Clinical Pharmacokinetic Evaluation of Optimized Liquisolid Tablets as a Potential Therapy for Male Sexual Dysfunction




Abstract
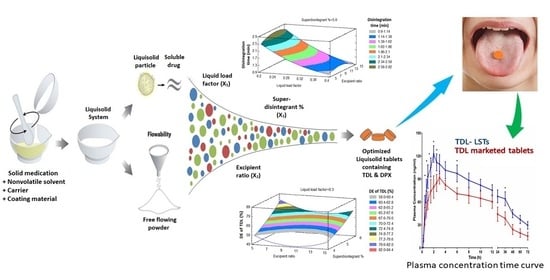
:
1. Introduction
2. Materials and Methods
2.1. Pre-Formulation Studies
2.1.1. Solubility Studies
2.1.2. Holding Capacity and Determination of the Liquid Load Factor (Lf)
2.1.3. Solid-State Characterization Studies
Differential Scanning Calorimetry (DSC)
Fourier Transform Infrared Spectroscopy (FT-IR)
Powder X-ray Diffraction (PXRD)
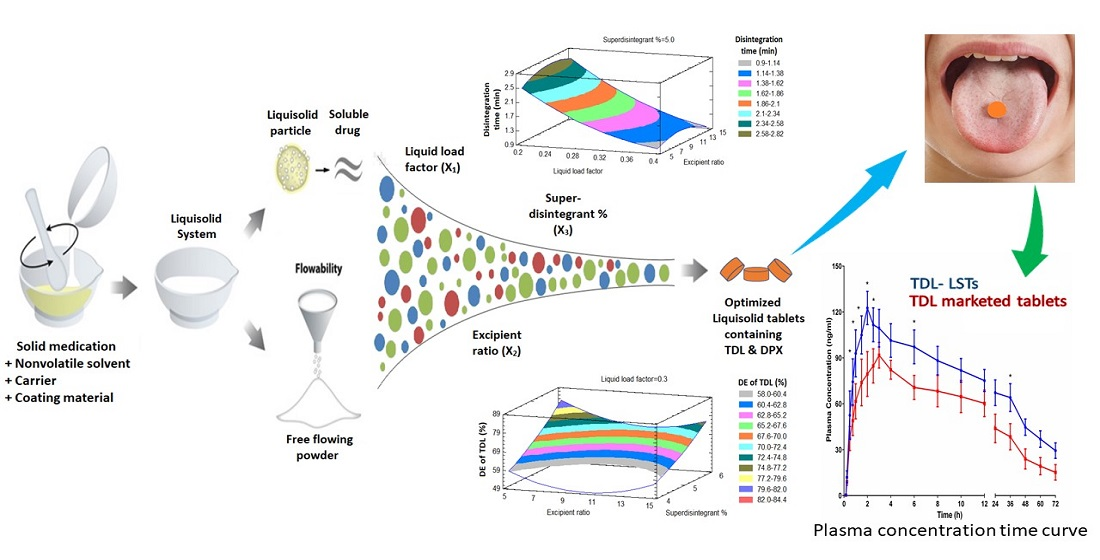
2.2. Formulation of TDL and DPX Liquisolid Tablets
2.3. Preparation of TDL and DPX Liquisolid Tablets
2.4. Pre-Compression Evaluation of the Liquisolid Powder Formulations
2.5. Post-Compression Evaluation of the Prepared Liquisolid Tablets
2.5.1. In-Vitro Disintegration Study
2.5.2. In-Vitro Dissolution Study
2.5.3. Mathematical Modeling of the Dissolution Data
Dissolution Rate (DR10)
Mean Dissolution Time (MDT)
Dissolution Efficiency after 60 Min (DE60)
2.6. Prediction of the Optimized Formulation
2.7. In-Vivo Pharmacokinetic Evaluation on Healthy Human Volunteers
2.7.1. Study Design and Conduct
2.7.2. Subjects
2.7.3. Blood Sampling
2.7.4. Chromatographic Conditions
2.7.5. Pharmacokinetic Data Analysis
2.8. Statistical Analysis
3. Results and Discussion
3.1. Pre-Formulation Studies
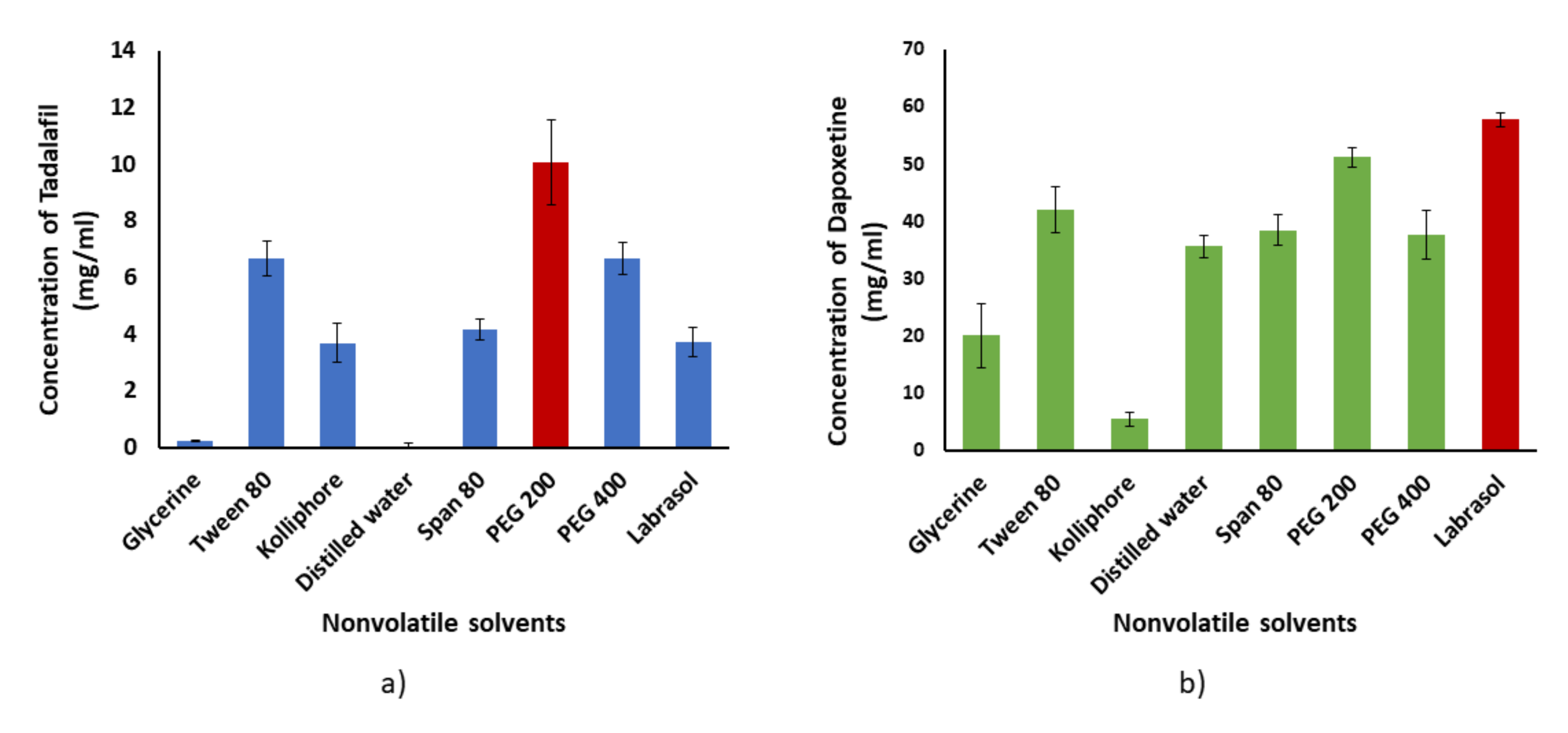
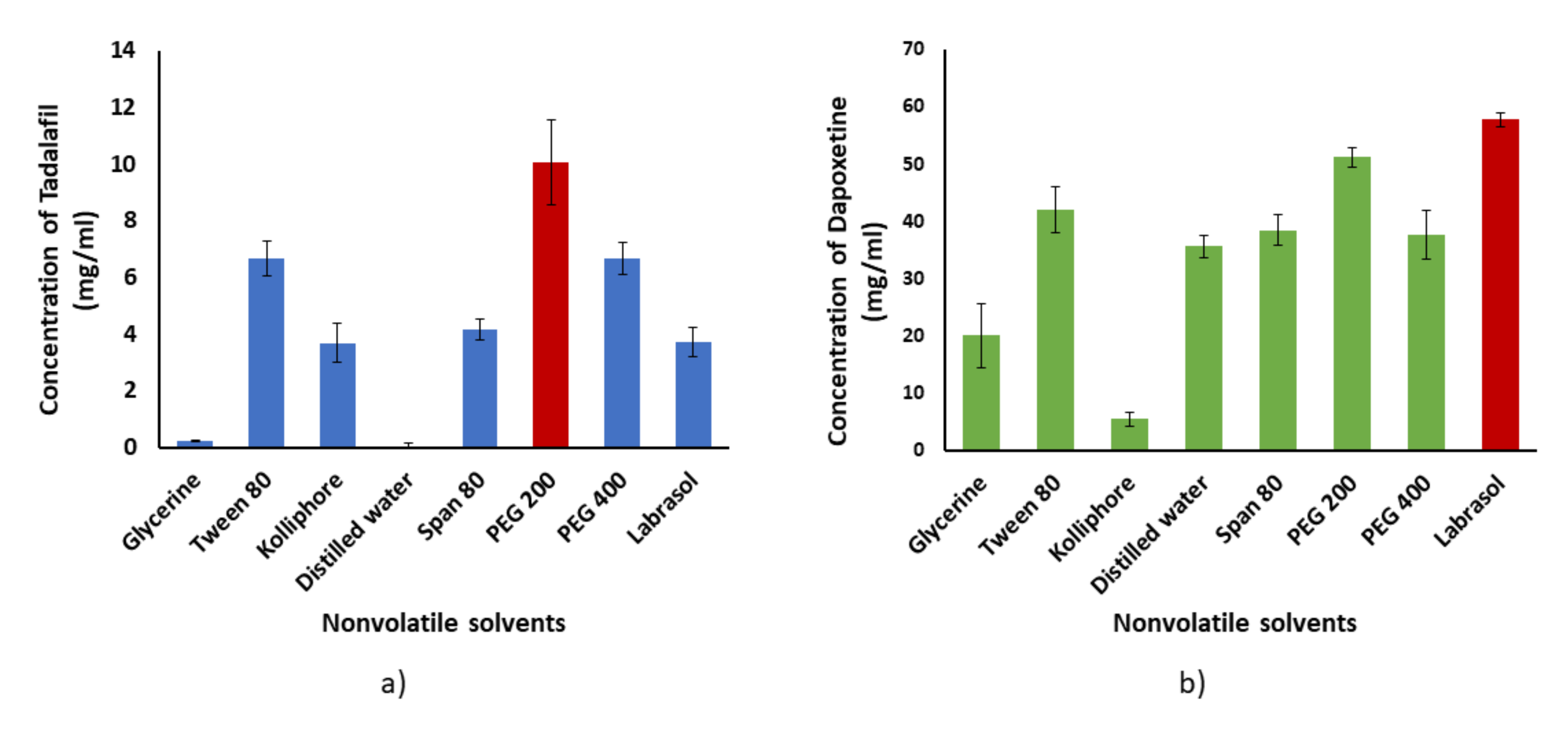
3.1.1. Solubility Study
3.1.2. Holding Capacity and Determination of Liquid Load Factor (Lf)
3.1.3. Solid-State Characterization Studies
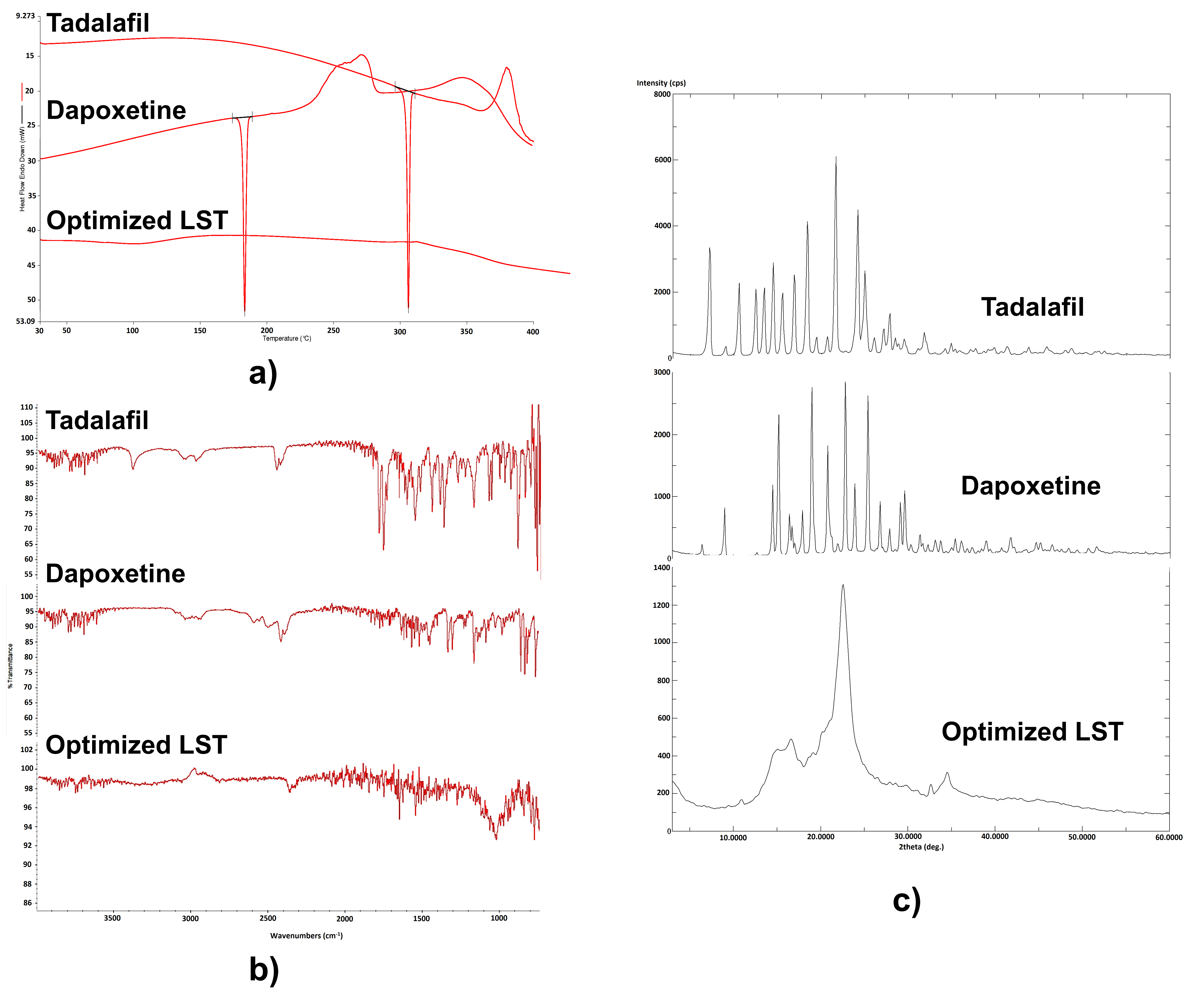
Differential Scanning Calorimetry (DSC)
Fourier Transform Infrared Spectroscopy (FTIR)
Powder X-ray Diffraction (PXRD)
3.2. Formulation and Evaluation of the Liquisolid Tablets
3.2.1. Pre-Compression Evaluation
3.2.2. Post-Compression Evaluation
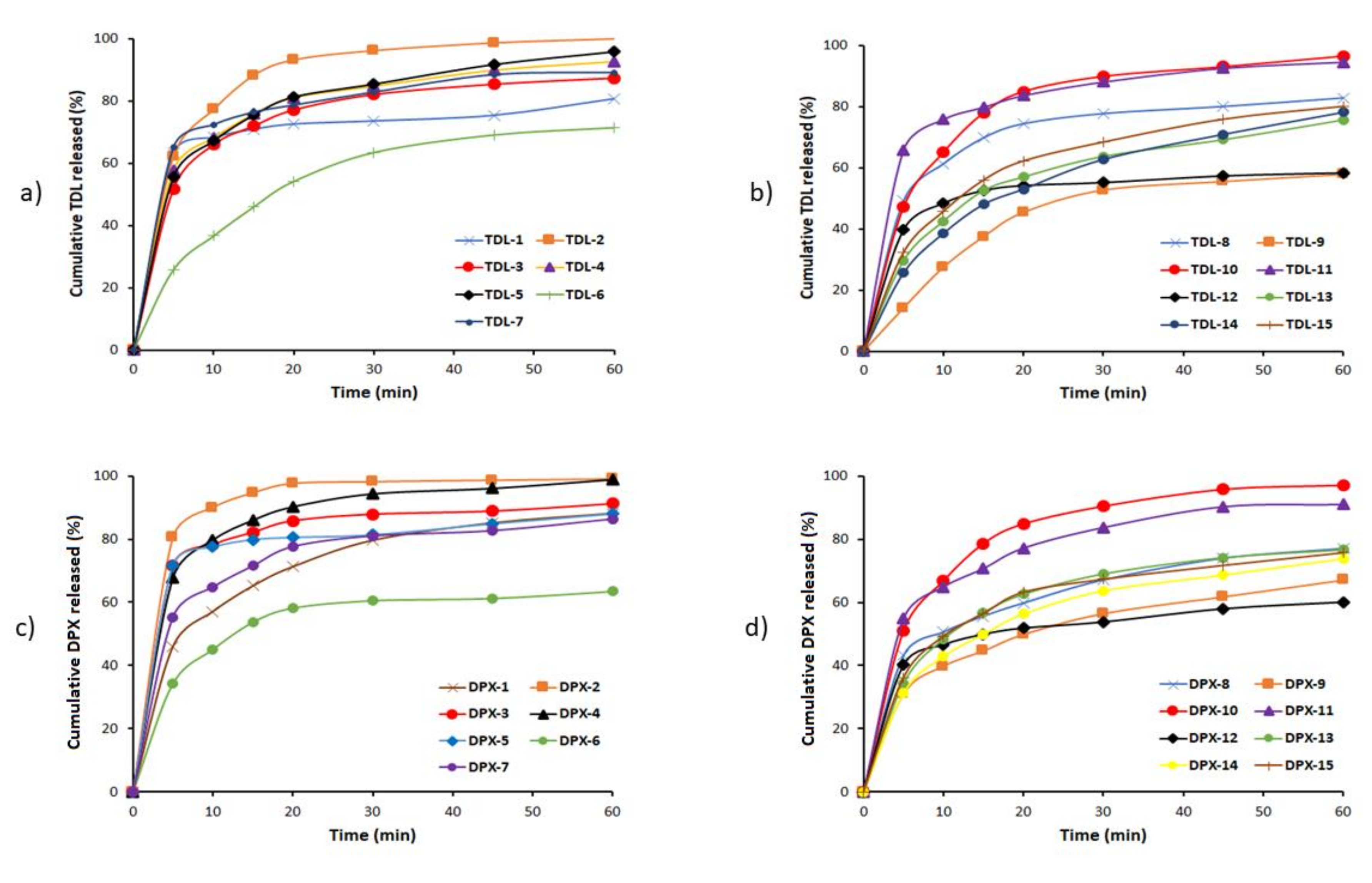
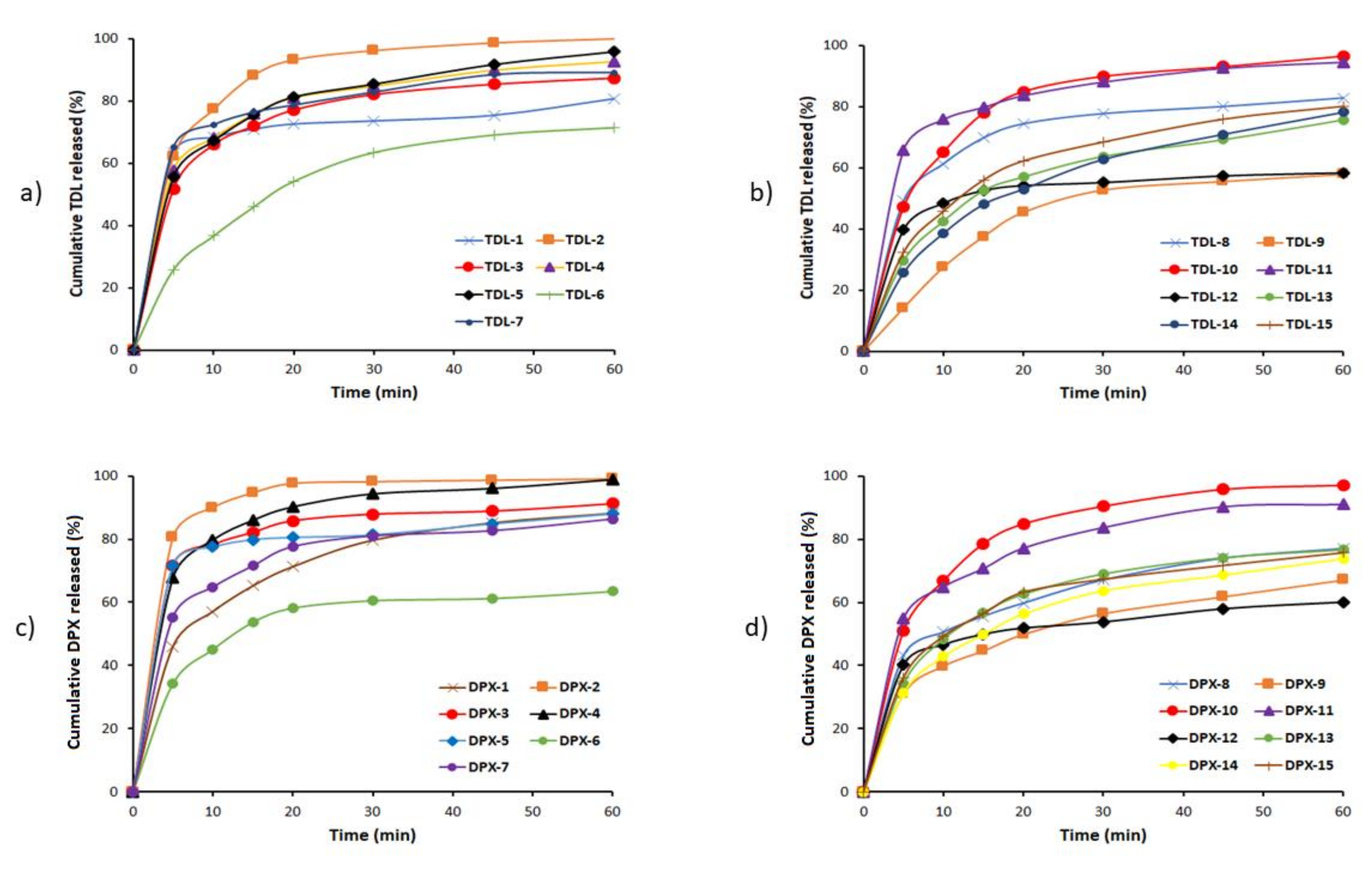
3.2.3. In-Vitro Dissolution Studies
3.2.4. Mathematical Modeling of the Release Data
3.3. Response Surface Methodology for Optimization of the Formulation
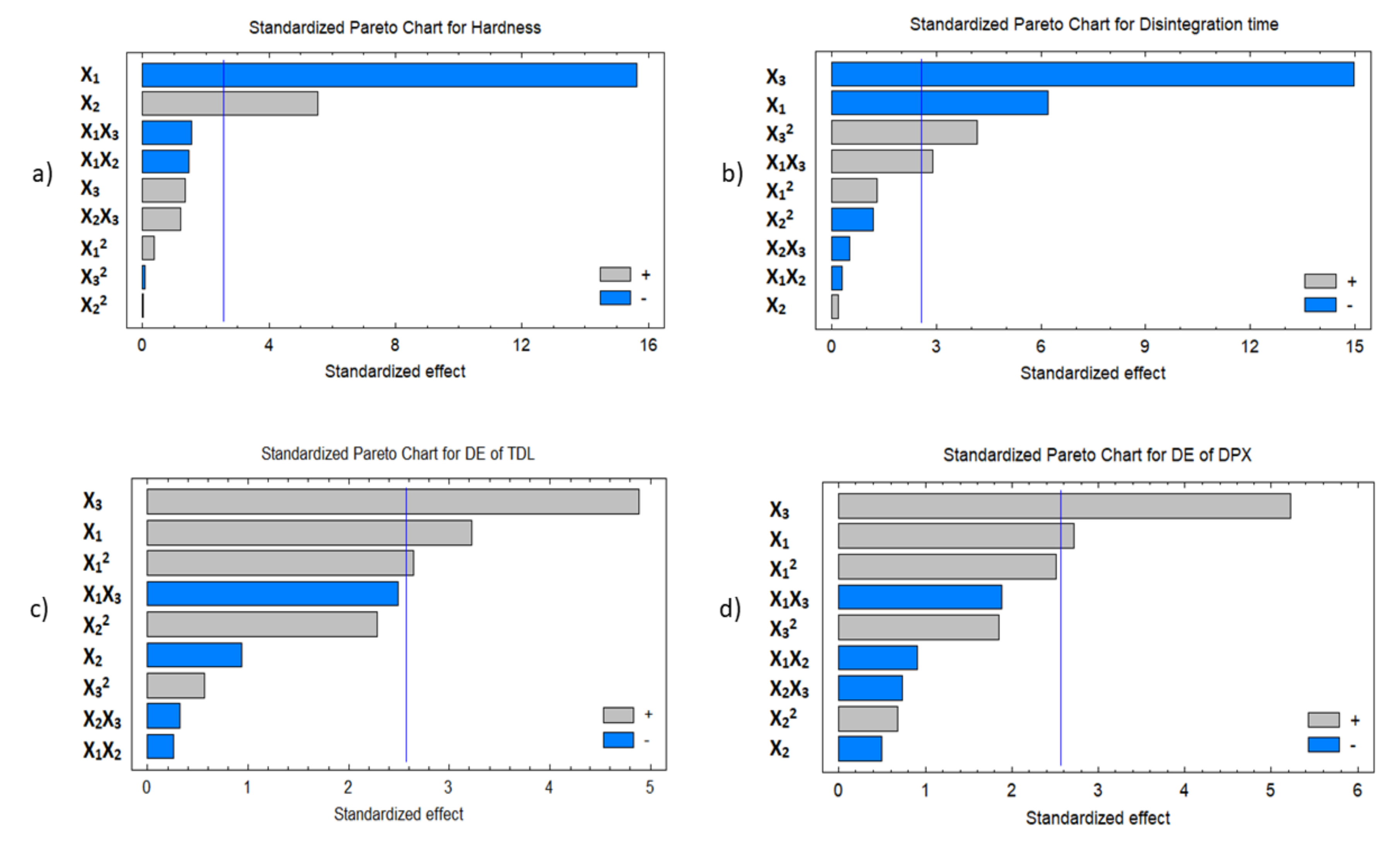
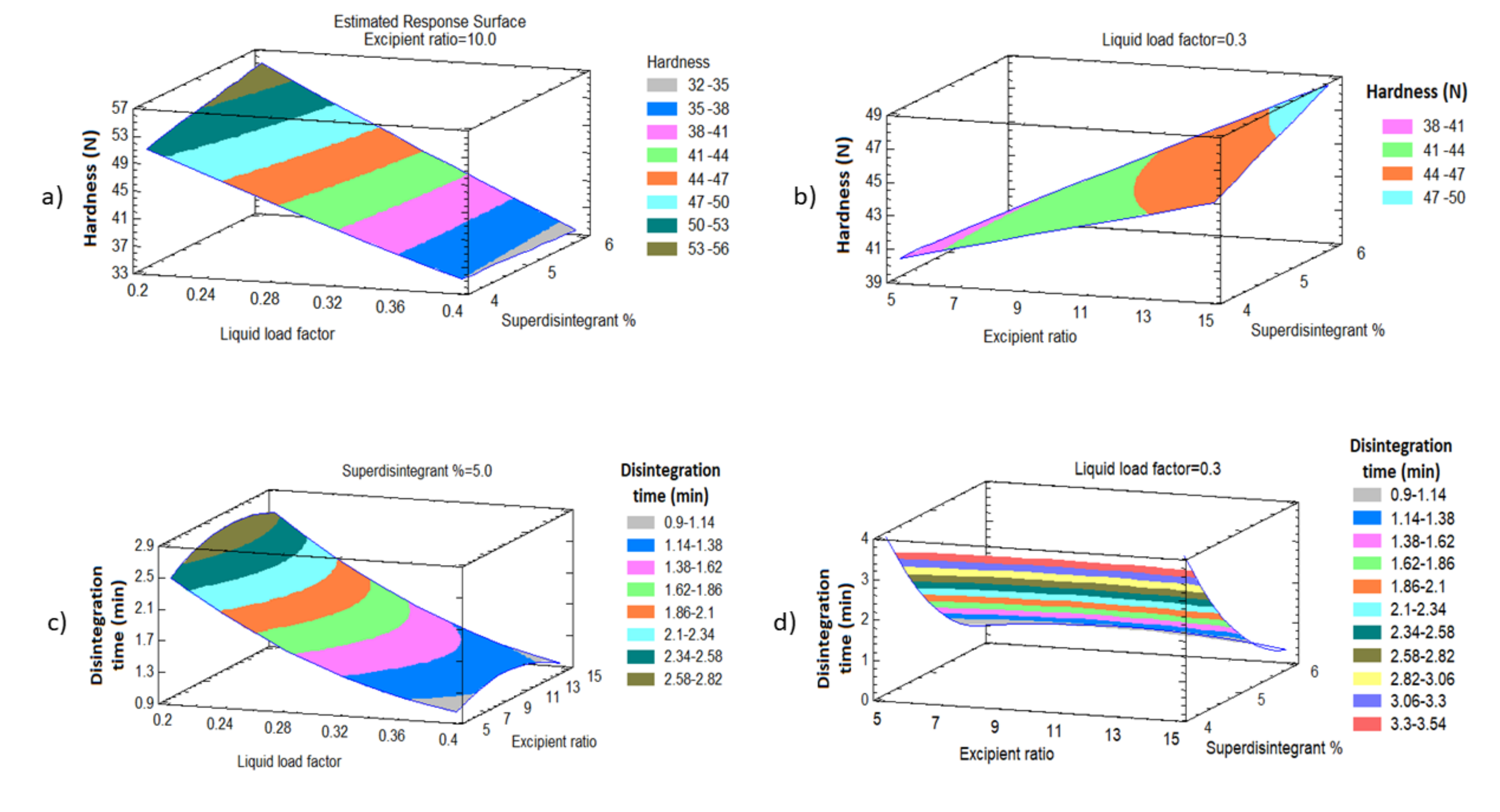
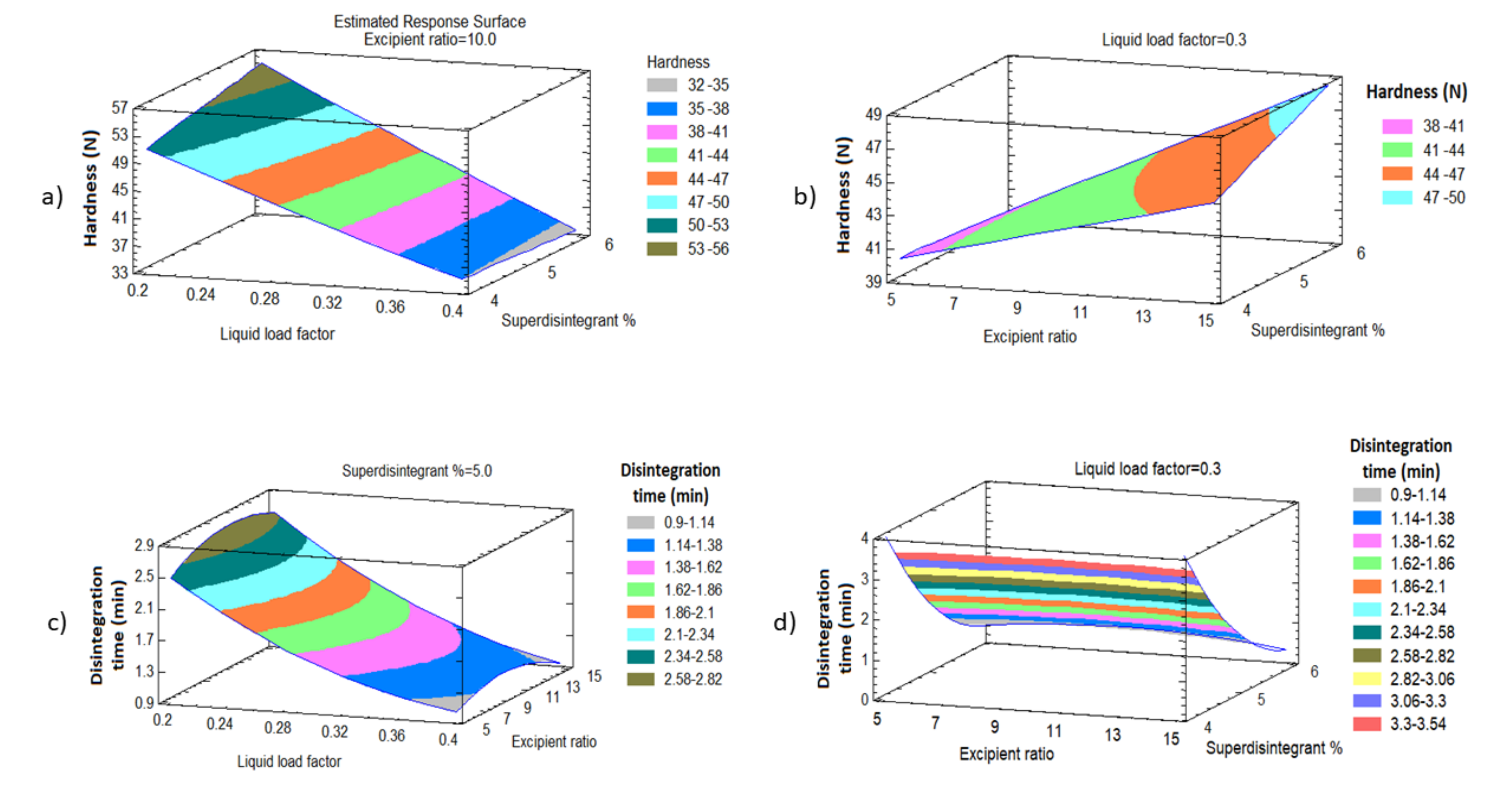
3.3.1. Effect of the Independent Variables on the Tablet Hardness (Y1)
3.3.2. Effect of the Independent Variables on Tablet Disintegration (Y2)
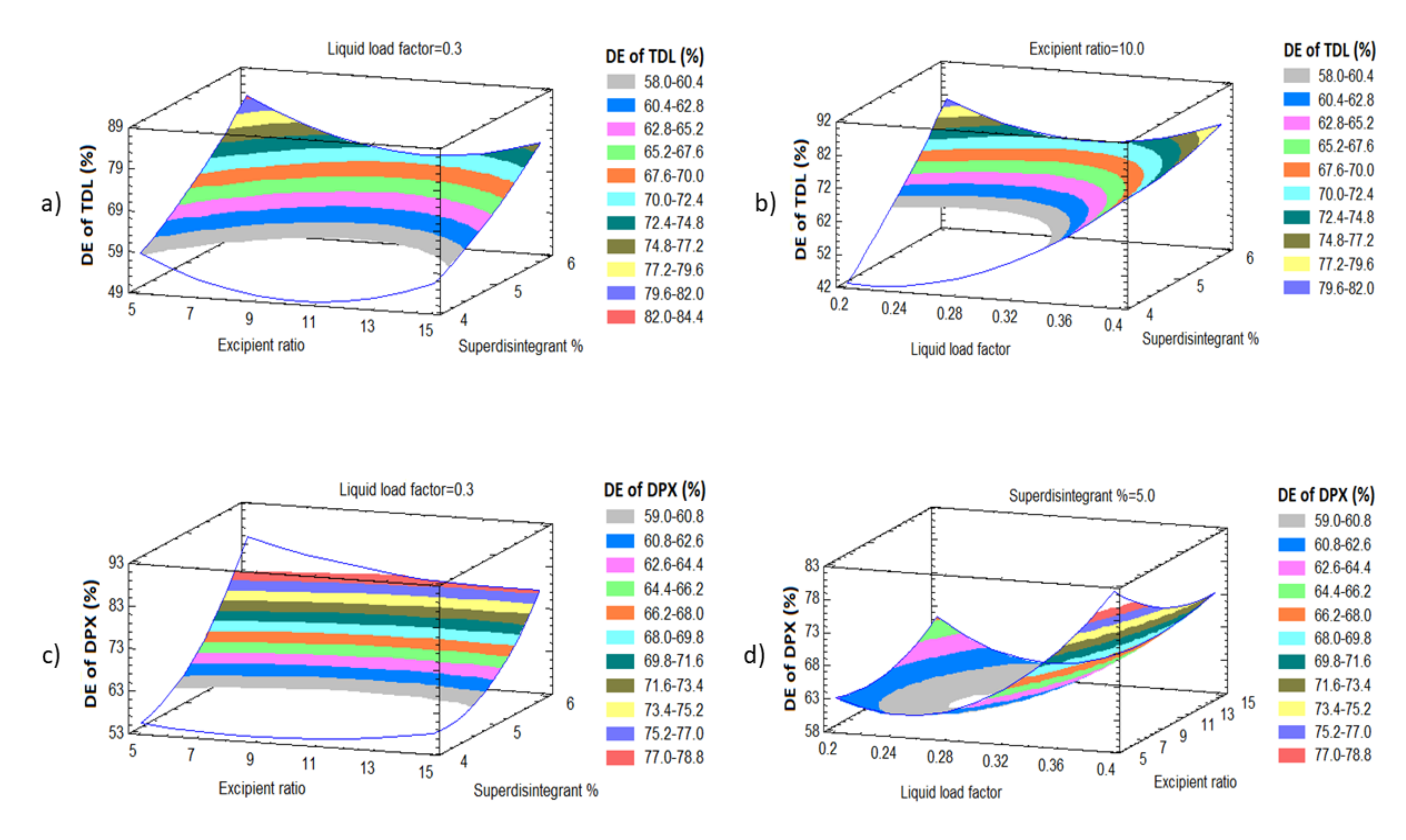
3.3.3. Effect of Independent Variables on the Dissolution Efficiency of TDL and DPX (Y3 and Y4)
3.4. Prediction of the Optimized Liquisolid Formulation
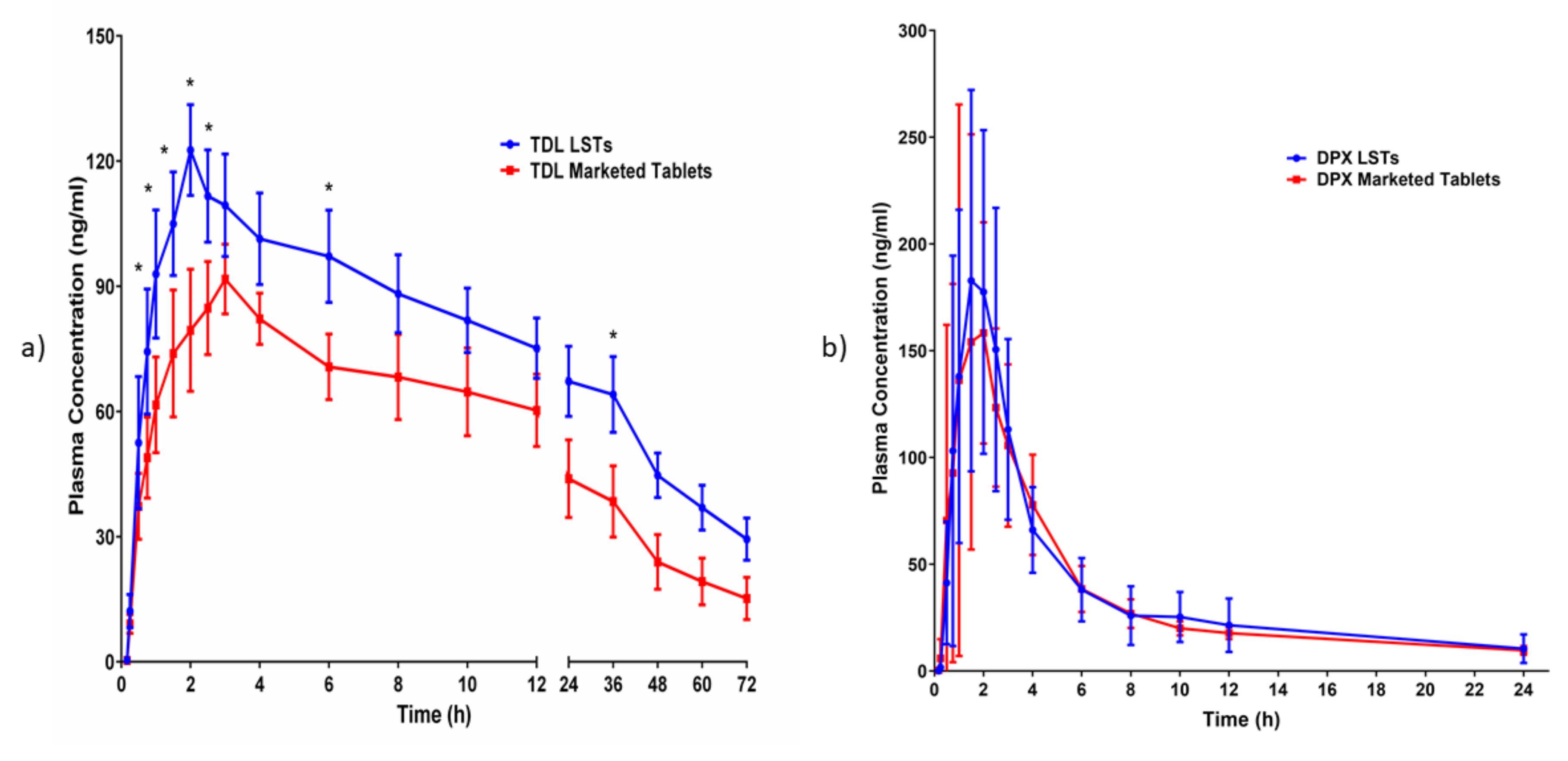
3.5. In-Vivo Pharmacokinetics Evaluation on Human Volunteers
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Avasthi, A.; Biswas, P. Pharmacotherapy of sexual dysfunctions: Current status. Indian J. Psychiatry 2004, 46, 213–220. [Google Scholar]
- Gao, J.; Zhang, X.; Su, P.; Liu, J.; Xia, L.; Yang, J.; Shi, K.; Tang, D.; Hao, Z.; Zhou, J.; et al. Prevalence and factors associated with the complaint of premature ejaculation and the four premature ejaculation syndromes: A large observational study in China. J. Sex. Med. 2013, 10, 1874–1881. [Google Scholar] [CrossRef]
- Serefoglu, E.C.; Yaman, O.; Cayan, S.; Asci, R.; Orhan, I.; Usta, M.F.; Ekmekcioglu, O.; Kendirci, M.; Semerci, B.; Kadioglu, A. Prevalence of the complaint of ejaculating prematurely and the four premature ejaculation syndromes: Results from the turkish society of andrology sexual health survey. J. Sex. Med. 2011, 8, 540–548. [Google Scholar] [CrossRef]
- Sangkum, P.; Badr, R.; Serefoglu, E.C.; Hellstrom, W.J.G. Dapoxetine and the treatment of premature ejaculation. Transl. Androl. Urol. 2013, 2, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.; Martyn-St James, M.; Kaltenthaler, E.; Dickinson, K.; Cantrell, A.; Wylie, K.; Frodsham, L.; Hood, C. Behavioral therapies for management of premature ejaculation: A systematic review. Sex. Med. 2015, 3, 174–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Hong, C.; Ren, L.L.; Yu, H.; Qiang, W. The role of dapoxetine hydrochloride on-demand for the treatment of men with premature ejaculation. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramezani, M.A.; Ahmadi, K.; Ghaemmaghami, A.; Marzabadi, E.A.; Pardakhti, F. Epidemiology of sexual dysfunction in iran: A systematic review and meta-analysis. Int. J. Prev. Med. 2015, 6. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, B.; Liu, D.-F.; Wang, X.-F.; Zhu, J.-C.; Jin, J.; Jiang, H. Medical management of erectile dysfunction in aging males: Is it too late to treat? Asian J. Androl. 2014, 16, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Rastrelli, G.; Cipriani, S.; Corona, G.; Vignozzi, L.; Maggi, M. Clinical characteristics of men complaining of premature ejaculation together with erectile dysfunction: A cross-sectional study. Andrology 2019, 7, 163–171. [Google Scholar] [CrossRef]
- Corona, G.; Rastrelli, G.; Limoncin, E.; Sforza, A.; Jannini, E.A.; Maggi, M. Interplay between premature ejaculation and erectile dysfunction: A Systematic review and Meta-analysis. J. Sex. Med. 2015, 12, 2291–2300. [Google Scholar] [CrossRef]
- Malavige, L.S.; Jayaratne, S.D.; Kathriarachchi, S.T.; Sivayogan, S.; Fernando, D.J.; Levy, J.C. Erectile dysfunction among men with diabetes is strongly associated with premature ejaculation and reduced libido. J. Sex. Med. 2008, 5, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.-K.; Chiang, P.-K.; Lu, C.-C.; Jiann, B.-P. The comorbidity between premature ejaculation and erectile dysfunction—A cross-sectional internet survey. Sex. Med. 2019, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, K.E. PDE5 inhibitors—Pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 2018, 175, 2554–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, D.; Wu, J.; Fan, X.; Dong, Q. Dapoxetine for the treatment of premature ejaculation: A meta-analysis of randomized controlled trials with trial sequential analysis. Ann. Saudi Med. 2018, 38, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Althof, S.E.; Abdo, C.H.N.; Dean, J.; Hackett, G.; McCabe, M.; McMahon, C.G.; Rosen, R.C.; Sadovsky, R.; Waldinger, M.; Becher, E.; et al. International Society for Sexual Medicine’s Guidelines for the Diagnosis and Treatment of Premature Ejaculation. J. Sex. Med. 2010, 7, 2947–2969. [Google Scholar] [CrossRef]
- Park, H.J.; Park, N.C.; Kim, T.N.; Baek, S.R.; Lee, K.M.; Choe, S. Discontinuation of dapoxetine treatment in patients with premature ejaculation: A 2-year prospective observational study. Sex. Med. 2017, 5, e99–e105. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.K.; Lee, S.H.; Cho, S.T.; Lee, Y.S.; Oh, C.Y.; Yoo, C.; Cho, J.S.; Lee, S.K.; Yang, D.Y. Comparison between on-demand dosing of dapoxetine alone and dapoxetine plus mirodenafil in patients with lifelong premature ejaculation: Prospective, randomized, double-blind, placebo-controlled, multicenter study. J. Sex. Med. 2013, 10, 2832–2841. [Google Scholar] [CrossRef]
- Bai, Y.; Pu, C.; Han, P.; Li, J.; Yuan, H.; Tang, Y.; Wang, X.; Wei, Q. Selective serotonin reuptake inhibitors plus phosphodiesterase-5 inhibitors for premature ejaculation: A systematic review and Meta-analysis. Urology 2015, 86, 758–765. [Google Scholar] [CrossRef]
- Mcmahon, C.G.; Giuliano, F.; Dean, J.; Hellstrom, W.J.G.; Bull, S.; Tesfaye, F.; Sharma, O.; Rivas, D.A.; Aquilina, J.W. Efficacy and safety of dapoxetine in men with premature ejaculation and concomitant erectile dysfunction treated with a phosphodiesterase type 5 inhibitor: Randomized, placebo-controlled, phase III study. J. Sex. Med. 2013, 10, 2312–2325. [Google Scholar] [CrossRef]
- Dresser, M.J.; Desai, D.; Gidwani, S.; Seftel, A.D.; Modi, N.B. Dapoxetine, a novel treatment for premature ejaculation, does not have pharmacokinetic interactions with phosphodiesterase-5 inhibitors. Int. J. Impot. Res. 2006, 18, 104–110. [Google Scholar] [CrossRef]
- Badr-Eldin, S.M.; Elkheshen, S.A.; Ghorab, M.M. Improving tadalafil dissolution via surfactant-enriched tablets approach: Statistical optimization, characterization, and pharmacokinetic assessment. J. Drug Deliv. Sci. Technol. 2017, 41, 197–205. [Google Scholar] [CrossRef]
- Badr-Eldin, S.M.; Elkheshen, S.A.; Ghorab, M.M. Inclusion complexes of tadalafil with natural and chemically modified beta-cyclodextrins. I: Preparation and in-vitro evaluation. Eur. J. Pharm. Biopharm. 2008, 70, 819–827. [Google Scholar] [CrossRef] [PubMed]
- El-Say, K.M.; Ahmed, O.A.A.; Aldawsari, H.M.; Badr-Eldin, S.M. Influence of different variables on the dissolution behavior of carvedilol from liquisolid compacts using response surface methodology. Dig. J. Nanomater. Biostructures 2019, 14, 879–894. [Google Scholar]
- Pandya, P.; Gattani, S.; Jain, P.; Khirwal, L.; Surana, S. Co-solvent evaporation method for enhancement of solubility and dissolution rate of poorly aqueous soluble drug simvastatin: In vitro-in vivo evaluation. AAPS PharmSciTech 2008, 9, 1247–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjay, P.D.; Deepak, M.; Bhanudas, S.R. Liquisolid technology: Technique for formulation with enhanced bioavailability. WORLD J. Pharm. Pharm. Sci. 2013, 3, 368–387. [Google Scholar]
- Patel, M.; Shah, A.; Patel, N.M.; Patel, M.R.; Patel, K.R. Nano suspension: A novel approach for drug delivery system. J. Pharm. Sci. Biosci. Res. 2011, 1, 1–10. [Google Scholar]
- Keck, C.M.; Müller, R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur. J. Pharm. Biopharm. 2006, 62, 3–16. [Google Scholar] [CrossRef]
- Erion, M.D.; van Poelje, P.D.; MacKenna, D.A.; Colby, T.J.; Montag, A.C.; Fujitaki, J.M.; Linemeyer, D.L.; Bullough, D.A. Liver-targeted drug delivery using hepdirect prodrugs. J. Pharmacol. Exp. Ther. 2005, 312, 554–560. [Google Scholar] [CrossRef]
- El-Say, K.M.; El-Sawy, H.S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar] [CrossRef]
- El-Say, K.M.; Ahmed, T.A.; Ahmed, O.A.A.; Hosny, K.M.; Abd-Allah, F.I. Self-nanoemulsifying lyophilized tablets for flash oral transmucosal delivery of vitamin K: Development and clinical evaluation. J. Pharm. Sci. 2017, 106, 2447–2456. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.A.; El-Say, K.M.; Hosny, K.M.; Aljaeid, B.M. Development of optimized self-nanoemulsifying lyophilized tablets (SNELTs) to improve finasteride clinical pharmacokinetic behavior. Drug Dev. Ind. Pharm. 2018, 44, 652–661. [Google Scholar] [CrossRef] [PubMed]
- El-Sayyad, N.M.E.-M.; Badawi, A.; Abdullah, M.E.; Abdelmalak, N.S. Dissolution enhancement of leflunomide incorporating self emulsifying drug delivery systems and liquisolid concepts. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 53–62. [Google Scholar] [CrossRef]
- Al-Subaie, M.M.; Hosny, K.M.; El-Say, K.M.; Ahmed, T.A.; Aljaeid, B.M. Utilization of nanotechnology to enhance percutaneous absorption of acyclovir in the treatment of herpes simplex viral infections. Int. J. Nanomed. 2015, 10, 3973–3985. [Google Scholar] [CrossRef] [Green Version]
- Aldawsari, H.M.; Elfaky, M.A.; Fahmy, U.A.; Aljaeid, B.M.; Alshareef, O.A.; El-Say, K.M. Development of a fluvastatin-loaded self-nanoemulsifying system to maximize therapeutic efficacy in human colorectal carcinoma cells. J. Drug Deliv. Sci. Technol. 2018, 46, 7–13. [Google Scholar] [CrossRef]
- Hausner, N.H. Flow properties of some pharmaceutical powders. Int. J. Powder Met. 1967, 3, 7–11. [Google Scholar]
- Carr, R. Evaluating flow properties of solids. Chem. Eng. 1965, 72, 163. [Google Scholar]
- The United States Pharmacopeial Convention. Uniformity of Dosage Units. Stage 6 Harmonization (905); The United States Pharmacopeial Convention: Silver Spring, MD, USA, 2011. [Google Scholar]
- Wagner, J.G. Interpretation of percent dissolved-time plots derived from in vitro testing of conventional tablets and capsules. J. Pharm. Sci. 1969, 58, 1253–1257. [Google Scholar] [CrossRef]
- Desai, S.; Singh, P.; Simonelli, A.; Higuchi, W. Investigation of factors influencing release of solid drug dispersed in inert matrices III. Quantitative studies involving the polyethylene plastic matrix. J. Pharm. Sci. 1966, 55, 1230–1234. [Google Scholar] [CrossRef]
- Langenbucher, F. Linearization of dissolution rate curves by the Weibull distribution. J. Pharm. Pharmacol. 1972, 24, 979–981. [Google Scholar] [CrossRef]
- Hixson, A.W.; Crowell, J.H. Dependence of reaction velocity upon surface and agitation. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Korsmeyer, R.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N. Mechanisms of potassium chloride release from compressed, hydrophilic, polymeric matrices: Effect of entrapped air. J. Pharm. Sci. 1983, 15, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.W.; Lonsdale, H.S. Controlled release: Mechanisms and release. In Controlled Release of Biological Active Agents; Taquary, A.C., Lacey, R.E., Eds.; Plenum Press: New York, NY, USA, 1974; pp. 15–71. [Google Scholar]
- Saeedi, M.; Akbari, J.; Morteza-Semnani, K.; Enayati-Fard, R.; Sar-Reshteh-Dar, S.; Soleymani, A. Enhancement of dissolution rate of indomethacin using liquisolid compacts. Iran. J. Pharm. Res. 2011, 10, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Nokhodchi, A.; Javadzadeh, Y.; Siahi-Shadbad, M.R.; Barzegar-Jalali, M. The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts. J. Pharm. Pharm. Sci. 2005, 8, 18–25. [Google Scholar]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Bernal, N.P.; Calpena, A.C.; Mallandrich, M.; Ruiz, A.; Clares, B. Development, physical-chemical stability, and release studies of four alcohol-free spironolactone suspensions for use in pediatrics. Dissolution Technol. 2014, 21, 19–30. [Google Scholar] [CrossRef]
- Chella, N.; Narra, N.; Rao, T.R. Preparation and characterization of liquisolid compacts for improved dissolution of telmisartan. J. Drug Deliv. 2014. [Google Scholar] [CrossRef]
- Ahuja, N.; Katare, O.P.; Singh, B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur. J. Pharm. Biopharm. 2007, 65, 26–38. [Google Scholar] [CrossRef]
- Staniforth, J.N.; Aulton, M.E. Powder flow. In Aulton Pharmaceutics: The Design and Manufacture of Medicines; Aulton, M.E., Ed.; Churchill Living Stone Elservier: London, UK, 2007; pp. 168–180. [Google Scholar]
- Zimmermann, I.; Eber, M.; Meyer, K. Nanomaterials as flow regulators in dry powders. Z. Phys. Chem. 2004, 218, 51–102. [Google Scholar] [CrossRef]
- Meyer, K.; Zimmermann, I. Effect of glidants in binary powder mixtures. Powder Technol. 2004, 139, 40–54. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, H.; Song, W.; Gu, D.; Hu, Q. Investigation of inclusion complex of cilnidipine with hydroxypropyl-β-cyclodextrin. Carbohydr. Polym. 2012, 90, 1719–1724. [Google Scholar] [CrossRef] [PubMed]
- Mateescu, C.; Popescu, A.M.; Radu, G.L.; Onisei, T.; Raducanu, A.E. Spectroscopic and spectrometric methods used for the screening of certain herbal food supplements suspected of adulteration. Adv. Pharm. Bull. 2017, 7, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanka, K.; Poienti, S.; Mohd, A.; Diwan, P. Improved oral delivery of clonazepam through liquisolid powder compact formulations: In-vitro and ex-vivo characterization. Powder Technol. 2014, 256, 336–344. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Ahmed, O.A.A.; Balata, G.F.; Almalki, W.H. Self-assembled biodegradable polymeric micelles to improve dapoxetine delivery across the blood–brain barrier. Int. J. Nanomed. 2018, 13, 3679–3687. [Google Scholar] [CrossRef] [Green Version]
- The United States Pharmacopeia, T.N.F. USP 28/NF 23; US Pharmacopoeial Convention Inc.: Rockville, MD, USA, 2005. [Google Scholar]
- Javadzadeh, Y.; Musaalrezaei, L.; Nokhodchi, A. Liquisolid technique as a new approach to sustain propranolol hydrochloride release from tablet matrices. Int. J. Pharm. 2008, 362, 102–108. [Google Scholar] [CrossRef]
- Patel, D.S.; Pipaliya, R.M.; Surti, N. Liquisolid tablets for dissolution enhancement of a hypolipidemic drug. Indian J. Pharm. Sci. 2015, 77, 290–298. [Google Scholar] [CrossRef] [Green Version]
- Sousa Lobo, J.M.; Costa, P. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Bushra, R.; Shoaib, M.H.; Ali, H.; Zafar, F.; Naeem, M.I.; Aslam, N.; Yousuf, R.I. Formulation design and Optimization of Aceclofenac Tablets (100 mg) using central composite design with response surface methodology. Lat. Am. J. Pharm. 2014, 33, 1009–1018. [Google Scholar]
- Elkordy, A.A.; Tan, X.N.; Essa, E.A. Spironolactone release from liquisolid formulations prepared with CapryolTM 90, Solutol® HS-15 and Kollicoat® SR 30 D as non-volatile liquid vehicles. Eur. J. Pharm. Biopharm. 2013, 83, 203–223. [Google Scholar] [CrossRef]
- Kornblum, S.S.; Stoopak, S.B. A new tablet disintegrating agent: Cross-linked polyvinylpyrrolidone. J. Pharm. Sci. 1973, 62, 43–49. [Google Scholar] [CrossRef]
- Yen, S.-Y.; Chen, C.-R.; Lee, M.-T.; Chen, L.-C. Investigation of dissolution enhancement of nifedipine by deposition on superdisintegrants. Drug Dev. Ind. Pharm. 1997, 23, 313–317. [Google Scholar] [CrossRef]
- El-Say, K.M.; Samy, A.M.; Fetouh, M.I.; Fetouh, I. Formulation and evaluation of rofecoxib liquisolid tablets. Int. J. Pharm. Sci. Rev. Res. 2010, 3, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Fahmy, R.; Kassem, M. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2008, 69, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Srinivasan, K.K.; Gowthamarajan, K.; Prakash, D.; Gaikwad, N.B.; Singare, D.S. Influence of formulation parameters on dissolution rate enhancement of glyburide using liquisolid technique. Drug Dev. Ind. Pharm. 2012, 38, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Asif, M.; Kumar, A.; Aggarval, A.; Pharmacy, D.J.C. BCS journal1.pdf. Int. J. PharmTech Res. 2010, 2, 1681–1690. [Google Scholar]
- Arzani, G.; Haeri, A.; Daeihamed, M.; Bakhtiari-Kaboutaraki, H.; Dadashzadeh, S. Niosomal carriers enhance oral bioavailability of carvedilol: Effects of bile salt-enriched vesicles and carrier surface charge. Int. J. Nanomed. 2015, 10, 4797–4813. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula Code | Avicel, Q | Silica, q | Liquid Medication, W | TDL | DPX | Methocel 3% | Magnesium Trisilicate 5% | Polyplasdone XL-10 | |
|---|---|---|---|---|---|---|---|---|---|
| PEG 200 | Labrasol | ||||||||
| (g) | |||||||||
| LS-1 | 5 | 0.3 | 0.795 | 0.795 | 0.1 | 0.6 | 0.22 | 0.379 | 0.455 |
| LS-2 | 5 | 0.3 | 1.06 | 1.06 | 0.1 | 0.6 | 0.24 | 0.406 | 0.406 |
| LS-3 | 5 | 0.3 | 0.53 | 0.53 | 0.1 | 0.6 | 0.24 | 0.353 | 0.353 |
| LS-4 | 5 | 0.5 | 0.55 | 0.55 | 0.1 | 0.6 | 0.21 | 0.365 | 0.438 |
| LS-5 | 5 | 1.0 | 1.2 | 1.2 | 0.1 | 0.6 | 0.27 | 0.455 | 0.455 |
| LS-6 | 5 | 0.5 | 1.1 | 1.1 | 0.1 | 0.6 | 0.27 | 0.420 | 0.504 |
| LS-7 | 5 | 0.5 | 1.1 | 1.1 | 0.1 | 0.6 | 0.25 | 0.420 | 0.336 |
| LS-8 | 5 | 1.0 | 0.9 | 0.9 | 0.1 | 0.6 | 0.25 | 0.425 | 0.510 |
| LS-9 | 5 | 1.0 | 0.9 | 0.9 | 0.1 | 0.6 | 0.25 | 0.425 | 0.340 |
| LS-10 | 5 | 1.0 | 0.795 | 0.795 | 0.1 | 0.6 | 0.24 | 0.414 | 0.331 |
| LS-11 | 5 | 1.0 | 0.55 | 0.55 | 0.1 | 0.6 | 0.23 | 0.390 | 0.312 |
| LS-12 | 5 | 1.0 | 0.6 | 0.6 | 0.1 | 0.6 | 0.23 | 0.395 | 0.395 |
| LS-13 | 5 | 0.5 | 0.825 | 0.825 | 0.1 | 0.6 | 0.23 | 0.392 | 0.392 |
| LS-14 | 5 | 0.5 | 0.825 | 0.825 | 0.1 | 0.6 | 0.23 | 0.392 | 0.392 |
| LS-15 | 5 | 0.5 | 0.825 | 0.825 | 0.1 | 0.6 | 0.23 | 0.392 | 0.392 |
| Formula Code | Pre-Compression Properties | Post-Compression Properties | ||||||||
| Hausner Ratio | Carr’s Index | Angle of Repose | Type of Flow | Friability (%) | Hardness (n) | Weight (mg) | Disintegration Time (min) | Content of TDL% | Content of DPX% | |
| LS-1 | 1.1 | 14.5 | 29 | Excellent | 0.461 | 58.02 | 200 | 2.54 | 95.2 | 95.8 |
| LS-2 | 1.2 | 19.7 | 26 | Good | 0.151 | 38.91 | 227 | 0.78 | 95.9 | 97.6 |
| LS-3 | 1.09 | 8.78 | 21 | Excellent | 0.357 | 56.44 | 199 | 1.04 | 99.6 | 99.8 |
| LS-4 | 1.1 | 5.1 | 26 | Excellent | 0.356 | 35.28 | 207 | 0.26 | 101.1 | 99.9 |
| LS-5 | 1.1 | 10 | 30 | Excellent | 0.334 | 47.14 | 259 | 0.57 | 97.3 | 98.2 |
| LS-6 | 1.2 | 12 | 35 | Good | 0.300 | 41.85 | 239 | 3.86 | 97.8 | 100.1 |
| LS-7 | 1.18 | 15.4 | 30 | Excellent | 0.294 | 36.75 | 235 | 1.13 | 102.2 | 99.3 |
| LS-8 | 1.19 | 16 | 34 | Fair | 0.253 | 48.51 | 244 | 2.32 | 98.1 | 100.1 |
| LS-9 | 1.21 | 17.5 | 36 | Fair | 0.245 | 49.69 | 238 | 6.29 | 99.0 | 97.3 |
| LS-10 | 1.15 | 13.4 | 31 | Good | 0.268 | 33.81 | 234 | 3.46 | 103.2 | 95.6 |
| LS-11 | 1.31 | 25 | 41 | Passable | 0.219 | 32.14 | 220 | 1.12 | 95.6 | 95.5 |
| LS-12 | 1.2 | 20 | 34 | Fair | 0.193 | 45.96 | 226 | 4.02 | 98.8 | 98.6 |
| LS-13 | 1.17 | 14 | 34 | Good | 0.170 | 44.2 | 223 | 1.86 | 101.3 | 102.2 |
| LS-14 | 1.14 | 12.5 | 31 | Good | 0.164 | 43.81 | 237 | 1.62 | 102.1 | 99.9 |
| LS-15 | 1.3 | 25 | 41 | Passable | 0.086 | 42.63 | 222 | 1.79 | 100.1 | 99.6 |
| Formula Code | Independent Variables | Dependent Variables | |||||
|---|---|---|---|---|---|---|---|
| X1 | X2 | X3 | Y1 | Y2 | Y3 | Y4 | |
| LS-1 | 0.2 | 15.0 | 5.0 | 58.02 | 2.54 | 70.285 | 71.858 |
| LS-2 | 0.3 | 5.0 | 6.0 | 38.91 | 0.78 | 87.871 | 91.812 |
| LS-3 | 0.2 | 10.0 | 6.0 | 56.44 | 1.04 | 74.851 | 82.016 |
| LS-4 | 0.4 | 10.0 | 6.0 | 35.28 | 0.26 | 78.682 | 86.741 |
| LS-5 | 0.3 | 15.0 | 6.0 | 47.14 | 0.57 | 79.418 | 78.323 |
| LS-6 | 0.3 | 5.0 | 4.0 | 41.85 | 3.86 | 55.255 | 54.192 |
| LS-7 | 0.4 | 15.0 | 5.0 | 36.75 | 1.13 | 78.256 | 74.047 |
| LS-8 | 0.2 | 5.0 | 5.0 | 48.51 | 2.32 | 70.918 | 62.051 |
| LS-9 | 0.2 | 10.0 | 4.0 | 49.69 | 6.29 | 44.375 | 51.427 |
| LS-10 | 0.4 | 10.0 | 4.0 | 33.81 | 3.46 | 80.555 | 81.886 |
| LS-11 | 0.4 | 5.0 | 5.0 | 32.14 | 1.12 | 82.183 | 76.889 |
| LS-12 | 0.3 | 15.0 | 4.0 | 45.96 | 4.02 | 51.630 | 51.115 |
| LS-13 | 0.3 | 10.0 | 5.0 | 44.20 | 1.86 | 57.564 | 61.947 |
| LS-14 | 0.3 | 10.0 | 5.0 | 43.81 | 1.62 | 56.571 | 57.015 |
| LS-15 | 0.3 | 10.0 | 5.0 | 42.63 | 1.79 | 62.234 | 61.171 |
| Factors | Hardness (Y1) | Disintegration Time (Y2), min | Dissolution Efficiency for TDL (Y3), % | Dissolution Efficiency for DPX (Y4), % | ||||
|---|---|---|---|---|---|---|---|---|
| Estimate | p-Value | Estimate | p-Value | Estimate | p-Value | Estimate | p-Value | |
| X1 | −18.671 | 0.0001 * | −1.555 | 0.0016 * | 14.791 | 0.0233 * | 13.020 | 0.0417 * |
| X2 | 6.615 | 0.0026 * | 0.045 | 0.8643 | −4.302 | 0.3913 | −2.420 | 0.6344 |
| X3 | 1.615 | 0.2337 | −3.745 | 0.0001 * | 22.415 | 0.0045 * | 25.009 | 0.0034 * |
| X12 | 0.648 | 0.7271 | 0.476 | 0.2529 | 17.814 | 0.0460 * | 17.711 | 0.0535 |
| X1 X2 | −2.451 | 0.2061 | −0.105 | 0.7786 | −1.647 | 0.8096 | −6.158 | 0.4044 |
| X1 X 3 | −2.641 | 0.1783 | 1.025 | 0.0339 * | −16.133 | 0.0553 | −12.721 | 0.1188 |
| X22 | −0.031 | 0.9863 | −0.434 | 0.2915 | 15.427 | 0.0710 | 4.788 | 0.5267 |
| X2 X3 | 2.061 | 0.2764 | −0.185 | 0.6234 | −2.128 | 0.7560 | −4.991 | 0.4939 |
| X32 | −0.131 | 0.9431 | 1.536 | 0.0087 * | 3.794 | 0.5983 | 13.042 | 0.1232 |
| R2 | 98.269 | 98.318 | 91.366 | 90.764 | ||||
| Adj. R2 | 95.152 | 95.290 | 75.825 | 74.139 | ||||
| Independent Variables | Optimum | Dependent Variables | Predicted Values | Observed Values | Residuals |
|---|---|---|---|---|---|
| Liquid load factor | 0.2 | Hardness | 55.2 | 54.1 | 1.1 |
| Powder excipient ratio | 11.82 | Disintegration time | 2.7 | 2.8 | 0.1 |
| Superdisintegrant concentration | 5.11 | Dissolution efficiency for TDL | 66.4 | 68.6 | −2.2 |
| Dissolution efficiency for DPX | 74.5 | 77.2 | −2.7 |
| PK Parameter | Unit | TDL | DPX | ||
|---|---|---|---|---|---|
| LSTs | Marketed Tablets | LSTs | Marketed Tablets | ||
| Lambda_z | 1/h | 0.104 ± 0.096 | 0.051 ± 0.034 | 0.065 ± 0.010 | 0.051 ± 0.006 |
| t1/2 | h | 18.523 ± 22.737 | 17.544 ± 8.557 | 10.951 ± 1.879 | 11.683 ± 1.989 |
| Tmax | h | 2 ± 0 | 3.0 ± 0 | 1.667 ± 0.289 | 1.833 ± 0.289 |
| Cmax | ng/ml | 122.612 * ± 10.876 | 91.719 ± 8.347 | 186.154 ± 83.741 | 171.063 ± 71.830 |
| AUC0–t | ng/mL∙h | 4484.953 * ± 408.147 | 2994.611 ± 591.332 | 919.633 ± 397.978 | 794.699 ± 195.442 |
| AUC0–inf | ng/mL∙h | 5231.316 ± 1579.022 | 3066.42 ± 573.078 | 1096.416 ± 521.708 | 936.702 ± 170.519 |
| AUMC0–inf | ng/mL∙h2 | 241,586.7 ± 175,386.7 | 97,771.74 ± 20,029.83 | 13,201.782 ± 9197.529 | 10,844.661 ± 983.277 |
| MRT0–inf | h | 42.650 ± 17.812 | 31.878 ± 2.155 | 11.492 ± 3.034 | 11.915 ± 2.817 |
| Vz/F | (mg)/(ng/mL) | 0.021 ± 0.022 | 0.042 ± 0.023 | 0.504 ± 0.251 | 0.562 ± 0.185 |
| Cl/F | (mg)/(ng/mL)/h | 0.001 ± 0.0002 | 0.002 ± 0.0003 | 0.033 ± 0.018 | 0.033 ± 0.006 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alotaibi, F.O.; Alhakamy, N.A.; Omar, A.M.; El-Say, K.M. Clinical Pharmacokinetic Evaluation of Optimized Liquisolid Tablets as a Potential Therapy for Male Sexual Dysfunction. Pharmaceutics 2020, 12, 1187. https://doi.org/10.3390/pharmaceutics12121187
Alotaibi FO, Alhakamy NA, Omar AM, El-Say KM. Clinical Pharmacokinetic Evaluation of Optimized Liquisolid Tablets as a Potential Therapy for Male Sexual Dysfunction. Pharmaceutics. 2020; 12(12):1187. https://doi.org/10.3390/pharmaceutics12121187
Chicago/Turabian StyleAlotaibi, Fayez O., Nabil A. Alhakamy, Abdelsattar M. Omar, and Khalid M. El-Say. 2020. "Clinical Pharmacokinetic Evaluation of Optimized Liquisolid Tablets as a Potential Therapy for Male Sexual Dysfunction" Pharmaceutics 12, no. 12: 1187. https://doi.org/10.3390/pharmaceutics12121187
APA StyleAlotaibi, F. O., Alhakamy, N. A., Omar, A. M., & El-Say, K. M. (2020). Clinical Pharmacokinetic Evaluation of Optimized Liquisolid Tablets as a Potential Therapy for Male Sexual Dysfunction. Pharmaceutics, 12(12), 1187. https://doi.org/10.3390/pharmaceutics12121187