An Enteric-Coated Polyelectrolyte Nanocomplex Delivers Insulin in Rat Intestinal Instillations When Combined with a Permeation Enhancer

 , , ,
, , ,  and
and 
Abstract
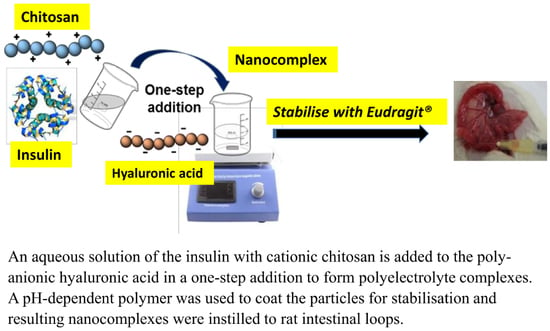
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Insulin-Associated Polyelectrolyte Nanoparticle Complexes (PECs)
2.3. Physicochemical Properties of PECs
2.4. Measurements of Insulin Loading in PECs and HPLC Analysis
2.5. Colloidal Stability of PECs
2.6. PEC Release Studies in Simulated Intestinal Fluids and Proteolytic Sensitivity
2.7. Cytotoxicity Assays of Uncoated Unloaded PECs: (3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) MTS, ATP, and Neutral Red Uptake Assays
2.8. Mucoadhesion of Uncoated, Unloaded Anionic and Cationic PECs
2.9. Rat Jejunal Instillations of Insulin-Associated PECs
2.10. Statistical Analysis
3. Results
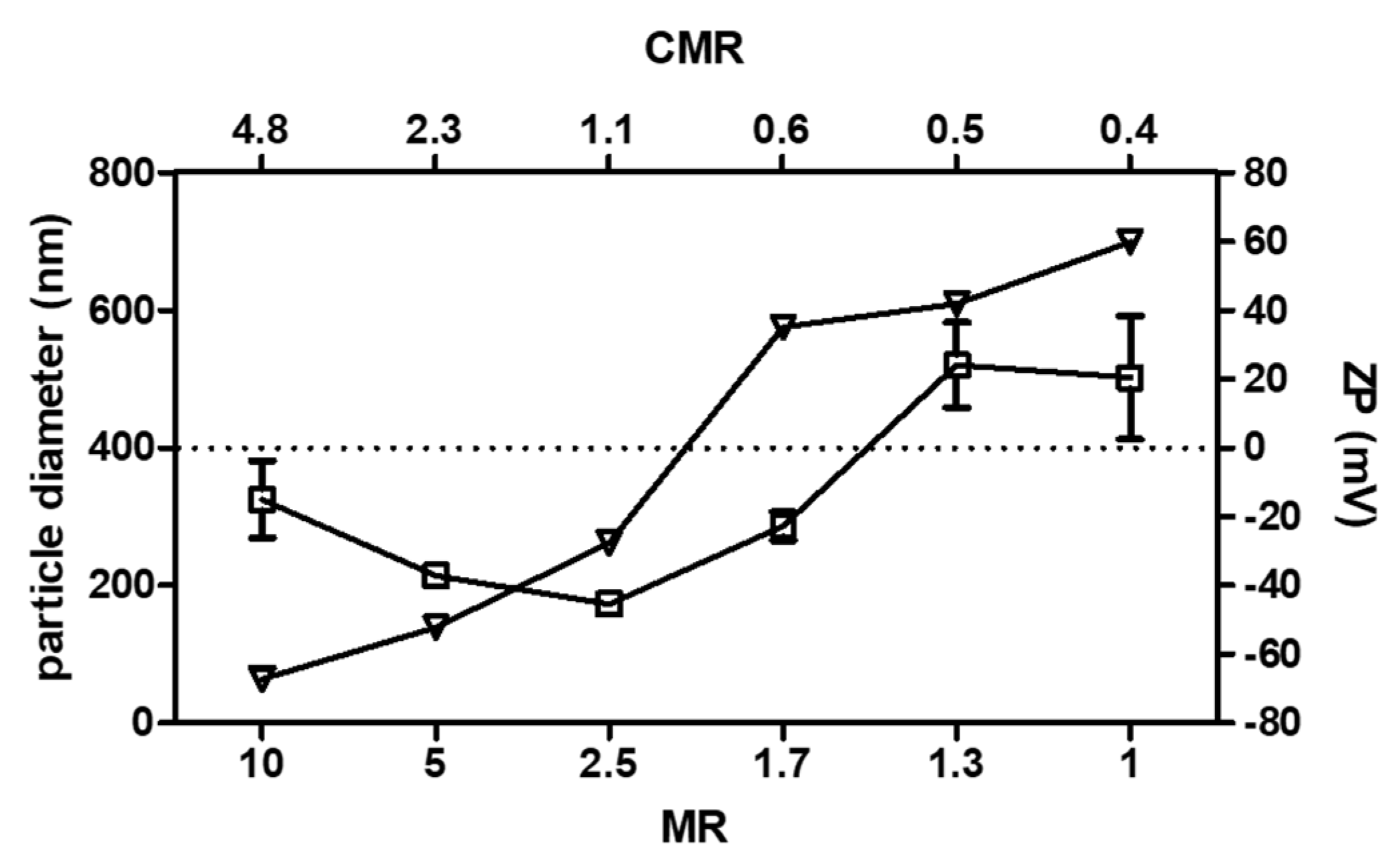
3.1. Selection of the Unloaded PEC Prototype
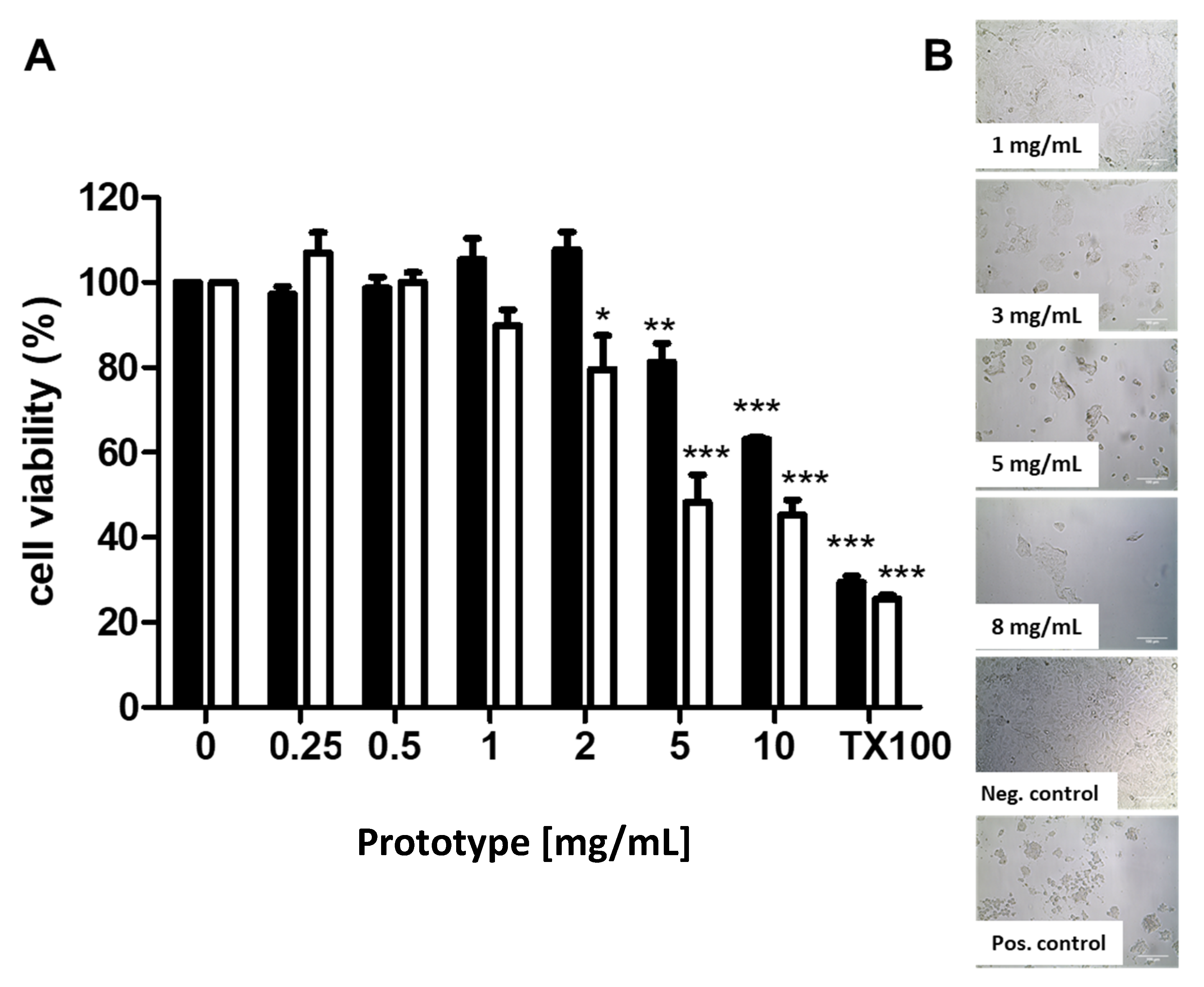
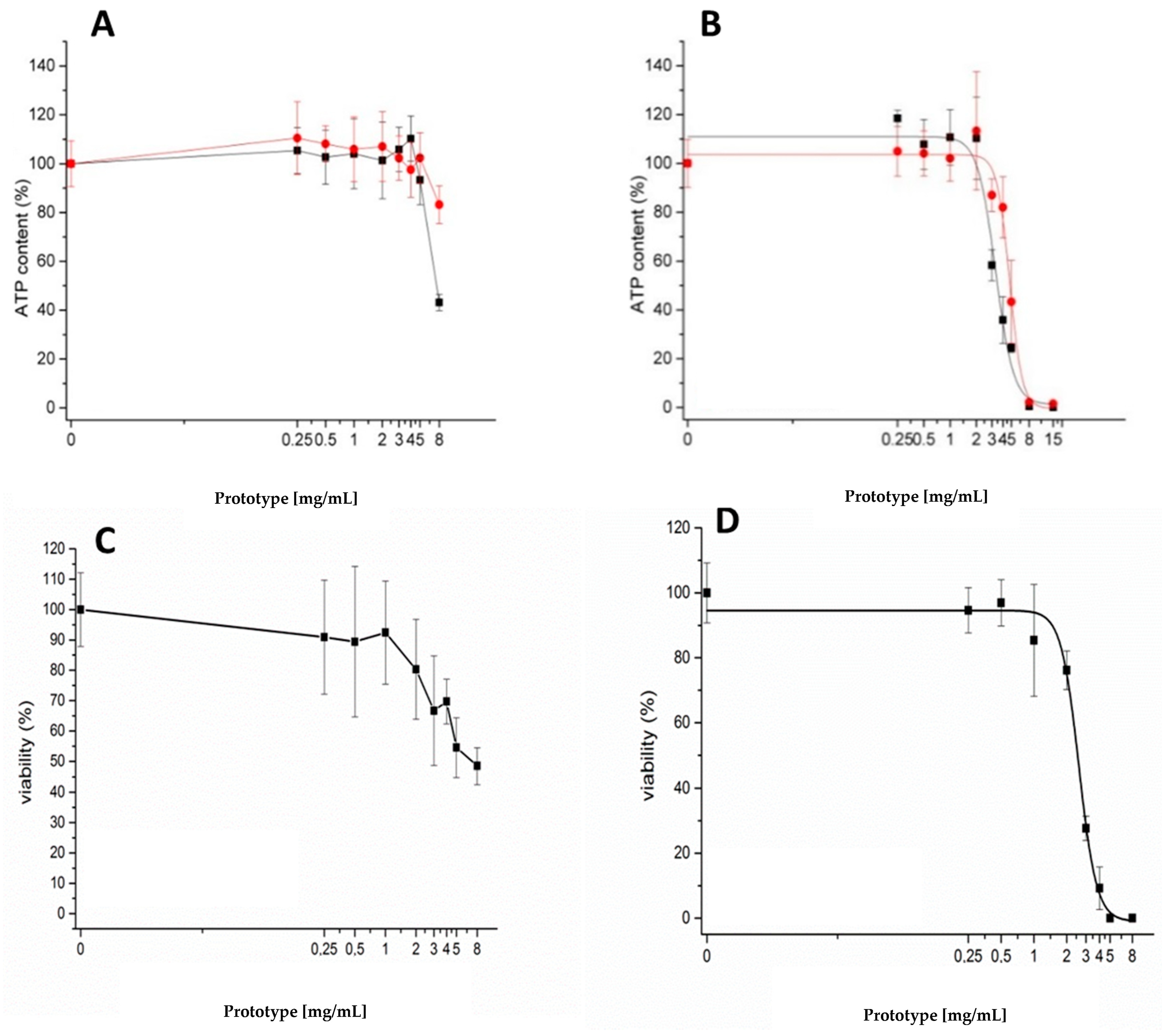
3.2. Cytotoxicity Assays in Caco-2 Cells
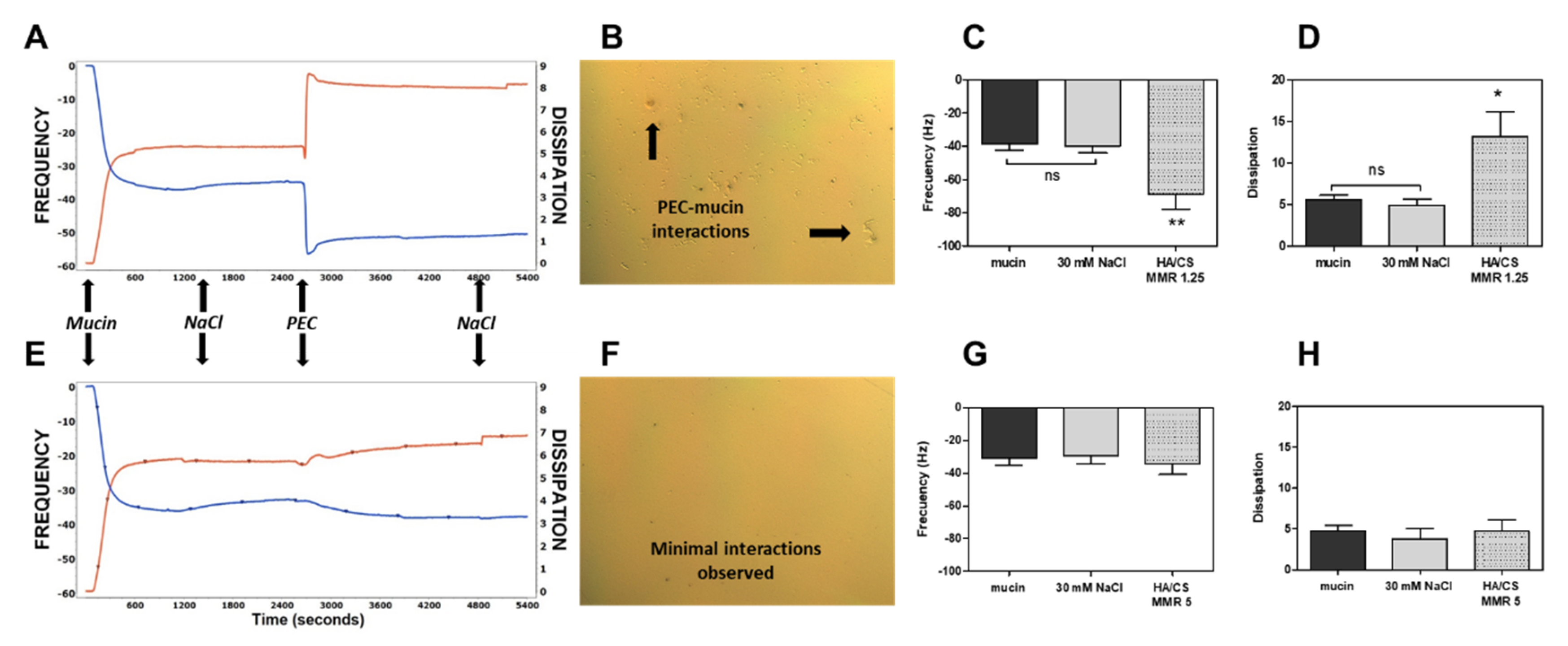
3.3. Mucoadhesion Studies
3.4. Insulin Loading of Uncoated and Coated PECs
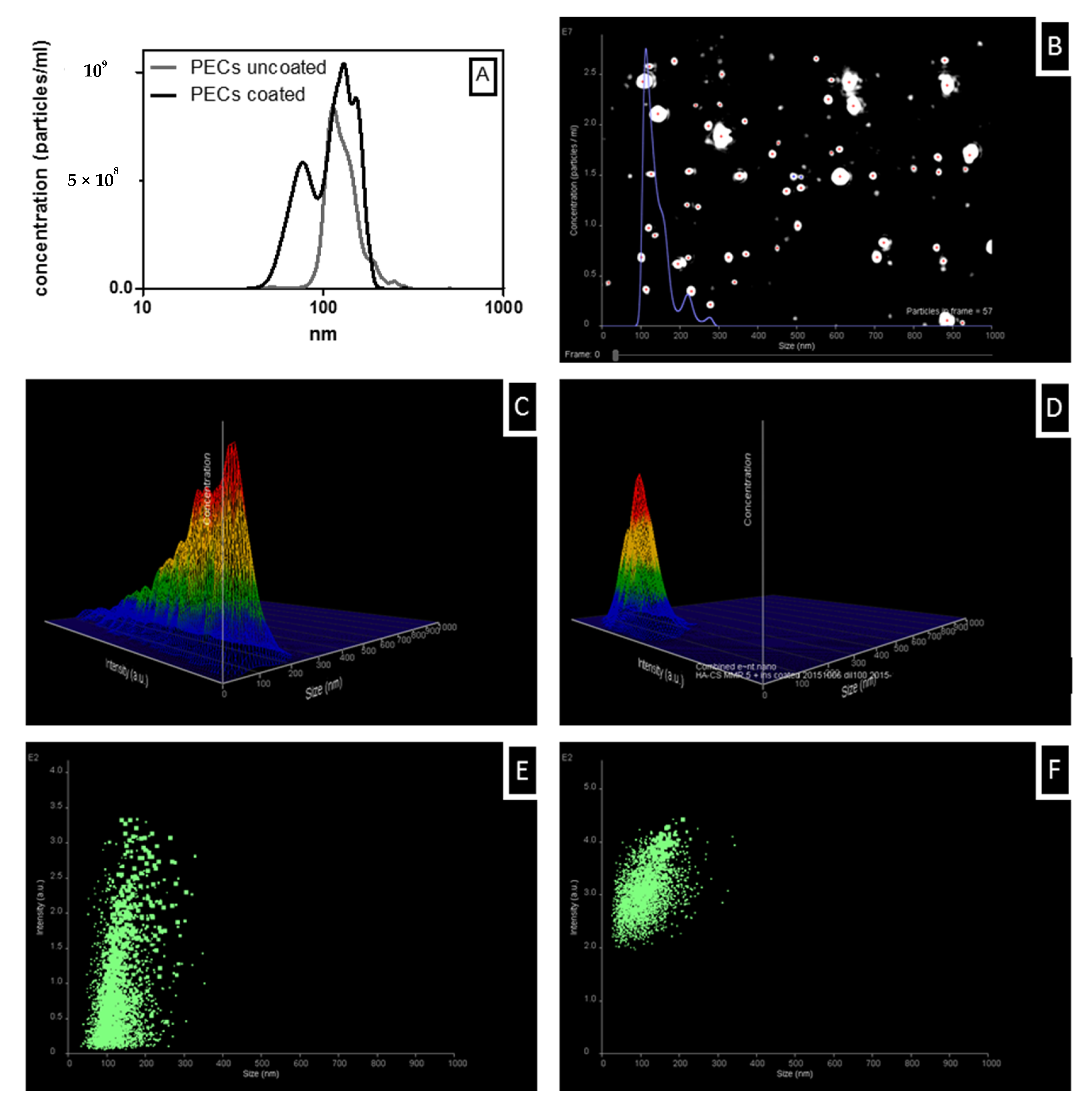
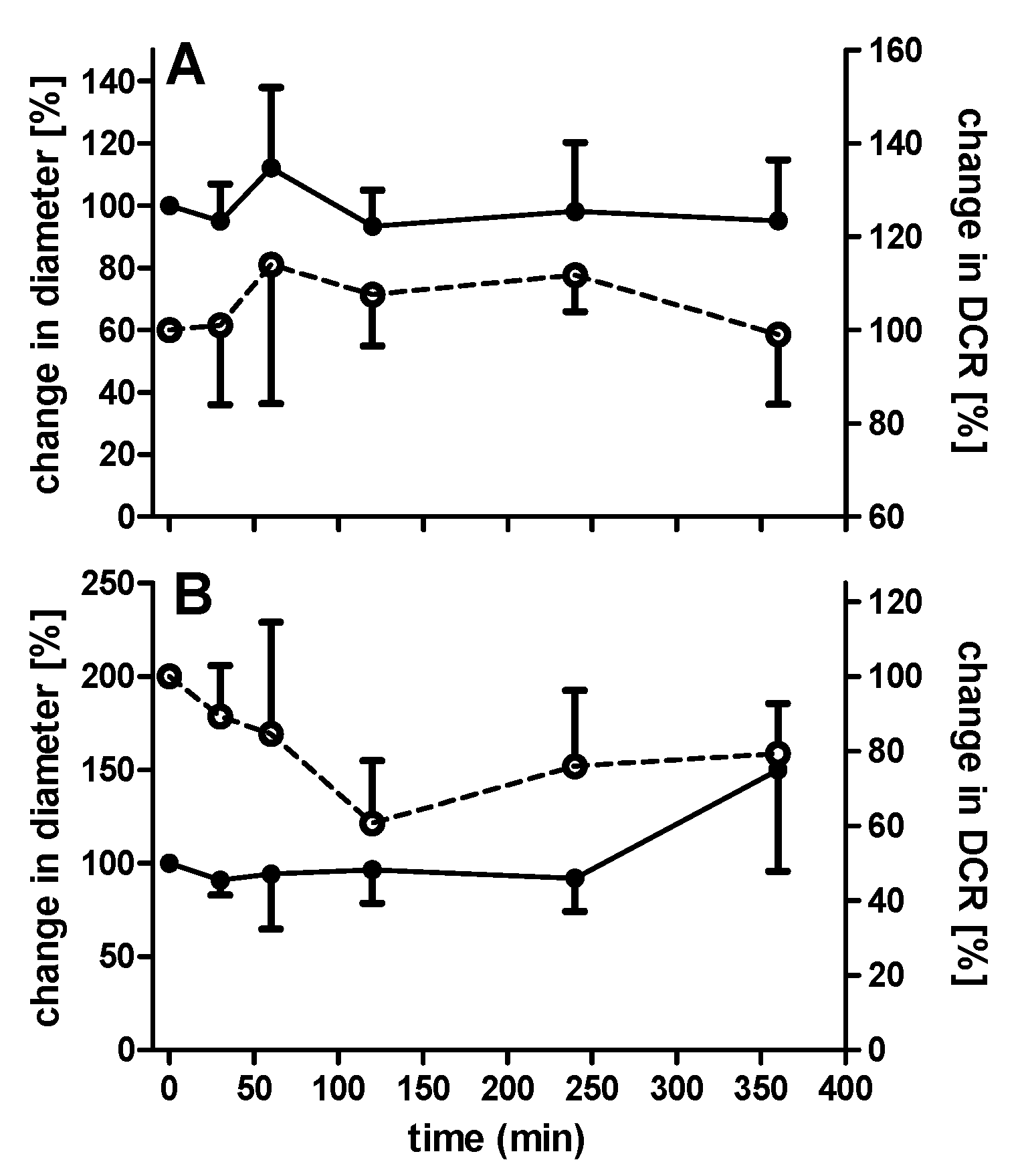
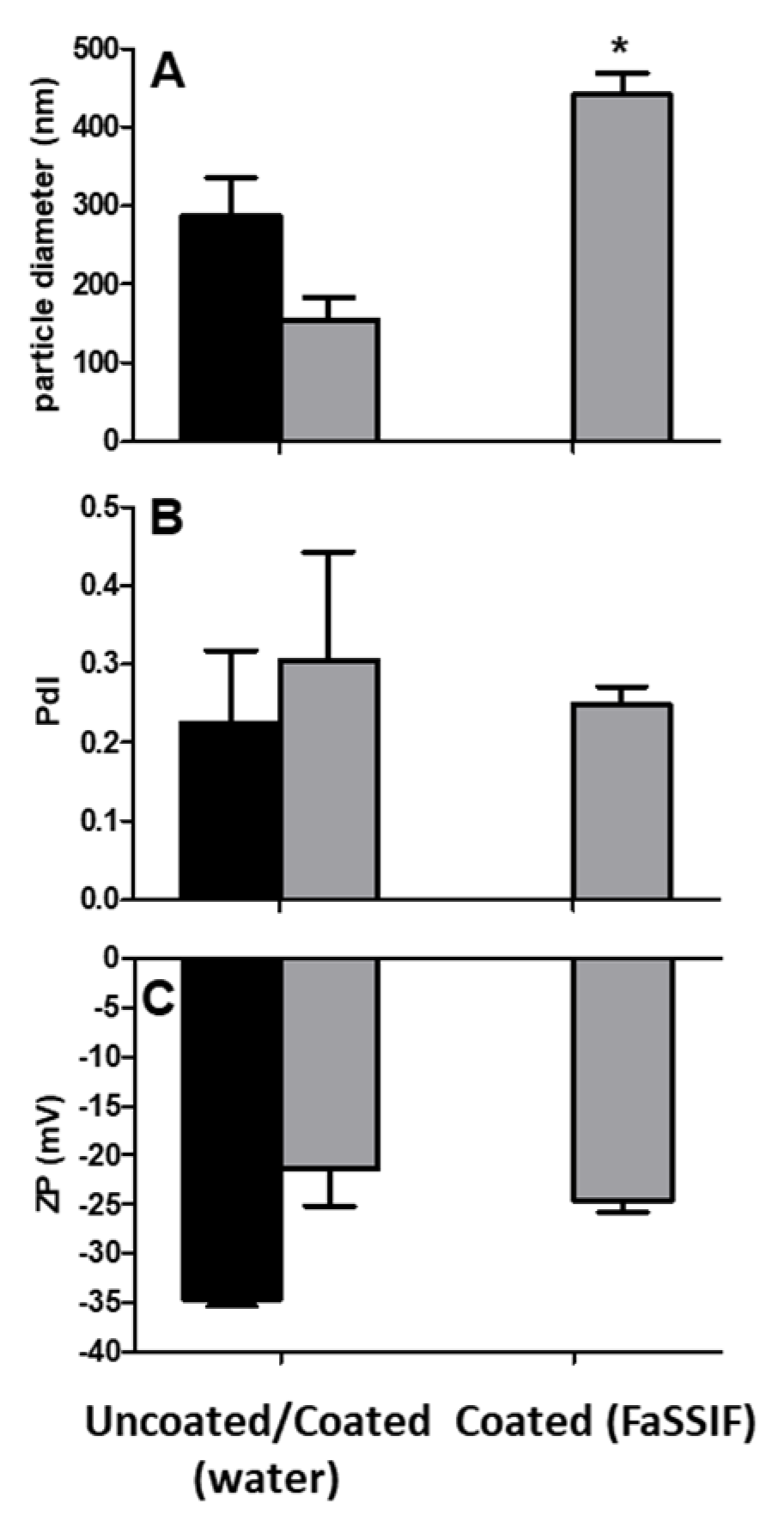
3.5. Colloidal Stability of Uncoated and Coated PECs
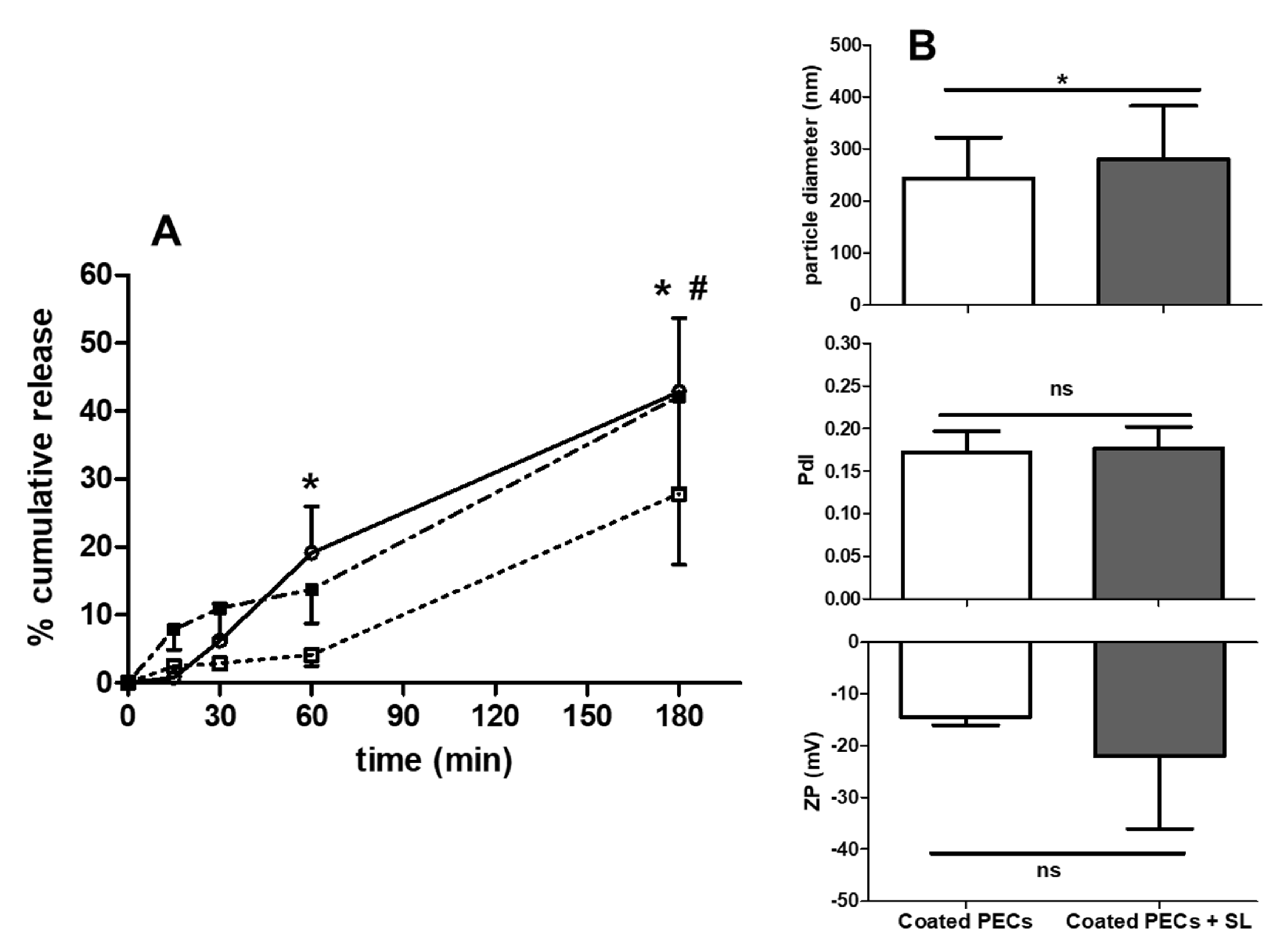
3.6. PEC Release of Insulin: Studies in FaSSIF-V2
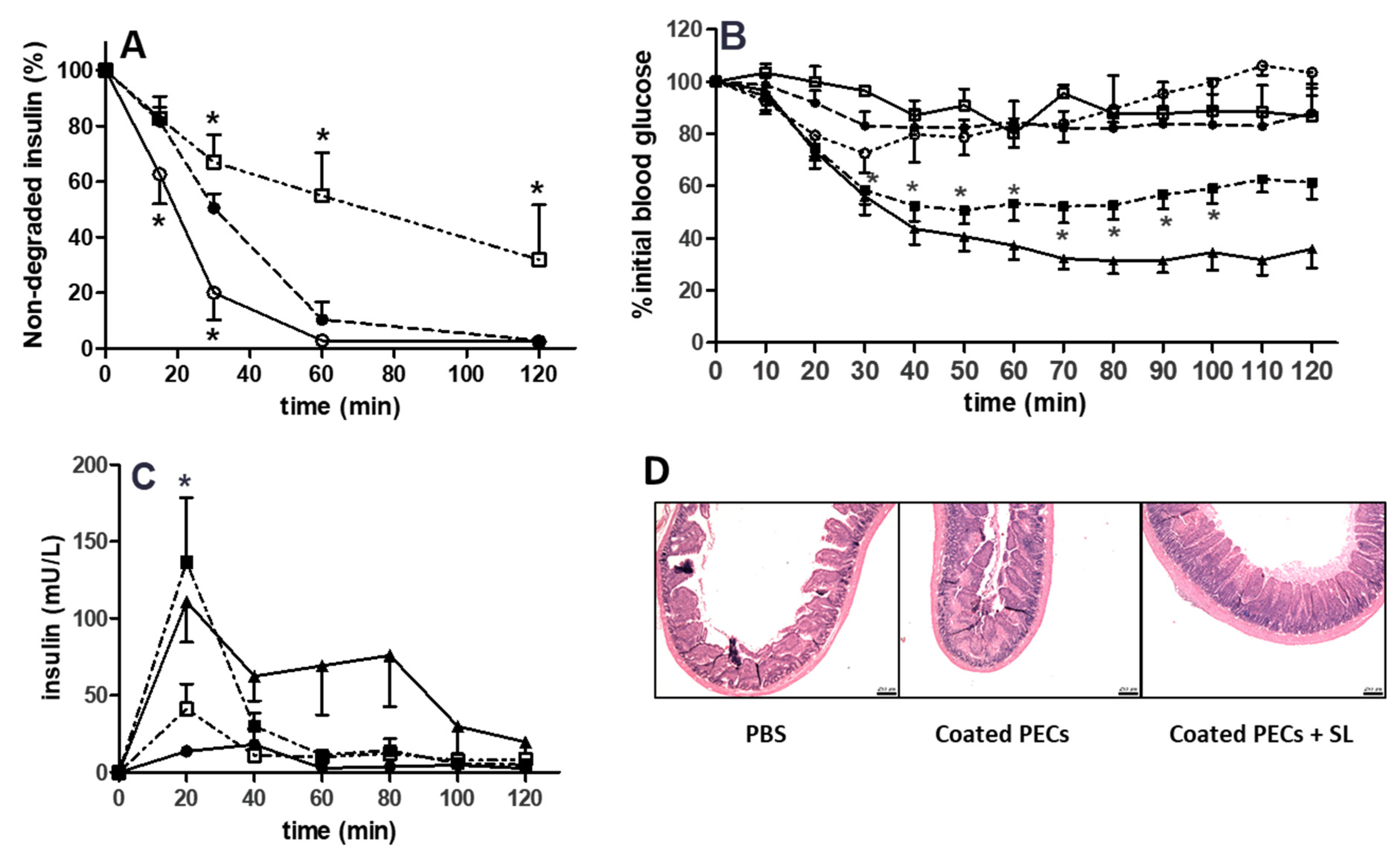
3.7. Proteolytic Sensitivity
3.8. Rat Intra-Jejunal Instillations of Insulin-Loaded PECs
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




References
- Hu, Q.; Luo, Y. Recent advances of polysaccharide-based nanoparticles for oral insulin delivery. Int. J. Biol. Macromol. 2018, 120, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.; Castro, P.M.; Madureira, A.R.; Sarmento, B.; Pintado, M. Recent insights in the use of nanocarriers for the oral delivery of bioactive proteins and peptides. Peptides 2018, 101, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Sonia, T.A.; Sharma, C.P. An overview of natural polymers for oral insulin delivery. Drug Discov. Today 2012, 17, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Nur, M.; Vasiljevic, T. Can natural polymers assist in delivering insulin orally? Int. J. Biol. Macromol. 2017, 103, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Potential of insulin nanoparticle formulations for oral delivery and diabetes treatment. J. Control. Release 2017, 264, 247–275. [Google Scholar] [CrossRef]
- Hartig, S.M.; Greene, R.R.; Dikov, M.M.; Prokop, A.; Davidson, J.M. Multifunctional nanoparticulate polyelectrolyte complexes. Pharm. Res. 2007, 24, 2353–2369. [Google Scholar] [CrossRef] [PubMed]
- Bourganis, V.; Karamanidou, T.; Kammona, O.; Kiparissides, C. Polyelectrolyte complexes as prospective carriers for the oral delivery of protein therapeutics. Eur. J. Pharm. Biopharm. 2017, 111, 44–60. [Google Scholar] [CrossRef]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic acid in the third millennium. Polymers 2018, 10, 701. [Google Scholar] [CrossRef]
- Han, L.; Zhao, Y.; Yin, L.; Li, R.; Liang, Y.; Huang, H.; Pan, S.; Wu, C.; Feng, M. Insulin-loaded pH-sensitive hyaluronic acid nanoparticles enhance transcellular delivery. AAPS PharmSciTech 2012, 13, 836–845. [Google Scholar] [CrossRef]
- Contreras-Ruiz, L.; de la Fuente, M.; Párraga, J.E.; López-García, A.; Fernández, I.; Seijo, B.; Sánchez, A.; Calonge, M.; Diebold, Y. Intracellular trafficking of hyaluronic acid-chitosan oligomer-based nanoparticles in cultured human ocular surface cells. Mol. Vis. 2011, 17, 279–290. [Google Scholar]
- Al Rubeaan, K.; Rafiullah, M.; Jayavanth, S. Oral insulin delivery systems using chitosan-based formulation: A review. Expert Opin. Drug Deliv. 2016, 13, 223–327. [Google Scholar] [CrossRef]
- Smith, J.; Wood, E.; Dornish, M. Effect of chitosan on epithelial cell tight junctions. Pharm. Res. 2004, 21, 43–49. [Google Scholar] [CrossRef]
- Vllasaliu, D.; Exposito-Harris, R.; Heras, A.; Casettari, L.; Garnett, M.; Illum, L.; Stolnik, S. Tight junction modulation by chitosan nanoparticles: Comparison with chitosan solution. Int. J. Pharm. 2010, 400, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Bilati, U.; Allémann, E.; Doelker, E. Strategic approaches for overcoming peptide and protein instability within biodegradable nano- and microparticles. Eur. J. Pharm. Biopharm. 2005, 59, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-Bailón, F.; Osorio-Revilla, G.; Gallardo-Velázquez, T. Microencapsulation of insulin using a W/O/W double emulsion followed by complex coacervation to provide protection in the gastrointestinal tract. J. Microencapsul. 2015, 32, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Hartig, S.M.; Carlesso, G.; Davidson, J.M.; Prokop, A. Development of improved nanoparticle polyelectrolyte complex physicochemistry by non-stoichiometric mixing of polyions with similar molecular weights. Biomacromolecules 2007, 8, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Schatz, C.; Domard, C.; Viton, C.; Pichot, C.; Delair, T. Versatile and efficient formation of colloids of biopolymer-based polyelectrolyte complexes. Biomacromolecules 2004, 5, 1882–1892. [Google Scholar] [CrossRef]
- Umerska, A.; Paluch, K.J.; Inkielewicz-Stępniak, I.; Santos-Martinez, M.J.; Corrigan, O.I.; Medina, C.; Tajber, L. Exploring the assembly process and properties of novel crosslinker-free hyaluronate-based polyelectrolyte complex nanocarriers. Int. J. Pharm. 2012, 436, 75–87. [Google Scholar] [CrossRef]
- Umerska, A.; Paluch, K.J.; Martinez, M.J.S.; Corrigan, O.I.; Medina, C.; Tajber, L. Self-assembled hyaluronate/protamine polyelectrolyte nanoplexes: Synthesis, stability, biocompatibility and potential use as peptide carriers. J. Biomed. Nanotechnol. 2014, 10, 3658–3673. [Google Scholar] [CrossRef]
- Umerska, A.; Paluch, K.J.; Santos-Martinez, M.J.; Medina, C.; Corrigan, O.I.; Tajber, L. Chondroitin-based nanoplexes as peptide delivery systems—Investigations into the self-assembly process, solid-state and extended release characteristics. Eur. J. Pharm. Biopharm. 2015, 93, 242–253. [Google Scholar] [CrossRef]
- Umerska, A.; Corrigan, O.I.; Tajber, L. Intermolecular interactions between salmon calcitonin, hyaluronate, and chitosan and their impact on the process of formation and properties of peptide-loaded nanoparticles. Int. J. Pharm. 2014, 477, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.M.; McMorrow, J.; Umerska, A.; Patel, H.B.; Kornerup, K.N.; Tajber, L.; Murphy, E.P.; Perretti, M.; Corrigan, O.I.; Brayden, D.J. An intra-articular salmon calcitonin-based nanocomplex reduces experimental inflammatory arthritis. J. Control. Release 2013, 167, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, B.; Ribeiro, A.J.; Veiga, F.; Sampaio, P.; Neufeld, R.J.; Ferreira, D. Alginate/chitosan nanoparticles are effective for oral insulin delivery. Pharm. Res. 2007, 24, 2198–2206. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Aniceto, D.; Abrantes, M.; Simões, S.; Branco, F.; Vitória, I.; Botelho, M.F.; Seiça, R.; Veiga, F.; Ribeiro, A. In vivo biodistribution of antihyperglycemic biopolymer-based nanoparticles for the treatment of type 1 and type 2 diabetes. Eur. J. Pharm. Biopharm. 2017, 113, 88–96. [Google Scholar] [CrossRef]
- Sung, H.-W.; Sonaje, K.; Liao, Z.-X. pH-responsive nanoparticles shelled with chitosan for oral delivery of insulin: From mechanism to therapeutic applications. Acc. Chem. Res. 2012, 45, 619–629. [Google Scholar] [CrossRef]
- Sonaje, K.; Chen, Y.J.; Chen, H.L.; Wey, S.P.; Juang, J.H.; Nguyen, H.N.; Hsu, C.W.; Lin, K.J.; Sung, H.W. Enteric-coated capsules filled with freeze-dried chitosan/poly(gamma-glutamic acid) nanoparticles for oral insulin delivery. Biomaterials 2010, 31, 3384–3394. [Google Scholar] [CrossRef]
- Yin, L.; Ding, J.; He, C.; Cui, L.; Tang, C.; Yin, C. Drug permeability and mucoadhesion properties of thiolated trimethyl chitosan nanoparticles in oral insulin delivery. Biomaterials 2009, 30, 5691–5700. [Google Scholar] [CrossRef]
- Umerska, A.; Tajber, L. Polyelectrolyte complexes as nanoparticulate drug delivery systems. Eur. Pharm. Rev. 2015, 20, 36–40. [Google Scholar]
- Thünemann, A.F.; Müller, M.; Dautzenberg, H.; Joanny, J.F.; Löwen, H. Polyelectrolyte complexes. Adv. Polym. Sci. 2004, 166, 113–171. [Google Scholar] [CrossRef]
- Plapied, L.; Duhem, N.; des Rieux, A.; Préat, V. Fate of polymeric nanocarriers for oral drug delivery. Curr. Opin. Colloid. Interface Sci. 2011, 16, 228–237. [Google Scholar] [CrossRef]
- McCartney, F.; Rosa, M.; Brayden, D.J. Evaluation of sucrose laurate as an intestinal permeation enhancer for macromolecules: Ex Vivo and In Vivo studies. Pharmaceutics 2019, 11, 565. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Seijo, B.; Alonso, M.J. Novel hyaluronan-based nanocarriers for transmucosal delivery of macromolecules. Macromol. Biosci. 2008, 8, 441–450. [Google Scholar] [CrossRef]
- Malloy, A.; Carr, B. Nanoparticle tracking analysis - the halo system. Part. Part. Syst. Charact. 2006, 23, 197–204. [Google Scholar] [CrossRef]
- Dash, R.N.; Habibuddin, M.; Sahoo, A.; Kothawade, A.; Sachin, N.; Chaudhari, M.R.; Mahadik, K.R. Factorial Approach for the development of stability indicating HPLC assay of recombinant human insulin: Application to its stability study. Curr. Pharm. Anal. 2013, 9, 318–329. [Google Scholar] [CrossRef]
- Jintapattanakit, A.; Junyaprasert, V.B.; Mao, S.; Sitterberg, J.; Bakowsky, U.; Kissel, T. Peroral delivery of insulin using chitosan derivatives: A comparative study of polyelectrolyte nanocomplexes and nanoparticles. Int. J. Pharm. 2007, 342, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Vertzoni, M.; Pastelli, E.; Psachoulias, D.; Kalantzi, L.; Reppas, C. Estimation of intragastric solubility of drugs: In what medium? Pharm. Res. 2007, 24, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Jantratid, E.; Jannsen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, D.O.; Healy, A.M.; Corrigan, O.I. Preparation and release of salbutamol from chitosan and chitosan co-spray dried compacts and multiparticulates. Eur. J. Pharm. Biopharm. 2006, 62, 295–305. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [CrossRef]
- Morciano, G.; Sarti, A.C.; Marchi, S.; Missiroli, S.; Falzoni, S.; Raffaghello, L.; Pistoia, V.; Giorgi, C.; Di Virgilio, F.; Pinton, P. Use of luciferase probes to measure ATP in living cells and animals. Nat. Protoc. 2017, 12, 1542–1562. [Google Scholar] [CrossRef] [PubMed]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Inkielewicz-Stepniak, I.; Tajber, L.; Behan, G.; Zhang, H.; Radomski, M.W.; Medina, C.; Santos-Martinez, M.J. The role of mucin in the toxicological impact of polystyrene nanoparticles. Materials 2018, 11, 724. [Google Scholar] [CrossRef]
- Presas, E.; McCartney, F.; Sultan, E.; Hunger, C.; Nellen, S.; Alvarez, C.V.; Werner, U.; Bazile, D.; Brayden, D.J.; O’Driscoll, C.M. Physicochemical, pharmacokinetic and pharmacodynamic analyses of amphiphilic cyclodextrin-based nanoparticles designed to enhance intestinal delivery of insulin. J. Control. Release 2018, 286, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, E.J.B.; Yoshida, S.; Kamei, N.; Iwamae, R.; Khafagy, E.S.; Olsen, J.; Rahbek, U.L.; Pedersen, B.L.; Takayama, K.; Takeda-Morishita, M. In Vivo proof of concept of oral insulin delivery based on a co-administration strategy with the cell-penetrating peptide penetratin. J. Control. Release 2014, 189, 19–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Wong, C.Y.; Luna, G.; Martinez, J.; Al-Salami, H.; Dass, C.R. Bio-nanotechnological advancement of orally administered insulin nanoparticles: Comprehensive review of experimental design for physicochemical characterization. Int. J. Pharm. 2019, 572, 118720. [Google Scholar] [CrossRef]
- Swenson, E.S.; Milisen, W.B.; Curatolo, W. Intestinal permeability enhancement: Efficacy, acute local toxicity, and reversibility. Pharm. Res. 1994, 11, 1132–1142. [Google Scholar] [CrossRef]
- Garcia-Fuentes, M.; Prego, C.; Torres, D.; Alonso, M.J. A comparative study of the potential of solid triglyceride nanostructures coated with chitosan or poly(ethylene glycol) as carriers for oral calcitonin delivery. Eur. J. Pharm. Sci. 2005, 25, 133–413. [Google Scholar] [CrossRef]
- Sladek, S.; Kearney, C.; Crean, D.; Brama, P.A.J.; Tajber, L.; Fawcett, K.; Labberte, M.C.; Leggett, B.; Brayden, D.J. Intra-articular delivery of a nanocomplex comprising salmon calcitonin, hyaluronic acid, and chitosan using an equine model of joint inflammation. Drug Deliv. Transl. Res. 2018, 8, 1421–1435. [Google Scholar] [CrossRef]
- Dul, M.; Paluch, K.J.; Kelly, H.; Healy, A.M.; Sasse, A.; Tajber, L. Self-assembled carrageenan/protamine polyelectrolyte nanoplexes—Investigation of critical parameters governing their formation and characteristics. Carbohydr. Polym. 2015, 123, 339–349. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Seijo, B.; Alonso, M.J. Novel hyaluronic acid-chitosan nanoparticles for ocular gene therapy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2016–2024. [Google Scholar] [CrossRef] [PubMed]
- Boddohi, S.; Moore, N.; Johnson, P.A.; Kipper, M.J. Polysaccharide-based polyelectrolyte complex nanoparticles from chitosan, heparin, and hyaluronan. Biomacromolecules 2009, 10, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, B.; Ribeiro, A.; Veiga, F.; Ferreira, D. Development and characterization of new insulin containing polysaccharide nanoparticles. Colloids Surf. B Biointerfaces 2006, 53, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Tedesco, E.; Benetti, F.; Mabondzo, A.; Montagner, I.M.; Marigo, I.; Gonzalez-Touceda, D.; Tovar, S.; Diéguez, C.; Santander-Ortega, M.J.; et al. Rational design of polyarginine nanocapsules intended to help peptides overcoming intestinal barriers. J. Control. Release 2017, 263, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.P.; Tømmeraas, T.; Vårum, K.M.; Østgaard, K. Electrophoretic light scattering studies of chitosans with different degrees of N-acetylation. Biomacromolecules 2001, 2, 1310–1314. [Google Scholar] [CrossRef]
- Cleland, R.L.; Wang, J.L.; Detweiler, D.M. Polyelectrolyte properties of sodium hyaluronate. 2. Potentiometric titration of hyaluronic acid. Macromolecules 1982, 15, 386–395. [Google Scholar] [CrossRef]
- Chen, R.H.; Tsaih, M.L.; Lin, W.C. Effects of chain flexibility of chitosan molecules on the preparation, physical, and release characteristics of the prepared capsule. Carbohydr. Polym. 1996, 31, 141–148. [Google Scholar] [CrossRef]
- Cegnar, M.; Kerc, J. Self-assembled polyelectrolyte nanocomplexes of alginate, chitosan and ovalbumin. Acta Chim. Slov. 2010, 57, 431–441. [Google Scholar]
- Voorhees, P.W. The theory of Ostwald ripening. J. Stat. Phys. 2005, 38, 231–252. [Google Scholar] [CrossRef]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Chern, C.S.; Lee, C.K.; Chang, C.J. Electrostatic interactions between amphoteric latex particles and proteins. Colloid Polym. Sci. 2004, 283, 257–264. [Google Scholar] [CrossRef]
- Bal, S.M.; Slütter, B.; van Riet, E.; Kruithof, A.C.; Ding, Z.; Kersten, G.F.A.; Jiskoot, W.; Bouwstra, J.A. Efficient induction of immune responses through intradermal vaccination with N-trimethyl chitosan containing antigen formulations. J. Control. Release 2010, 142, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, H.; Kjoniksen, A.L.; Hiorth, M. Stability of chitosan nanoparticles cross-linked with tripolyphosphate. Biomacromolecules 2012, 13, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.; Gonçalves, I.C.; Nardecchia, S.; Amaral, I.F.; Barbosa, M.A.; Martins, M.C.L. Modulation of stability and mucoadhesive properties of chitosan microspheres for therapeutic gastric application. Int. J. Pharm. 2013, 454, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Santander-Ortega, M.J.; Jódar-Reyes, A.B.; Csaba, N.; Bastos-González, D.; Ortega-Vinuesa, J.L. Colloidal stability of Pluronic F68-coated PLGA nanoparticles: A variety of stabilisation mechanisms. J. Colloid Interface Sci. 2006, 302, 522–529. [Google Scholar] [CrossRef]
- Tobío, M.; Sánchez, A.; Vila, A.; Soriano, I.; Evora, C.; Vila-Jato, J.L.; Alonso, M.J. The role of PEG on the stability in digestive fluids and in vivo fate of PEG-PLA nanoparticles following oral administration. Colloids Surf. B Biointerfaces 2000, 18, 315–323. [Google Scholar] [CrossRef]
- Harde, H.; Agrawal, A.K.; Jain, S. Tetanus toxoids loaded glucomannosylated chitosan based nanohoming vaccine adjuvant with improved oral stability and immunostimulatory response. Pharm. Res. 2015, 32, 122–134. [Google Scholar] [CrossRef]
- Verheul, R.J.; Slütter, B.; Bal, S.M.; Bouwstra, J.A.; Jiskoot, W.; Hennink, W.E. Covalently stabilized trimethyl chitosan-hyaluronic acid nanoparticles for nasal and intradermal vaccination. J. Control. Release 2011, 156, 50–56. [Google Scholar] [CrossRef]
- Momenzadeh, S.; Sadeghi, A.; Vatandoust, N.; Salehi, R. Evaluation of in vivo transfection efficiency of Eudragit-coated nanoparticles of chitosan-DNA: A pH-sensitive system prepared for oral DNA delivery. Iran. Red Crescent Med. J. 2015, 17, e16761. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, X.; Zhang, Y.; Li, G.; Cai, C.; Xu, J.; Tang, X. Thiolated Eudragit-based nanoparticles for oral insulin delivery: Preparation, characterization, and evaluation using intestinal epithelial cells In Vitro. Macromol. Biosci. 2014, 14, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Tiyaboonchai, W.; Limpeanchob, N. Formulation and characterization of amphotericin B-chitosan-dextran sulfate nanoparticles. Int. J. Pharm. 2007, 329, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Shrestha, N.; Correia, A.; Shahbazi, M.A.; Sarmento, B.; Hirvonen, J.; Veiga, F.; Seiça, R.; Ribeiro, A.; Santos, H.A. Dual chitosan/albumin-coated alginate/dextran sulfate nanoparticles for enhanced oral delivery of insulin. J. Control. Release 2016, 232, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Weiyuan, J.K. Drug release kinetics and transport mechanisms of non- degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Moustafine, R.I.; Margulis, E.B.; Sibgatullina, L.F.; Kemenova, V.A.; Mooter, A.; Van Den, G. Comparative evaluation of interpolyelectrolyte complexes of chitosan with Eudragit L100 and Eudragit L100-55 as potential carriers for oral controlled drug delivery. Eur. J. Pharm. Biopharm. 2008, 70, 155–225. [Google Scholar] [CrossRef]
- Calcetti, P.; Salmaso, S.; Walker, G.; Bernkop-Schnürch, A. Development and in vivo evaluation of an oral insulin-PEG delivery system. Eur. J. Pharm. Sci. 2004, 22, 315–323. [Google Scholar] [CrossRef]
- Thompson, C.J.; Tetley, L.; Uchegbu, I.F.; Cheng, W.P. The complexation between novel comb shaped amphiphilic polyallylamine and insulin—Towards oral insulin delivery. Int. J. Pharm. 2009, 376, 46–55. [Google Scholar] [CrossRef]
- Braia, M.; Tubio, G.; Nerli, B.; Loh, W.; DRomanini, D. Analysis of the interactions between Eudragit® L100 and porcine pancreatic trypsin by calorimetric techniques. Int. J. Biol. Macromol. 2012, 50, 180–186. [Google Scholar] [CrossRef]
- Hong, R.; Fischer, N.O.; Verma, A.; Goodman, C.M.; Emrick, T.; Rotello, V.M. Control of protein structure and function through surface recognition by tailored nanoparticle scaffolds. J. Am. Chem. Soc. 2004, 126, 739–743. [Google Scholar] [CrossRef]
- McCartney, F. Use of Permeation Enhancers and Nanotechnology to Increase Intestinal Peptide Permeability. Ph.D. Thesis, University College Dublin, Dublin, Ireland, 2016. [Google Scholar]
- Henze, L.J.; Koehl, N.J.; O’Shea, P.; Kostewicz, E.S.; Holm, R.; Griffin, B.T. The pig as a preclinical model for predicting oral bioavailability and in vivo performance of pharmaceutical oral dosage forms: A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 581–602. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PEC | Loading (µg/mL) | Diameter (nm) | PdI | ZP (mV) | AE (%) | PL (%) | FL (µg/mg) |
|---|---|---|---|---|---|---|---|
| Uncoated | 100 | 232 ± 36 | 0.15 ± 0.06 | −33 ± 5 | 82 ± 6 | 8 ± 1 | 83 ± 7 |
| 500 | 295 ± 58 | 0.28 ± 0.13 | −36 ± 3 | 80 ± 2 | 31 ± 1 | 285 ± 13 | |
| Coated | 100 | 154 ± 27 | 0.16 ± 0.07 | −26 ± 8 | 95 | 1.5 | 17 |
| 500 | 116 ± 28 | 0.21 ± 0.04 | −23 ± 3 | 97 ± 1 | 6 ± 0 | 58 ± 5 |
| PEC | Batches | Particle Diameter (nm) | PdI | ZP (mV) | Cf | |||
|---|---|---|---|---|---|---|---|---|
| Before | After | Before | After | Before | After | |||
| Unloaded | 1 | 175 | 298 | 0.16 | 0.16 | −48 | −53 | 6.71 |
| 500 µg/mL insulin, uncoated | 4 | 200 (± 24) | 307 (± 74) | 0.23 (± 0.04) | 0.25 (± 0.05) | −49 (± 1) | −54 (± 2) | 3.17 (± 1.48) |
| 500 µg/mL insulin, coated | 2 | 87 | 105 | 0.15 | 0.24 | −37 | −59 | 4.05 |
| Treatment | AUC (0–120 min) | Tmax (min) | Cmax (mU/L) | %F(0–120min) |
|---|---|---|---|---|
| Insulin (s.c.) | 6936 ± 2436 | 20 ± 0 | 111 ± 26 | - |
| Coated PECs | 1725 ± 411 | 30 ± 10 | 45 ± 14 | 0.5 |
| Uncoated PECs + SL | 3301 ± 784 | 23 ± 3 | 66 ± 16 | 1.0 |
| Coated PECs + SL | 4001 ± 2031 | 23 ± 3 | 142 ± 39 | 1.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sladek, S.; McCartney, F.; Eskander, M.; Dunne, D.J.; Santos-Martinez, M.J.; Benetti, F.; Tajber, L.; Brayden, D.J. An Enteric-Coated Polyelectrolyte Nanocomplex Delivers Insulin in Rat Intestinal Instillations When Combined with a Permeation Enhancer. Pharmaceutics 2020, 12, 259. https://doi.org/10.3390/pharmaceutics12030259
Sladek S, McCartney F, Eskander M, Dunne DJ, Santos-Martinez MJ, Benetti F, Tajber L, Brayden DJ. An Enteric-Coated Polyelectrolyte Nanocomplex Delivers Insulin in Rat Intestinal Instillations When Combined with a Permeation Enhancer. Pharmaceutics. 2020; 12(3):259. https://doi.org/10.3390/pharmaceutics12030259
Chicago/Turabian StyleSladek, Svenja, Fiona McCartney, Mena Eskander, David J. Dunne, Maria Jose Santos-Martinez, Federico Benetti, Lidia Tajber, and David J. Brayden. 2020. "An Enteric-Coated Polyelectrolyte Nanocomplex Delivers Insulin in Rat Intestinal Instillations When Combined with a Permeation Enhancer" Pharmaceutics 12, no. 3: 259. https://doi.org/10.3390/pharmaceutics12030259
APA StyleSladek, S., McCartney, F., Eskander, M., Dunne, D. J., Santos-Martinez, M. J., Benetti, F., Tajber, L., & Brayden, D. J. (2020). An Enteric-Coated Polyelectrolyte Nanocomplex Delivers Insulin in Rat Intestinal Instillations When Combined with a Permeation Enhancer. Pharmaceutics, 12(3), 259. https://doi.org/10.3390/pharmaceutics12030259