Optimization of Liposomes for Antigen Targeting to Splenic CD169+ Macrophages

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Liposome Preparation and Charactarisation
2.2. Analysis of Peptide Encapsulation
2.3. CD169 Fc ELISA
2.4. Liposome Binding to TSn Cells
2.5. Enzymatic Spleen Digestion
2.6. In Vitro Liposome Uptake
2.7. In Vivo Liposome Uptake
2.8. Fluorescent Microscopy
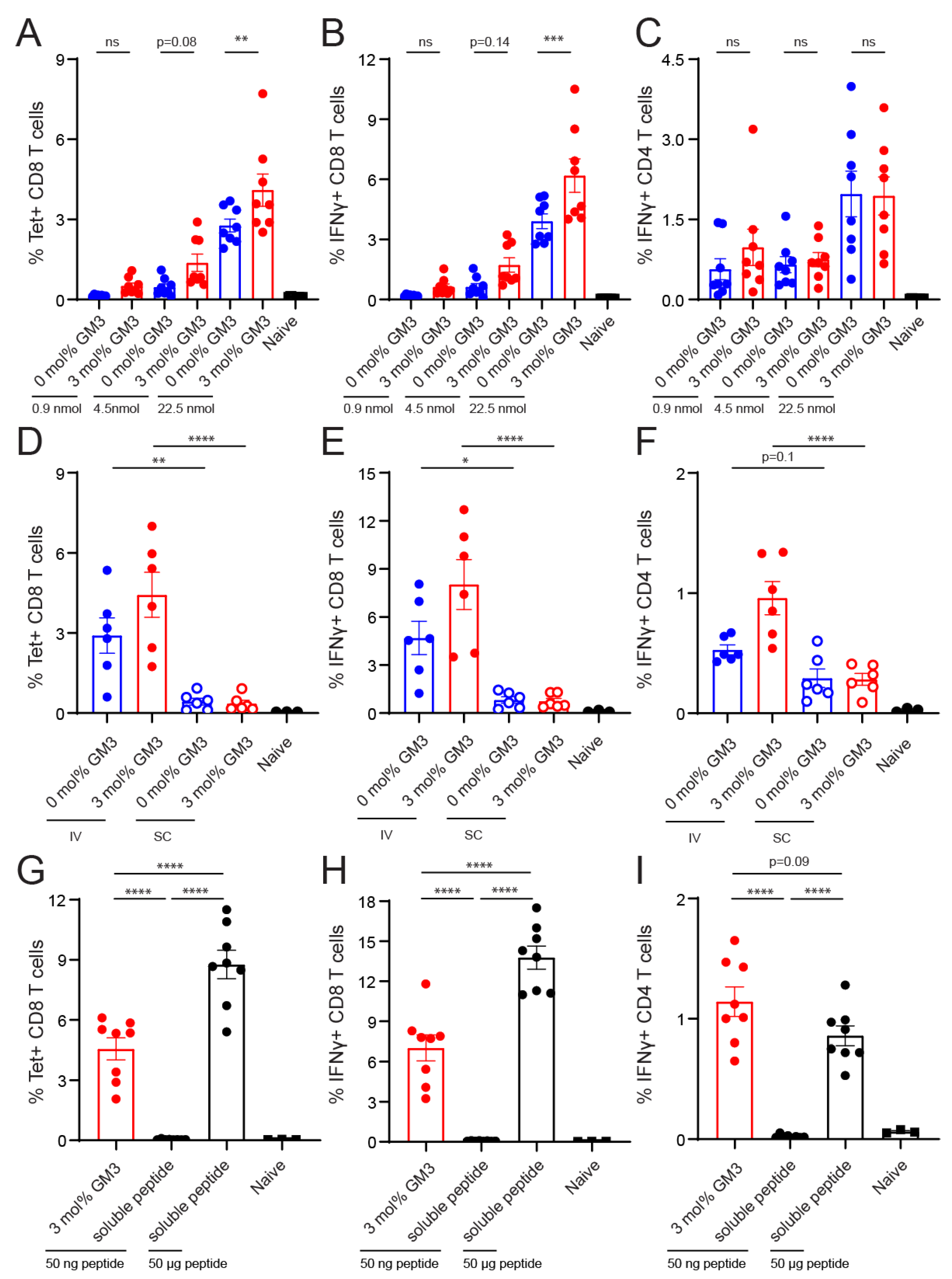
2.9. In Vivo T-Cell Priming
2.10. Flow Cytometry and Antibodies
2.11. Animal Experiments
2.12. Statistical Analysis
3. Results
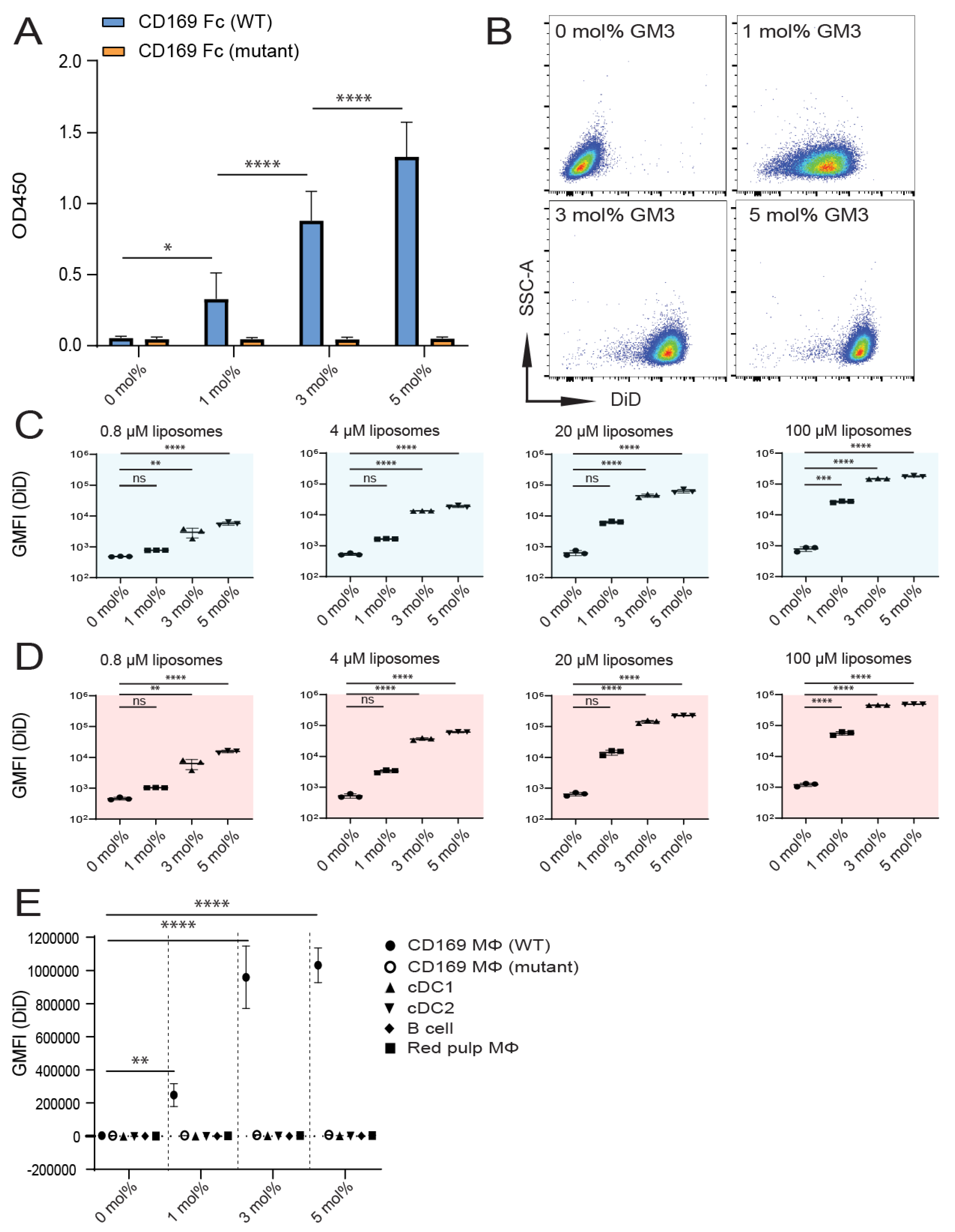
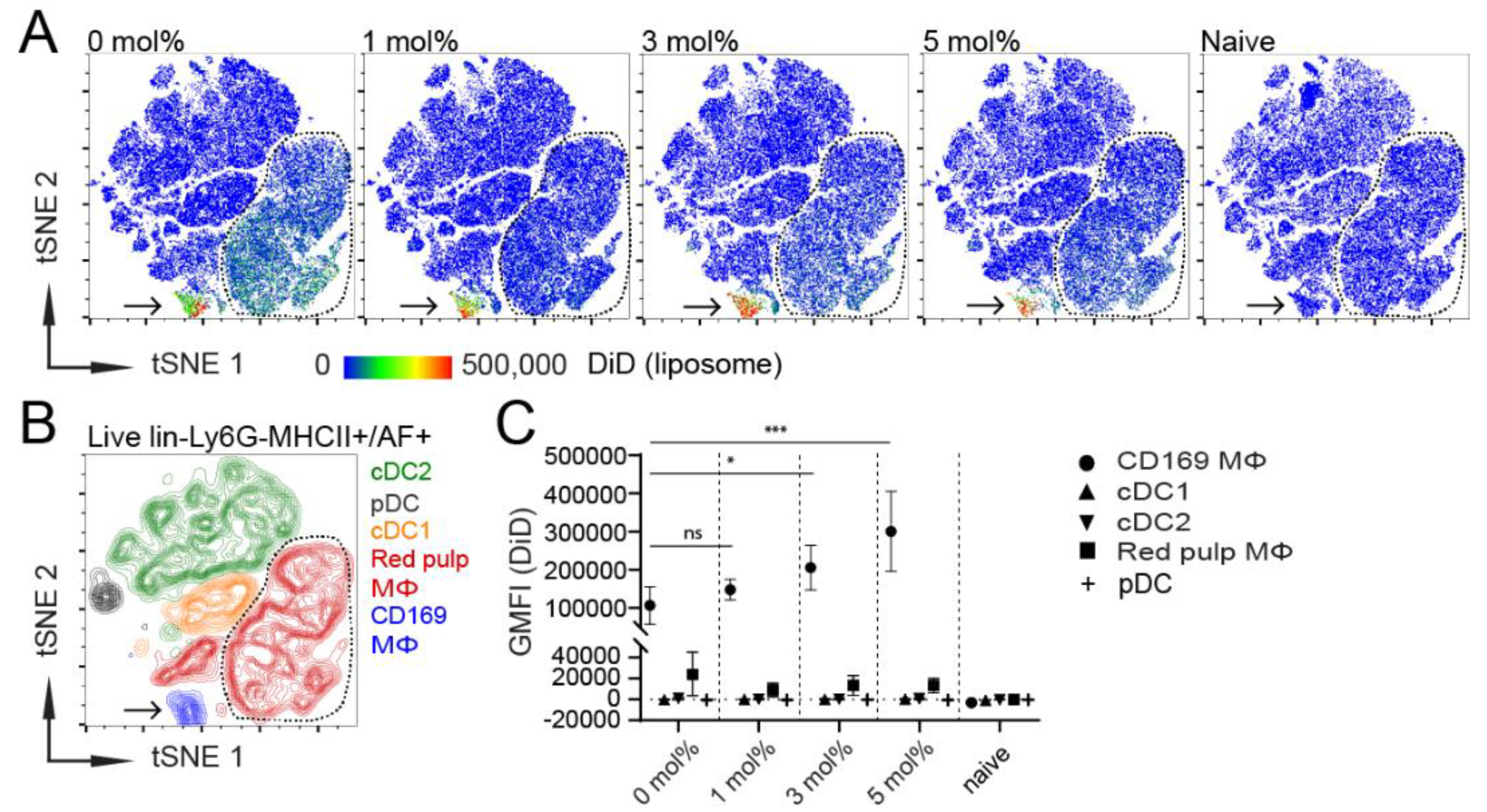
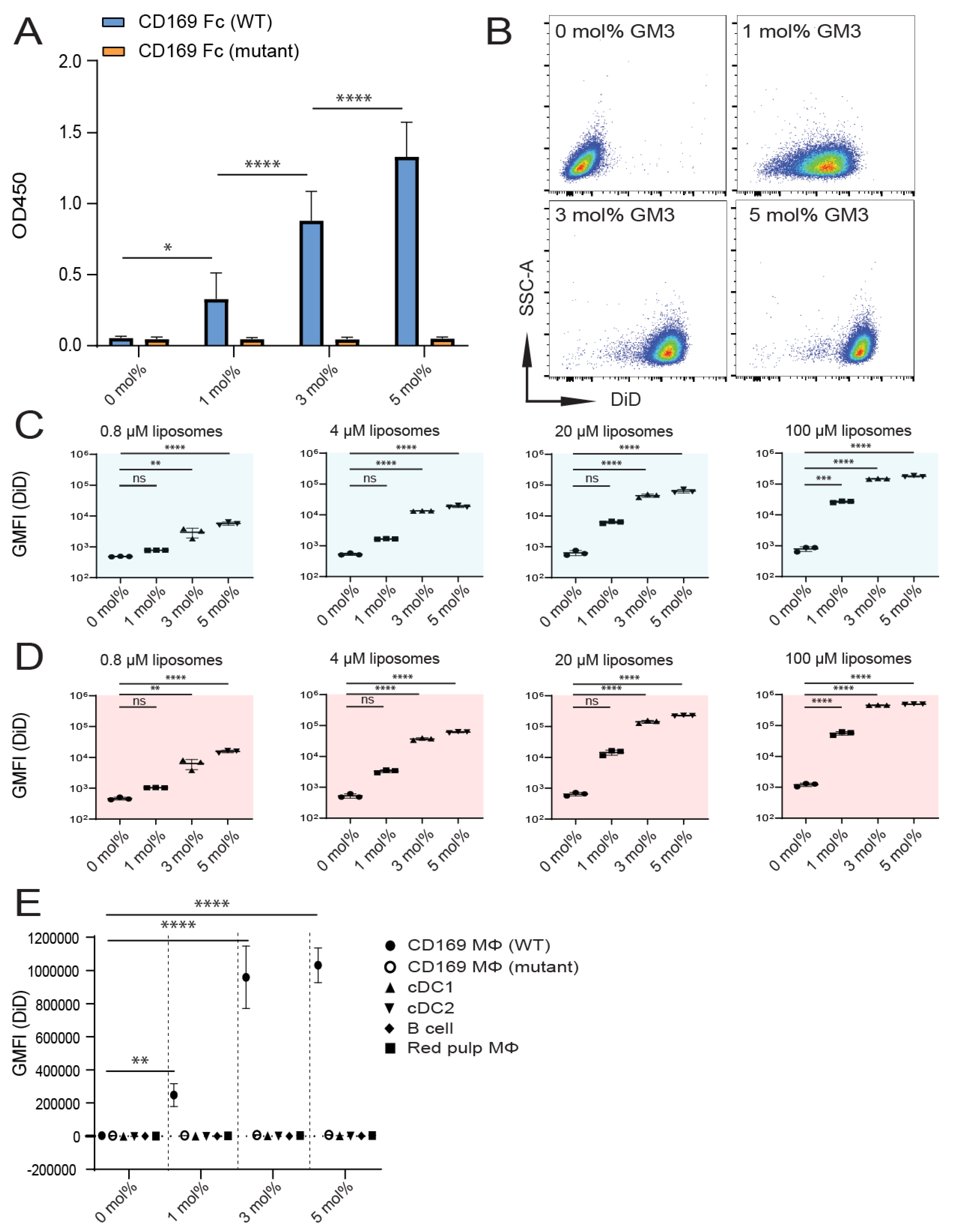
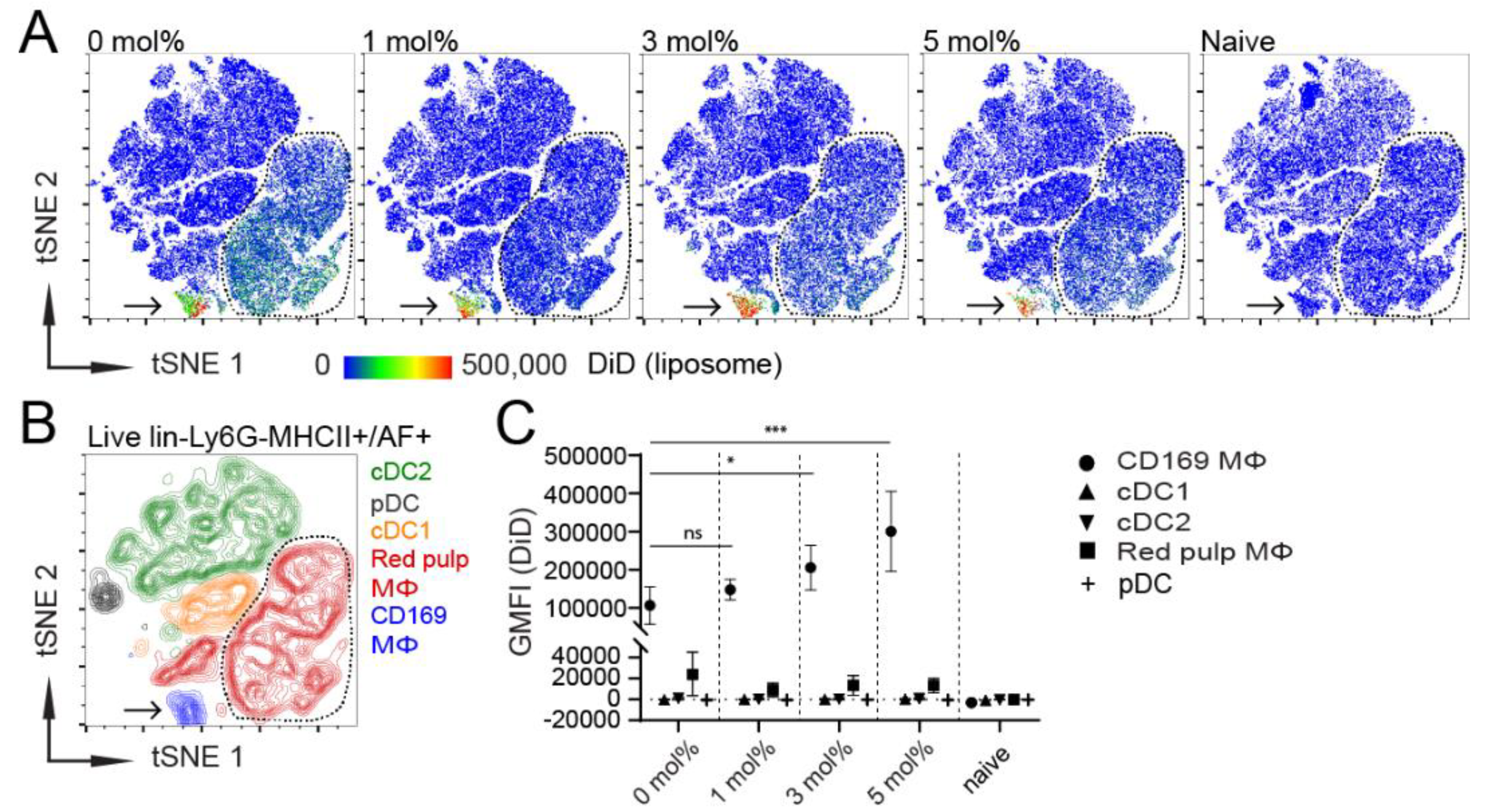
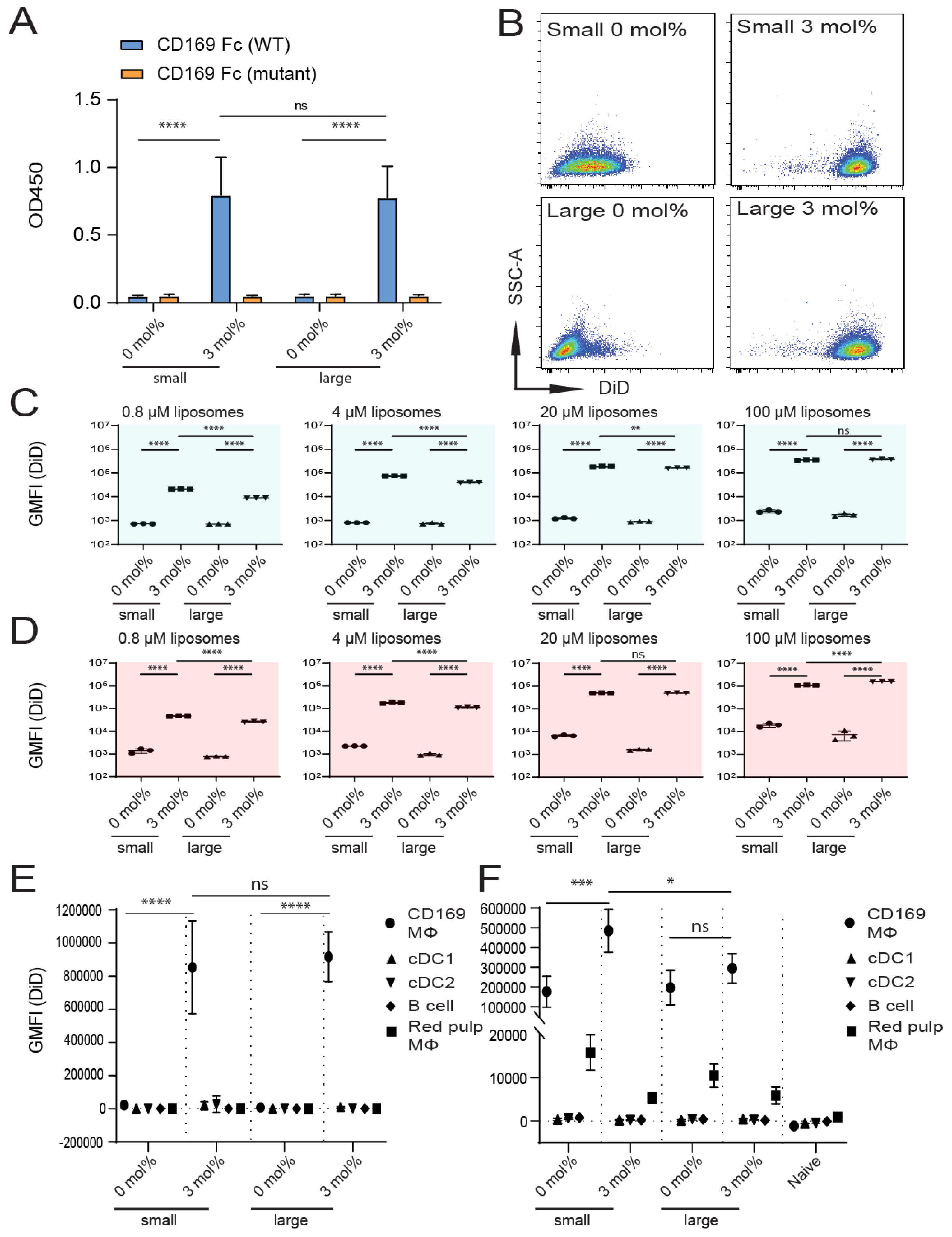
3.1. Influence of GM3 Incorporation
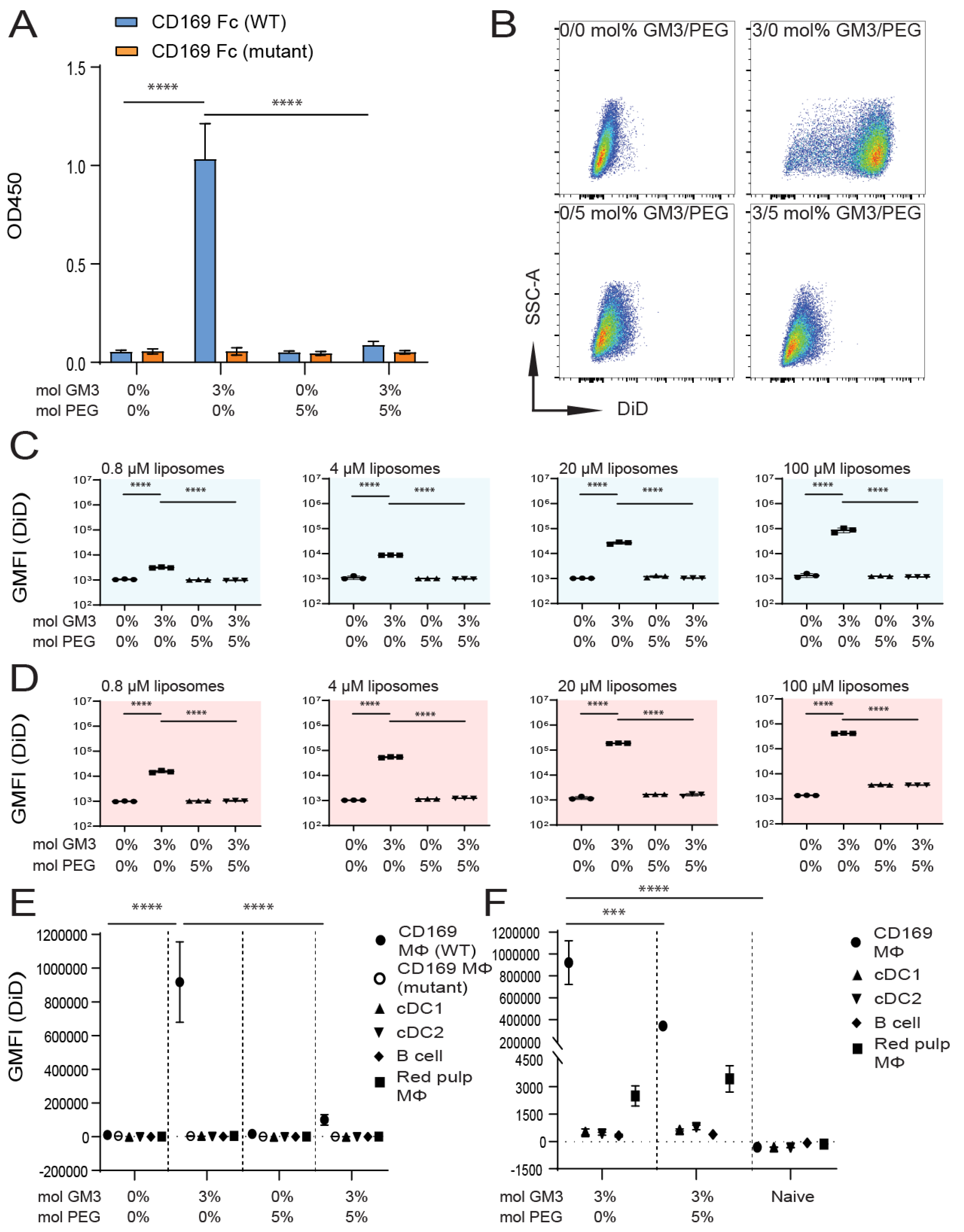
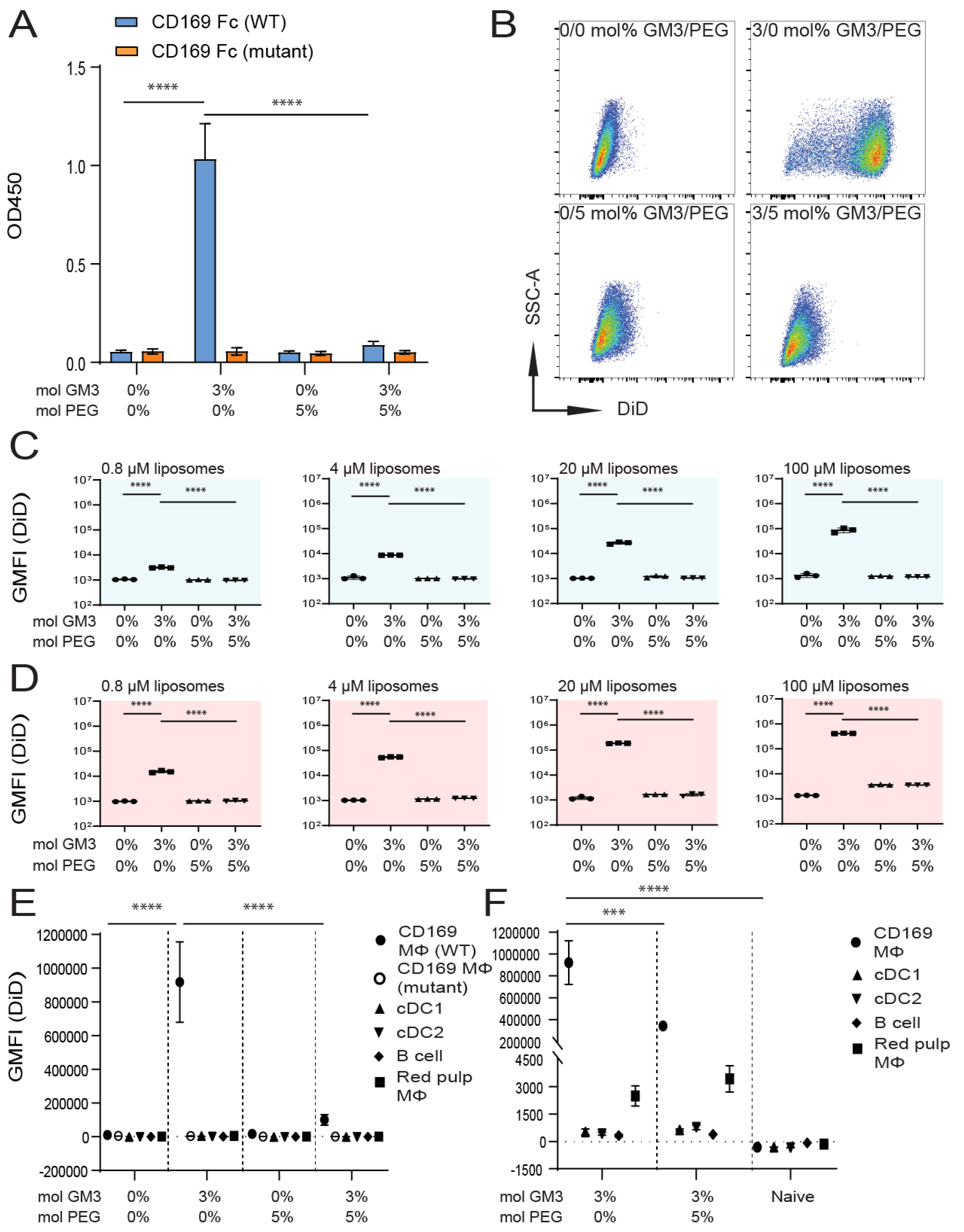
3.2. Influence of Liposomal PEG
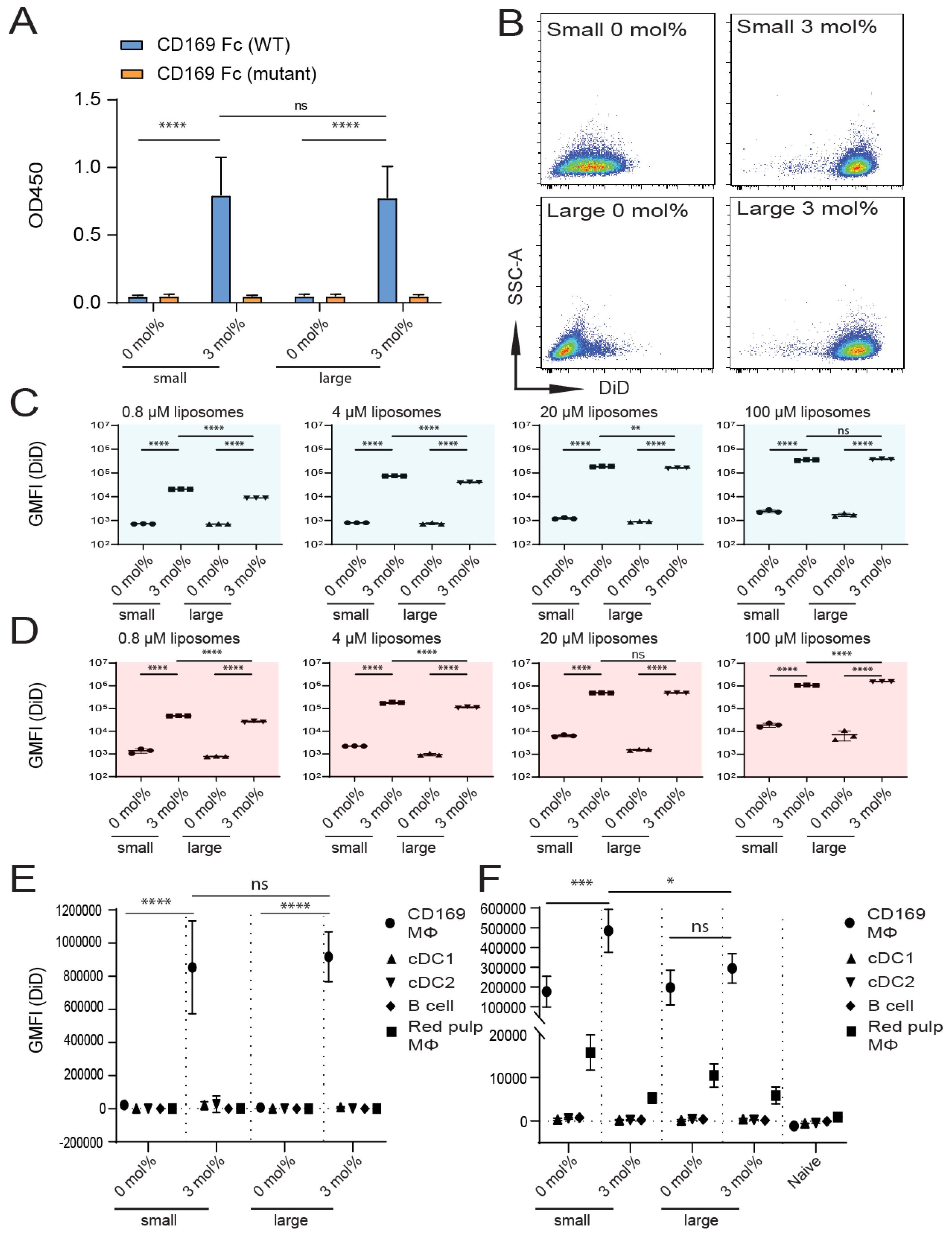
3.3. Influence of Liposomal Size
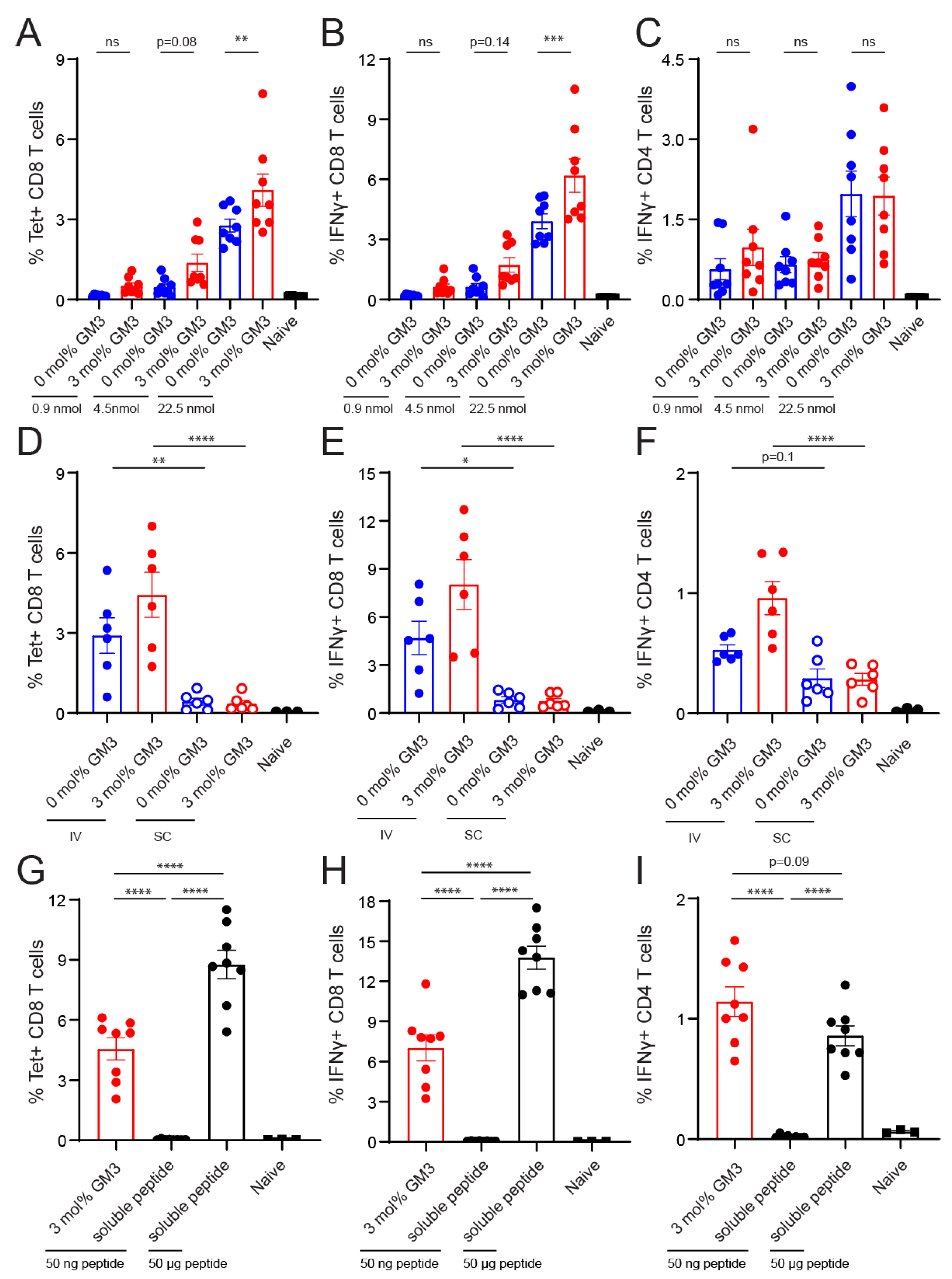
3.4. Immunogenicity of Liposomes Containing Antigen
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef] [PubMed]
- Haen, S.P.; Löffler, M.W.; Rammensee, H.G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.-M.; Buqué, A.; Senovilla, L.; Baracco, E.E.; Bloy, N.; Castoldi, F.; Abastado, J.-P.; Agostinis, P.; et al. Classification of current anticancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef] [Green Version]
- Finn, O.J. The dawn of vaccines for cancer prevention. Nat. Rev. Immunol. 2018, 18, 183–194. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Vara Perez, M.; Schaaf, M.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Dendritic cell-based anticancer immunotherapy. Oncoimmunology 2017, 6, e1328341. [Google Scholar] [CrossRef] [PubMed]
- Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, A.A., Jr. Dendritic Cells in Anticancer Vaccination: Rationale for Ex Vivo Loading or In Vivo Targeting. Cancers 2020, 12, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J.M. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.-G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef] [Green Version]
- Hirosue, S.; Vokali, E.; Raghavan, V.R.; Rincon-Restrepo, M.; Lund, A.W.; Corthésy-Henrioud, P.; Capotosti, F.; Halin Winter, C.; Hugues, S.; Swartz, M.A. Steady-state antigen scavenging, cross-presentation, and CD8+ T cell priming: A new role for lymphatic endothelial cells. J. Immunol. 2014, 192, 5002–5011. [Google Scholar] [CrossRef] [Green Version]
- Van Hall, T.; van der Burg, S.H. Mechanisms of peptide vaccination in mouse models: Tolerance, immunity, and hyperreactivity. Adv. Immunol. 2012, 114, 51–76. [Google Scholar]
- Khadke, S.; Roces, C.B.; Cameron, A.; Devitt, A.; Perrie, Y. Formulation and manufacturing of lymphatic targeting liposomes using microfluidics. J. Control. Release 2019, 307, 211–220. [Google Scholar] [CrossRef]
- Perrie, Y.; Crofts, F.; Devitt, A.; Griffiths, H.R.; Kastner, E.; Nadella, V. Designing liposomal adjuvants for the next generation of vaccines. Adv. Drug Deliv. Rev. 2016, 99 Pt A, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Boks, M.A.; Ambrosini, M.; Bruijns, S.C.; Kalay, H.; van Bloois, L.; Storm, G.; Garcia-Vallejo, J.J.; van Kooyk, Y. MPLA incorporation into DC-targeting glycoliposomes favours anti-tumour T cell responses. J. Control Release 2015, 216, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.A.; Bruijns, S.C.M.; Ambrosini, M.; Kalay, H.; van Bloois, L.; Storm, G.; Gruijl, T.; van Kooyk, Y. In situ Delivery of Tumor Antigen- and Adjuvant-Loaded Liposomes Boosts Antigen-Specific T-Cell Responses by Human Dermal Dendritic Cells. J. Investig. Dermatol. 2015, 135, 2697–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stolk, D.A.; de Haas, A.; Vree, J.; Duinkerken, S.; Lübbers, J.; van de Ven, R.; Ambrosini, M.; Kalay, H.; Bruijns, S.; van der Vliet, H.J.; et al. Lipo-Based Vaccines as an Approach to Target Dendritic Cells for Induction of T- and iNKT Cell Responses. Front. Immunol. 2020, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.D.; Unger, W.J.; Storm, G.; van Kooyk, Y.; Mastrobattista, E. Targeting tumor antigens to dendritic cells using particulate carriers. J. Control Release 2012, 161, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, J.; Lopez-Venegas, M.A.; Affandi, A.J.; den Haan, J.M.M. CD169+ Macrophages Capture and Dendritic Cells Instruct: The Interplay of the Gatekeeper and the General of the Immune System. Front. Immunol. 2018, 9, 2472. [Google Scholar] [CrossRef] [Green Version]
- Saunderson, S.C.; Dunn, A.C.; Crocker, P.R.; McLellan, A.D. CD169 mediates the capture of exosomes in spleen and lymph node. Blood 2014, 123, 208–216. [Google Scholar] [CrossRef]
- Collins, B.E.; Kiso, M.; Hasegawa, A.; Tropak, M.B.; Roder, J.C.; Crocker, P.R.; Schnaar, R.L. Binding specificities of the sialoadhesin family of I-type lectins. Sialic acid linkage and substructure requirements for binding of myelin-associated glycoprotein, Schwann cell myelin protein, and sialoadhesin. J. Biol. Chem. 1997, 272, 16889–16895. [Google Scholar] [CrossRef] [Green Version]
- Crocker, P.R.; Kelm, S.; Dubois, C.; Martin, B.; McWilliam, A.S.; Shotton, D.M.; Paulson, J.C.; Gordon, S. Purification and properties of sialoadhesin, a sialic acid-binding receptor of murine tissue macrophages. EMBO J. 1991, 10, 661–1669. [Google Scholar] [CrossRef]
- Xu, F.; Bandara, A.; Akiyama, H.; Eshaghi, B.; Stelter, D.; Keyes, T.; Straub, J.E.; Gummuluru, S.; Reinhard, B.M. Membrane-wrapped nanoparticles probe divergent roles of GM3 and phosphatidylserine in lipid-mediated viral entry pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E9041–E9050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Feizpour, A.; Ramirez, N.-G.P.; Wu, L.; Akiyama, H.; Xu, F.; Gummuluru, S.; Reinhard, B.M. Glycosphingolipid-functionalized nanoparticles recapitulate CD169-dependent HIV-1 uptake and trafficking in dendritic cells. Nat. Commun. 2014, 5, 4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wubbolts, R.; Leckie, R.S.; Veenhuizen, P.T.; Schwarzmann, G.; Möbius, W.; Hoernschemeyer, J.; Slot, J.W.; Geuze, H.J.; Stoorvogel, W. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J. Biol. Chem. 2003, 278, 10963–10972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorente, A.; Skotland, T.; Sylvänne, T.; Kauhanen, D.; Róg, T.; Orłowski, A.; Vattulainen, I.; Ekroos, K.; Sandvig, K. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2013, 1831, 1302–1309. [Google Scholar] [CrossRef]
- Akiyama, H.; Miller, C.; Patel, H.V.; Hatch, S.C.; Archer, J.; Ramirez, N.-G.P.; Gummuluru, S. Virus particle release from glycosphingolipid-enriched microdomains is essential for dendritic cell-mediated capture and transfer of HIV-1 and henipavirus. J. Virol. 2014, 88, 8813–8825. [Google Scholar] [CrossRef] [Green Version]
- Puryear, W.B.; Yu, X.; Ramirez, N.P.; Reinhard, B.M.; Gummuluru, S. HIV-1 incorporation of host-cell-derived glycosphingolipid GM3 allows for capture by mature dendritic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 7475–7480. [Google Scholar] [CrossRef] [Green Version]
- Van Dinther, D.; Veninga, H.; Revet, M.; Hoogterp, L.; Olesek, K.; Grabowska, J.; Borg, E.G.F.; Kalay, H.; van Kooyk, Y.; den Haan, J.M.M. Comparison of Protein and Peptide Targeting for the Development of a CD169-Based Vaccination Strategy Against Melanoma. Front. Immunol. 2018, 9, 1997. [Google Scholar] [CrossRef]
- Van Dinther, D.; Veninga, H.; Iborra, S.; Borg, E.G.F.; Hoogterp, L.; Olesek, K.; Beijer, M.R.; Schetters, S.T.T.; Kalay, H.; Garcia-Vallejo, J.J.; et al. Functional CD169 on Macrophages Mediates Interaction with Dendritic Cells for CD8+ T Cell Cross-Priming. Cell Rep. 2018, 22, 1484–1495. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, N.; Vela, J.L.; Nycholat, C.M.; Rademacher, C.; Khurana, A.; van Rooijen, N.; Crocker, P.R.; Kronenberg, M.; Paulson, J.C. Targeted delivery of lipid antigen to macrophages via the CD169/sialoadhesin endocytic pathway induces robust invariant natural killer T cell activation. Proc. Natl. Acad. Sci. USA 2013, 110, 7826–7831. [Google Scholar] [CrossRef] [Green Version]
- Edgar, L.J.; Kawasaki, N.; Nycholat, C.M.; Paulson, J.C. Targeted Delivery of Antigen to Activated CD169+ Macrophages Induces Bias for Expansion of CD8+ T Cells. Cell Chem. Biol. 2019, 26, 131–136.e4. [Google Scholar] [CrossRef]
- Klaas, M.; Oetke, C.; Lewis, L.E.; Erwig, L.P.; Heikema, A.P.; Easton, A.; Willison, H.J.; Crocker, P.R. Sialoadhesin Promotes Rapid Proinflammatory and Type I IFN Responses to a Sialylated Pathogen, Campylobacter jejuni. J. Immunol. 2012, 189, 2414–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rempel, H.; Calosing, C.; Sun, B.; Pulliam, L. Sialoadhesin expressed on IFN-induced monocytes binds HIV-1 and enhances infectivity. PLoS ONE 2008, 3, e1967. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Tang, P.S.; Chan, W.C.W. The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, G.T.; Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. Ligand-targeted liposome design: Challenges and fundamental considerations. Trends Biotechnol. 2014, 32, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [Green Version]
- Affandi, A.J.; Grabowska, J.; Olesek, K.; Lopez Venegas, M.; Barbaria, A.; Rodríguez, E.; Mulder, P.P.G.; Pijffers, H.J.; Ambrosini, M.; Kalay, H.; et al. Selective tumor antigen vaccine delivery to human CD169+ antigen-presenting cells using ganglioside-liposomes. Proc. Natl. Acad. Sci. USA 2020, 117, 27528–27539. [Google Scholar] [CrossRef]
- Xu, F.; Reiser, M.; Yu, X.; Gummuluru, S.; Wetzler, L.; Reinhard, B.M. Lipid-Mediated Targeting with Membrane-Wrapped Nanoparticles in the Presence of Corona Formation. ACS Nano 2016, 10, 1189–1200. [Google Scholar] [CrossRef] [Green Version]
- Szebeni, J.; Storm, G. Complement activation as a bioequivalence issue relevant to the development of generic liposomes and other nanoparticulate drugs. Biochem. Biophys. Res. Commun. 2015, 468, 490–497. [Google Scholar] [CrossRef]
- Cataldi, M.; Vigliotti, C.; Mosca, T.; Cammarota, M.; Capone, D. Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. Int. J. Mol. Sci. 2017, 18, 1249. [Google Scholar] [CrossRef] [Green Version]
- Moghimi, S.M.; Hunter, A.C.; Andresen, T.L. Factors Controlling Nanoparticle Pharmacokinetics: An Integrated Analysis and Perspective. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomed. (Lond. Engl.) 2016, 11, 673–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, N.; Leroux, J.C. The journey of a drug-carrier in the body: An anatomo-physiological perspective. J. Control Release 2012, 161, 152–163. [Google Scholar] [CrossRef]
- Tang, J.; Kuai, R.; Yuan, W.; Drake, L.; Moon, J.J.; Schwendeman, A. Effect of size and pegylation of liposomes and peptide-based synthetic lipoproteins on tumor targeting. Nanomedicine 2017, 13, 1869–1878. [Google Scholar] [CrossRef] [Green Version]
- Samuelsson, E.; Shen, H.; Blanco, E.; Ferrari, M.; Wolfram, J. Contribution of Kupffer cells to liposome accumulation in the liver. Colloids Surf. B Biointerfaces 2017, 158, 356–362. [Google Scholar] [CrossRef]
- Aarntzen, E.H.J.G.; De Vries, I.J.M.; Lesterhuis, W.J.; Schuurhuis, D.; Jacobs, J.F.M.; Bol, K.; Schreibelt, G.; Mus, R.; De Wilt, J.H.W.; Haanen, J.B.A.G.; et al. Targeting CD4+ T-Helper Cells Improves the Induction of Antitumor Responses in Dendritic Cell–Based Vaccination. Cancer Res. 2013, 73, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Backer, R.; Schwandt, T.; Greuter, M.; Oosting, M.; Jüngerkes, F.; Tüting, T.; Boon, L.; O’Toole, T.; Kraal, G.; Limmer, A.; et al. Effective collaboration between marginal metallophilic macrophages and CD8+ dendritic cells in the generation of cytotoxic T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Varypataki, E.M.; Benne, N.; Bouwstra, J.; Jiskoot, W.; Ossendorp, F. Efficient Eradication of Established Tumors in Mice with Cationic Liposome-Based Synthetic Long-Peptide Vaccines. Cancer Immunol. Res. 2017, 5, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Lynn, G.M.; Sedlik, C.; Baharom, F.; Zhu, Y.; Ramirez-Valdez, R.A.; Coble, V.L.; Tobin, K.; Nichols, S.R.; Itzkowitz, Y.; Zaidi, N.; et al. Peptide–TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat. Biotechnol. 2020, 38, 320–332. [Google Scholar] [CrossRef]
- Alloatti, A.; Kotsias, F.; Pauwels, A.-M.; Carpier, J.-M.; Jouve, M.; Timmerman, E.; Pace, L.; Vargas, P.; Maurin, M.; Gehrmann, U.; et al. Toll-like Receptor 4 Engagement on Dendritic Cells Restrains Phago-Lysosome Fusion and Promotes Cross-Presentation of Antigens. Immunity 2015, 43, 1087–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair-Gupta, P.; Baccarini, A.; Tung, N.; Seyffer, F.; Florey, O.; Huang, Y.; Banerjee, M.; Overholtzer, M.; Roche, P.A.; Tampé, R.; et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 2014, 158, 506–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Miyabe, H.; Hyodo, M.; Sato, Y.; Hayakawa, Y.; Harashima, H. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J. Control Release 2015, 216, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Theoretical Molecular Mass (Da) | Ionization Mode | Observed [M ± 3H]3± (m/z) | Observed Molecular Mass (Da) |
|---|---|---|---|---|---|
| OVA247–279 | NH2-DEVSGLEQLESIINFEKLT EWTSSNVMEERKIK-COOH | 3883.3 | Positive | 1295.3 | 3882.9 |
| 5-FAM-Lys-OVA247–279 | NH2-5-FAM-Lys-DEVSGLEQLESIINFEKLTEWTSSNVMEERKIK-COOH | 4369.8 | Negative | 1455.3 | 4368.9 |
| GM3 Amount Incorporated (mol%) | Mean Size (nm) | Mean PDI | Mean Zeta Potential (mV) |
|---|---|---|---|
| 0 mol% GM3 | 169 ± 0.3 | 0.13 ± 0.02 | −61.0 ± 2.7 |
| 1 mol% GM3 | 178 ± 2.2 | 0.17 ± 0.004 | −58.5 ± 1.4 |
| 3 mol% GM3 | 154 ± 0.8 | 0.17 ± 0.02 | −67.3 ± 2.4 |
| 5 mol% GM3 | 158 ± 0.9 | 0.17 ± 0.006 | −64.9 ± 3.0 |
| GM3/PEG Amount Incorporated (mol%) | Mean Size (nm) | Mean PDI | Mean Zeta Potential (mV) |
|---|---|---|---|
| 0 mol% GM3/0 mol% PEG | 191 ± 3.9 | 0.1 ± 0.02 | −50.4 ± 2.3 |
| 3 mol% GM3/0 mol% PEG | 176 ± 1.5 | 0.1 ± 0.001 | −53.2 ± 4.9 |
| 0 mol% GM3/5 mol% PEG | 159 ± 5.9 | 0.1 ± 0.05 | −24.6 ± 0.7 |
| 3 mol% GM3/5 mol% PEG | 164 ± 3.7 | 0.1 ± 0.02 | −26.9 ± 1.3 |
| Liposome Size and GM3 Amount Incorporated (mol%) | Mean Size (nm) | Mean PDI | Mean Zeta Potential (mV) |
|---|---|---|---|
| Small 0 mol% GM3 | 180 ± 0.9 | 0.1 ± 0.04 | −49.6 ± 1.0 |
| Small 3 mol% GM3 | 167 ± 4.1 | 0.1 ± 0.02 | −52.6 ± 2.6 |
| Large 0 mol% GM3 | 385 ± 12.4 | 0.2 ± 0.03 | −44.8 ± 1.0 |
| Large 3 mol% GM3 | 390 ± 12.7 | 0.2 ± 0.06 | −46.2 ± 0.8 |
| GM3 Amount Incorporated (mol%) | Mean Size (nm) | Mean PDI | Mean Zeta-Potential (mV) | Loading Efficiency (%) | Loading Capacity (ng Peptide/nmol Phospholipid) |
|---|---|---|---|---|---|
| 0 mol% GM3 | 231 ± 6.1 | 0.2 ± 0.05 | −64.8 ± 2.3 | 0.40 ± 0.01 | 2.40 ± 0.04 |
| 3 mol% GM3 | 224 ± 15.5 | 0.2 ± 0.05 | −44.5 ± 1.8 | 0.43 ± 0.02 | 2.41 ± 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nijen Twilhaar, M.K.; Czentner, L.; Grabowska, J.; Affandi, A.J.; Lau, C.Y.J.; Olesek, K.; Kalay, H.; van Nostrum, C.F.; van Kooyk, Y.; Storm, G.; et al. Optimization of Liposomes for Antigen Targeting to Splenic CD169+ Macrophages. Pharmaceutics 2020, 12, 1138. https://doi.org/10.3390/pharmaceutics12121138
Nijen Twilhaar MK, Czentner L, Grabowska J, Affandi AJ, Lau CYJ, Olesek K, Kalay H, van Nostrum CF, van Kooyk Y, Storm G, et al. Optimization of Liposomes for Antigen Targeting to Splenic CD169+ Macrophages. Pharmaceutics. 2020; 12(12):1138. https://doi.org/10.3390/pharmaceutics12121138
Chicago/Turabian StyleNijen Twilhaar, Maarten K., Lucas Czentner, Joanna Grabowska, Alsya J. Affandi, Chun Yin Jerry Lau, Katarzyna Olesek, Hakan Kalay, Cornelus F. van Nostrum, Yvette van Kooyk, Gert Storm, and et al. 2020. "Optimization of Liposomes for Antigen Targeting to Splenic CD169+ Macrophages" Pharmaceutics 12, no. 12: 1138. https://doi.org/10.3390/pharmaceutics12121138
APA StyleNijen Twilhaar, M. K., Czentner, L., Grabowska, J., Affandi, A. J., Lau, C. Y. J., Olesek, K., Kalay, H., van Nostrum, C. F., van Kooyk, Y., Storm, G., & Haan, J. M. M. d. (2020). Optimization of Liposomes for Antigen Targeting to Splenic CD169+ Macrophages. Pharmaceutics, 12(12), 1138. https://doi.org/10.3390/pharmaceutics12121138