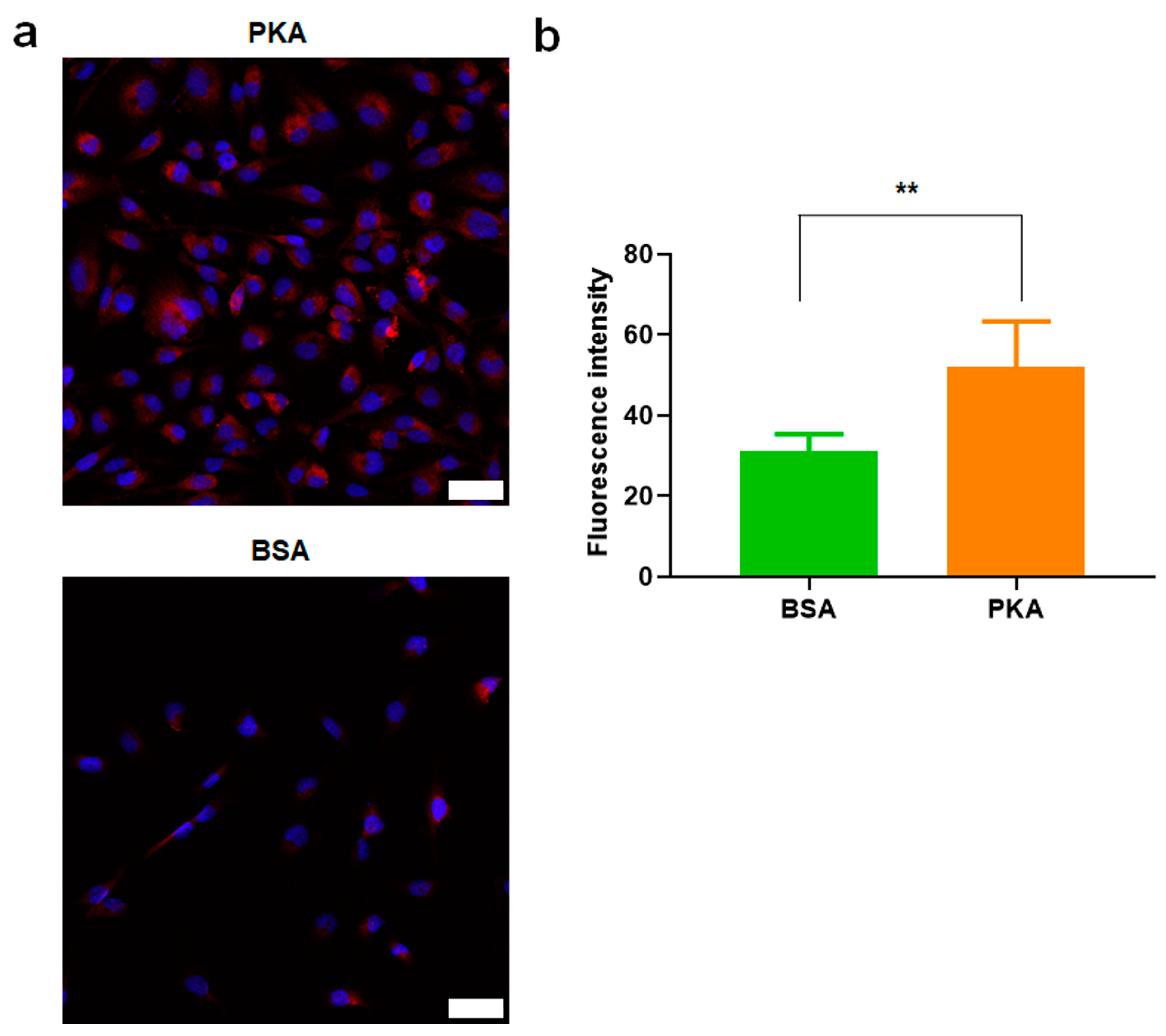
3.2.2. PKA Was Upregulated by PKA-Lipid Nanoparticles Delivery
3.3. Analysis of the Effects of PKA-Lipid Nanoparticles Delivery on EMT-Associated Marker Expression and CSCs
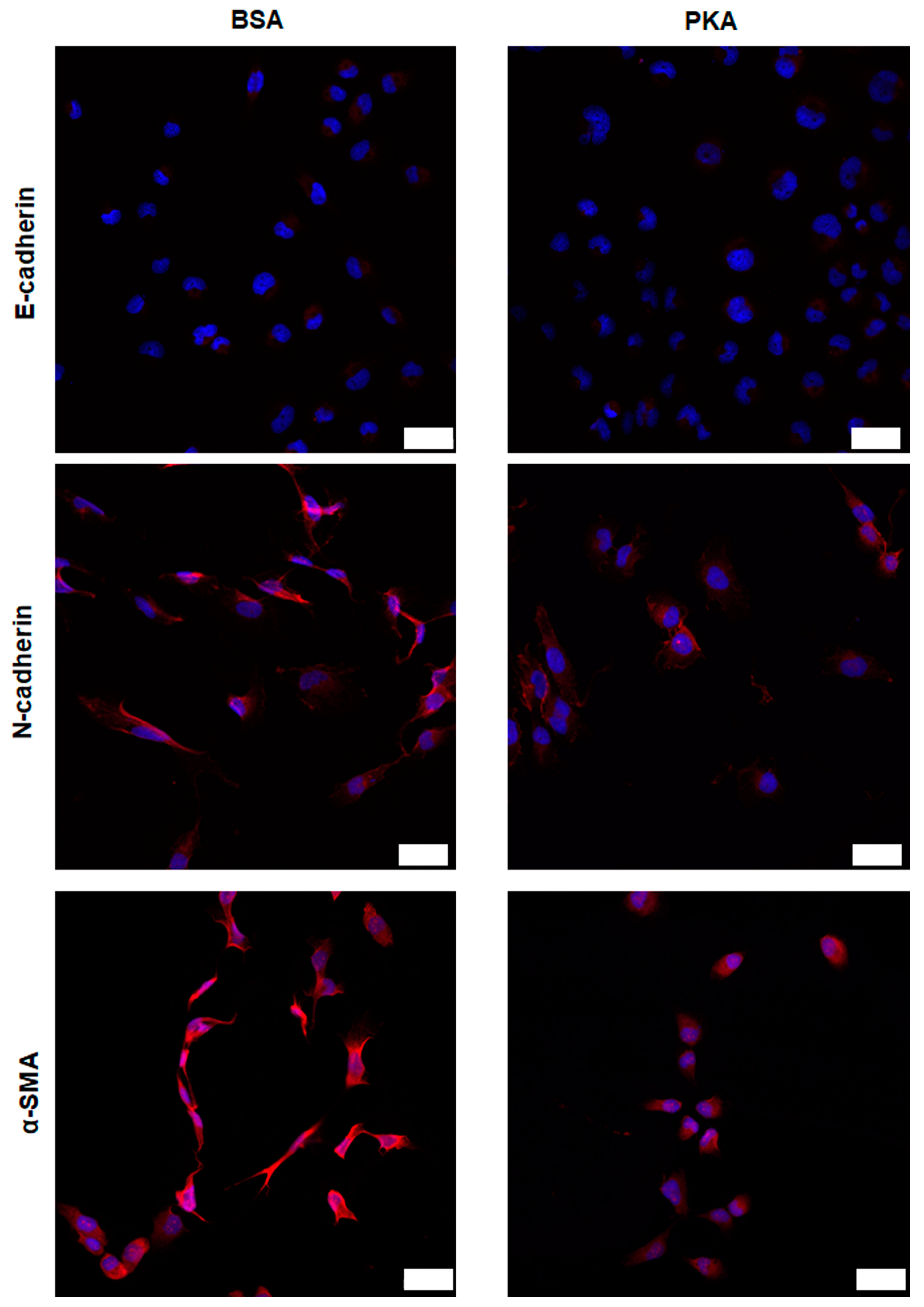
3.3.1. Expression Analysis of E-Cadherin, N-Cadherin, and α-SMA after PKA Delivery Using CLSM
3.3.2. Analysis of the Effects of PKA Enzyme Delivery on CSCs
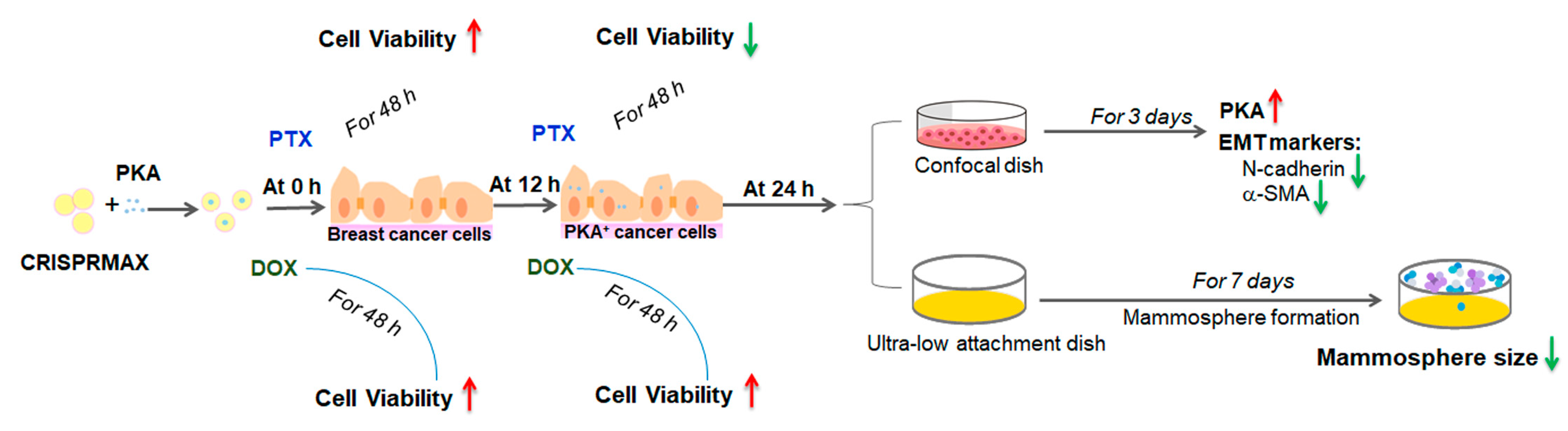
3.4. Analysis of the Synergy Effects of CRISPRMAX-Based PKA Delivery along with Chemotherapy Drugs
3.4.1. Analysis of Synergy Effect of CRISPRMAX-Based PKA Delivery and DOX in Breast Cancer Cells
3.4.2. Analysis of Synergy Effect of CRISPRMAX-PKAs Delivery and PTX in Breast Cancer Cells
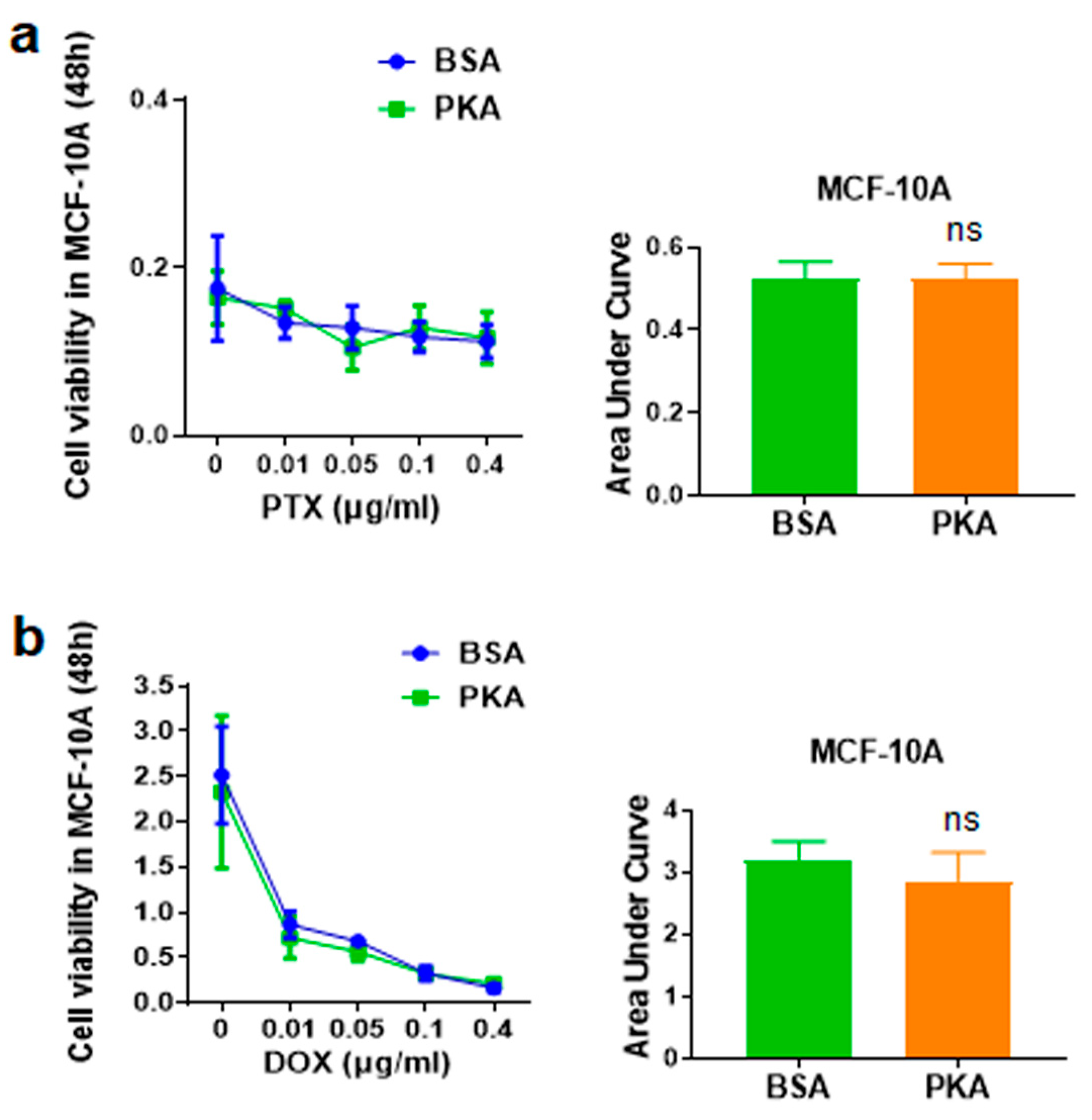
3.4.3. Analysis of the Synergy Effect of CRISPRMAX-PKA Delivery and PTX in Normal Breast Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Desai, A.; Yan, Y.; Gerson, S.L. Concise Reviews: Cancer Stem Cell Targeted Therapies: Toward Clinical Success. Stem Cells Transl. Med. 2019, 8, 75–81. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Voon, D.C.-C.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. The EMT spectrum and therapeutic opportunities. Mol. Oncol. 2017, 11, 878–891. [Google Scholar] [CrossRef]
- Vellard, M. The enzyme as drug: Application of enzymes as pharmaceuticals. Curr. Opin. Biotechnol. 2003, 14, 444–450. [Google Scholar] [CrossRef]
- Wang, X.; Yin, L.; Yang, L.; Zheng, Y.; Liu, S.; Yang, J.; Cui, H.; Wang, H. Silencing ubiquitin-conjugating enzyme 2C inhibits proliferation and epithelial–mesenchymal transition in pancreatic ductal adenocarcinoma. FEBS J. 2019, 286, 4889–4909. [Google Scholar] [CrossRef]
- Lucena, M.C.; Carvalho-Cruz, P.; Donadio, J.L.; Oliveira, I.A.; De Queiroz, R.M.; Marinho-Carvalho, M.M.; Sola-Penna, M.; De Paula, I.F.; Gondim, K.C.; McComb, M.E.; et al. Epithelial Mesenchymal Transition Induces Aberrant Glycosylation through Hexosamine Biosynthetic Pathway Activation. J. Biol. Chem. 2016, 291, 12917–12929. [Google Scholar] [CrossRef]
- Kotiyal, S.; Bhattacharya, S. Breast cancer stem cells, EMT and therapeutic targets. Biochem. Biophys. Res. Commun. 2014, 453, 112–116. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef]
- Xin, Y.; Yin, M.; Zhao, L.; Meng, F.; Luo, L. Recent progress on nanoparticle-based drug delivery systems for cancer therapy. Cancer Biol. Med. 2017, 14, 228–241. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Z.; Chen, H.; Gao, Y. Nanoparticle-based drug delivery systems for controllable photodynamic cancer therapy. Eur. J. Pharm. Sci. 2020, 144, 105213. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Koudelka, S.; Turánek, J. Liposomal paclitaxel formulations. J. Control. Release 2012, 163, 322–334. [Google Scholar] [CrossRef]
- Dave, V.; Yadav, R.B.; Kushwaha, K.; Yadav, S.; Sharma, S.; Agrawal, U. Lipid-polymer hybrid nanoparticles: Development & statistical optimization of norfloxacin for topical drug delivery system. Bioact. Mater. 2017, 2, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-N.; Zeng, Q.; Wang, H.-Y.; Zhang, B.; Li, S.-T.; Nan, X.; Cao, N.; Fu, C.-J.; Yan, X.-L.; Jia, Y.L.; et al. MicroRNA-125b attenuates epithelial-mesenchymal transitions and targets stem-like liver cancer cells through small mothers against decapentaplegic 2 and 4. Hepatology 2015, 62, 801–815. [Google Scholar] [CrossRef]
- Zhao, P.; Hou, X.; Yan, J.; Du, S.; Xue, Y.; Li, W.; Xiang, G.; Dong, Y. Long-term storage of lipid-like nanoparticles for mRNA delivery. Bioact. Mater. 2020, 5, 358–363. [Google Scholar] [CrossRef]
- Wolfram, J.; Ferrari, M. Clinical cancer nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef]
- Yu, X.; Liang, X.; Xie, H.; Kumar, S.; Ravinder, N.; Potter, J.; Jeu, X.D.M.D.; Chesnut, J.D. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol. Lett. 2016, 38, 919–929. [Google Scholar] [CrossRef]
- Lim, D.; Sreekanth, V.; Cox, K.J.; Law, B.K.; Wagner, B.K.; Karp, J.M.; Choudhary, A. Engineering designer beta cells with a CRISPR-Cas9 conjugation platform. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Ghasemi, R.; Struthers, H.; Wilson, E.R.; Spencer, D.H. Contribution of CTCF binding to transcriptional activity at the HOXA locus in NPM1-mutant AML cells. Leukemia 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Zhang, X.; Zhang, H.; Qu, X.; Chen, D.; Servos, M.R.; Mäkilä, E.; Salonen, J.; Santos, H.A.; Hai, M.; et al. Inhibition of Multidrug Resistance of Cancer Cells by Co-Delivery of DNA Nanostructures and Drugs Using Porous Silicon Nanoparticles@Giant Liposomes. Adv. Funct. Mater. 2015, 25, 3330–3340. [Google Scholar] [CrossRef]
- Bauleth-Ramos, T.; Shahbazi, M.-A.; Liu, D.; Fontana, F.; Correia, A.; Figueiredo, P.; Zhang, H.B.; Martins, J.P.; Hirvonen, J.T.; Granja, P.; et al. Nutlin-3a and Cytokine Co-loaded Spermine-Modified Acetalated Dextran Nanoparticles for Cancer Chemo-Immunotherapy. Adv. Funct. Mater 2017, 27, 1703303. [Google Scholar] [CrossRef]
- Zhang, H.; Cui, W.; Qu, X.; Wu, H.; Qu, L.; Zhang, X.; Mäkilä, E.; Salonen, J.; Zhu, Y.; Yang, Z.; et al. Photothermal-responsive nanosized hybrid polymersome as versatile therapeutics codelivery nanovehicle for effective tumor suppression. Proc. Natl. Acad. Sci. USA 2019, 116, 7744–7749. [Google Scholar] [CrossRef] [PubMed]
- Kankala, R.K.; Zhang, H.; Liu, C.; Kanubaddi, K.R.; Lee, C.; Wang, S.; Cui, W.; Santos, H.A.; Lin, K.; Chen, A. Metal Species–Encapsulated Mesoporous Silica Nanoparticles: Current Advancements and Latest Breakthroughs. Adv. Funct. Mater. 2019, 29, 1902652. [Google Scholar] [CrossRef]
- Yan, J.; Liu, C.; Wu, Q.; Zhou, J.; Xu, X.; Zhang, L.; Wang, D.; Yang, F.; Zhang, H. Mineralization of ph-sensitive doxorubicin prodrug in zif-8 to enable targeted delivery to solid tumors. Anal. Chem. 2020, 92, 11453–11461. [Google Scholar] [CrossRef]
- Zhang, Y.; Tu, J.; Wang, D.; Zhu, H.; Maity, S.K.; Qu, X.; Bogaert, B.; Pei, H.; Zhang, H. Programmable and multifunctional DNA-based materials for biomedical applications. Adv. Mater. 2018, 30, e1703658. [Google Scholar] [CrossRef]
- Auvinen, H.; Zhang, H.; Nonappa; Kopilow, A.; Niemela, E.H.; Nummelin, S.; Correia, A.; Santos, H.A.; Linko, V.; Kostiainen, M.A. Protein coating of DNA nanostructures for enhanced stability and immunocompatibility. Adv. Healthc. Mater. 2017, 6, 1700692. [Google Scholar] [CrossRef]
- Kong, F.; Zhang, H.; Qu, X.; Zhang, X.; Chen, D.; Ding, R.; Mäkilä, E.; Salonen, J.; Santos, H.A.; Hai, M. Gold Nanorods, DNA Origami, and Porous Silicon Nanoparticle-functionalized Biocompatible Double Emulsion for Versatile Targeted Therapeutics and Antibody Combination Therapy. Adv. Mater. 2016, 28, 10195–10203. [Google Scholar] [CrossRef]
- Yan, X.-L.; Fu, C.-J.; Chen, L.; Qin, J.-H.; Zeng, Q.; Yuan, H.-F.; Nan, X.; Chen, H.-X.; Zhou, J.-N.; Lin, Y.-L.; et al. Mesenchymal stem cells from primary breast cancer tissue promote cancer proliferation and enhance mammosphere formation partially via EGF/EGFR/Akt pathway. Breast Cancer Res. Treat. 2012, 132, 153–164. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, H.; Wang, D.; Zeng, Q.; Fan, Z.; Xi, J.; Nan, X.; He, L.; Zhou, J.-N.; Pei, X.; et al. A new protocol for long-term culture of a specific subpopulation of liver cancer stem cells enriched by cell surface markers. FEBS Open Bio 2020, 10, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Brgles, M.; Jurašin, D.D.; Sikirić, M.D.; Frkanec, R.; Tomasić, J. Entrapment of Ovalbumin into Liposomes—Factors Affecting Entrapment Efficiency, Liposome Size, and Zeta Potential. J. Liposome Res. 2008, 18, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Blick, T.; Hugo, H.J.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial Mesenchymal Transition Traits in Human Breast Cancer Cell Lines Parallel the CD44hi/CD24lo/- Stem Cell Phenotype in Human Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2010, 15, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Shaw, F.L.; Harrison, H.; Spence, K.; Ablett, M.P.; Simões, B.M.; Farnie, G.; Clarke, R.B. A Detailed Mammosphere Assay Protocol for the Quantification of Breast Stem Cell Activity. J. Mammary Gland. Biol. Neoplasia 2012, 17, 111–117. [Google Scholar] [CrossRef]
- Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res. 2002, 62, 5443–5450. [Google Scholar]
- Avramis, V.I.; Sencer, S.; Periclou, A.P.; Sather, H.; Bostrom, B.C.; Cohen, L.J.; Ettinger, A.G.; Ettinger, L.J.; Franklin, J.; Gaynon, P.S.; et al. A randomized comparison of native escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: A children’s cancer group study. Blood 2002, 99, 1986–1994. [Google Scholar] [CrossRef]
- Kurre, H.A.; Ettinger, A.G.; Veenstra, D.L.; Gaynon, P.S.; Franklin, J.; Sencer, S.F.; Reaman, G.H.; Lange, B.J.; Holcenberg, J.S. A Pharmacoeconomic Analysis of Pegaspargase Versus Native Escherichia Coli l-Asparaginase for the Treatment of Children with Standard-Risk, Acute Lymphoblastic Leukemia: The Children’s Cancer Group Study (CCG-1962). J. Pediatr. Hematol. 2002, 24, 175–181. [Google Scholar] [CrossRef]
- Ali, H.S.; Boshra, M.S.; El Meteini, M.S.; Matboli, M.; Matboli, M. lncRNA- RP11-156p1.3, novel diagnostic and therapeutic targeting via CRISPR/Cas9 editing in hepatocellular carcinoma. Genomics 2020, 112, 3306–3314. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Yu, Z.L.X.L.H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Brito, D.A.; Yang, Z.; Rieder, C. Microtubules do not promote mitotic slippage when the spindle assembly checkpoint cannot be satisfied. J. Cell Biol. 2008, 182, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2012, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Sanz-Moreno, V. What does not kill you makes you stronger: Surviving anti-cancer therapies by cytoskeletal remodeling and Myosin II reactivation. Mol. Cell. Oncol. 2020, 7, 1735911. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.-M.; Bera, R.; Chiou, C.-Y.; Yu, M.-C.; Chen, T.-C.; Chen, C.-W.; Wang, T.-R.; Chiang, W.-L.; Chai, S.-P.; Chia-Wei, C.; et al. Actin cytoskeleton remodeling drives epithelial-mesenchymal transition for hepatoma invasion and metastasis in mice. Hepatology 2018, 67, 2226–2243. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-J.; Xu, P. Smart Mesoporous Silica Nanoparticles for Protein Delivery. Nanomaterials 2019, 9, 511. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Q.; Lin, J.; Cai, Z.; Liao, G.; Wang, K.; Bai, L.; Zhao, P.; Yu, Z.; Yu, Y.Z. Recent Advance in Polymer Based Microspheric Systems for Controlled Protein and Peptide Delivery. Curr. Med. Chem. 2019, 26, 2285–2296. [Google Scholar] [CrossRef]
- Liang, K.; Ricco, R.; Doherty, C.M.; Styles, M.J.; Bell, S.; Kirby, N.; Mudie, S.; Haylock, D.; Hill, A.J.; Doonan, C.J.; et al. Biomimetic mineralization of metal-organic frameworks as protective coatings for biomacromolecules. Nat. Commun. 2015, 6, 7240. [Google Scholar] [CrossRef]
- Wu, X.; Yue, H.; Zhang, Y.; Gao, X.; Li, X.; Wang, L.; Cao, Y.; Hou, M.; An, H.; Zhang, L.; et al. Packaging and delivering enzymes by amorphous metal-organic frameworks. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Davarani, F.H.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef]
- Dang, Y.; Guan, J. Nanoparticle-based drug delivery systems for cancer therapy. Smart Mater. Med. 2020, 1, 10–19. [Google Scholar] [CrossRef]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Crist, R.M.; Clogston, J.D.; McNeil, S.E. Zeta potential: A case study of cationic, anionic, and neutral liposomes. Anal. Bioanal. Chem. 2017, 409, 5779–5787. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Kemper, A.; Weissmann, C.; Reyher, H.-J.; Brandt, R. Monitoring Cytoskeletal Dynamics in Living Neurons Using Fluorescence Photoactivation. Methods Enzymol. 2012, 505, 3–21. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| CRISPRMAX-PKAs | 349.8 ± 21.65 | 0.232 ± 0.172 | −8.38 ± 0.337 |
| CRISPRMAX-BSAs | 344.5 ± 45.54 | 0.343 ± 0.051 | −7.87 ± 0.0777 |
| Null CRISPRMAX | 345.56 ± 33.30 | 0.382 ± 0.231 | −8.33 ± 0.403 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.-N.; Rautio, T.-C.; Liu, C.; Xu, X.-Y.; Wang, D.-Q.; Guo, Y.; Eriksson, J.; Zhang, H. Delivery of Protein Kinase A by CRISPRMAX and Its Effects on Breast Cancer Stem-Like Properties. Pharmaceutics 2021, 13, 11. https://doi.org/10.3390/pharmaceutics13010011
Zhou J-N, Rautio T-C, Liu C, Xu X-Y, Wang D-Q, Guo Y, Eriksson J, Zhang H. Delivery of Protein Kinase A by CRISPRMAX and Its Effects on Breast Cancer Stem-Like Properties. Pharmaceutics. 2021; 13(1):11. https://doi.org/10.3390/pharmaceutics13010011
Chicago/Turabian StyleZhou, Jun-Nian, Tzu-Chen Rautio, Chang Liu, Xiao-Yu Xu, Dong-Qing Wang, Yong Guo, John Eriksson, and Hongbo Zhang. 2021. "Delivery of Protein Kinase A by CRISPRMAX and Its Effects on Breast Cancer Stem-Like Properties" Pharmaceutics 13, no. 1: 11. https://doi.org/10.3390/pharmaceutics13010011
APA StyleZhou, J.-N., Rautio, T.-C., Liu, C., Xu, X.-Y., Wang, D.-Q., Guo, Y., Eriksson, J., & Zhang, H. (2021). Delivery of Protein Kinase A by CRISPRMAX and Its Effects on Breast Cancer Stem-Like Properties. Pharmaceutics, 13(1), 11. https://doi.org/10.3390/pharmaceutics13010011