Enhancement of Aqueous Solubility and Dissolution of Celecoxib through Phosphatidylcholine-Based Dispersion Systems Solidified with Adsorbent Carriers


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Phosphatidylcholine (PC)-Based Solid Dispersions (SDs)
2.3. Evaluation of Apparent Aqueous Solubility of Celecoxib (CLC) Incorporated in PC-Based Dispersion Formulations
2.4. High-Performance Liquid Chromatography (HPLC) Analysis of CLC
2.5. Wicking Test of Hydrophilic Diluents and Disintegrants as a Water Absorption Enhancer
2.6. Assessment of Flowability of PC-Based Dispersion Formulations Powderized with Neusilin® US2
2.7. In Vitro Drug Dissolution Study
2.8. Differential Scanning Calorimetry (DSC)
2.9. X-Ray Powder Diffraction (XRD)
2.10. Fourier Transform Infrared (FT-IR) Spectroscopy
2.11. Statistical Analysis
3. Results and discussion
3.1. Apparent Aqueous Solubility of CLC Incorporated in PC-Based Dispersion Formulations
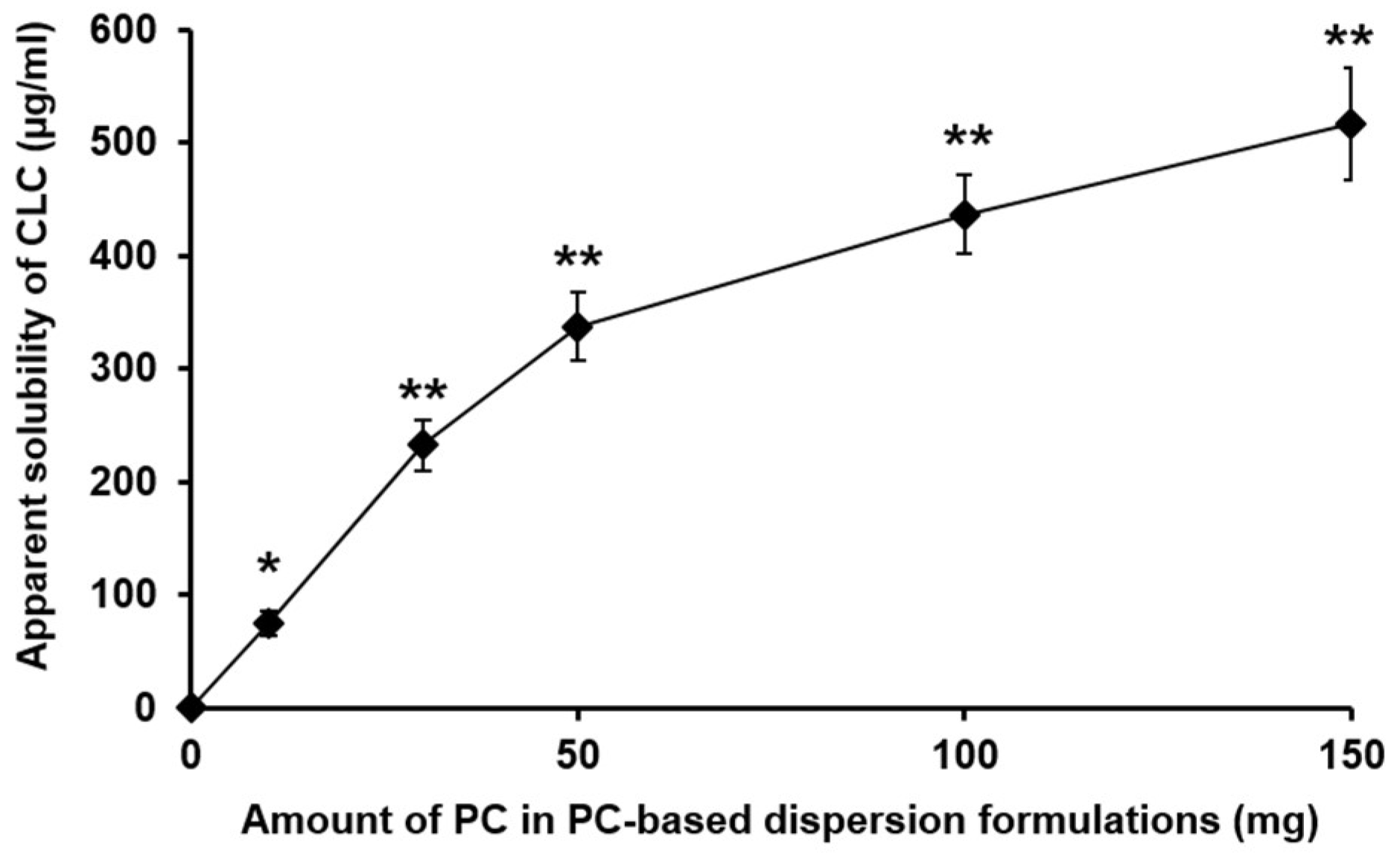
3.1.1. The Effect of the Amount of PC in PC-Based Dispersion Formulations on the Apparent Aqueous Solubility of CLC
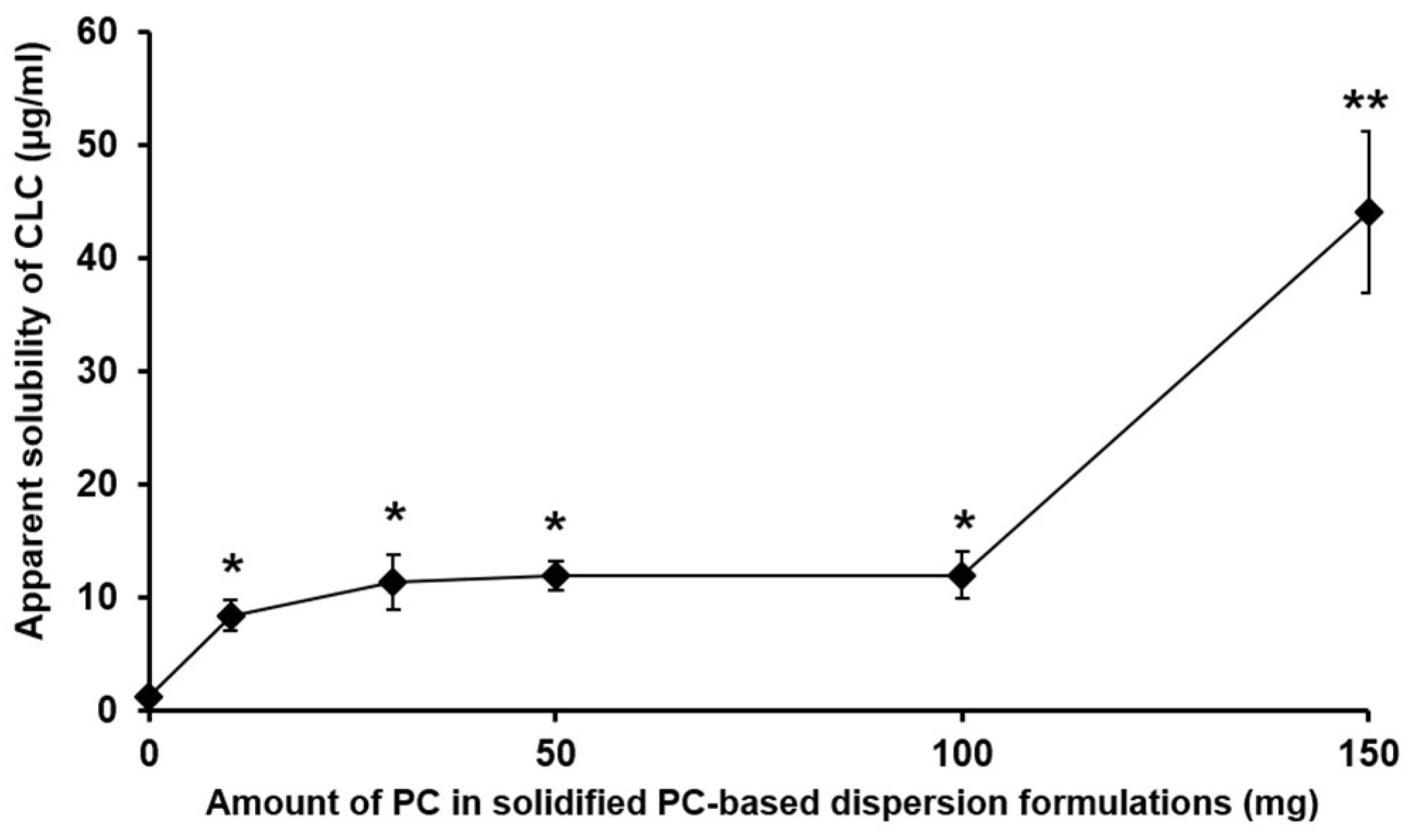
3.1.2. The Apparent Aqueous Solubility of CLC Evaluated from PC-Based Dispersion Formulations Powderized with the Adsorbent Carrier
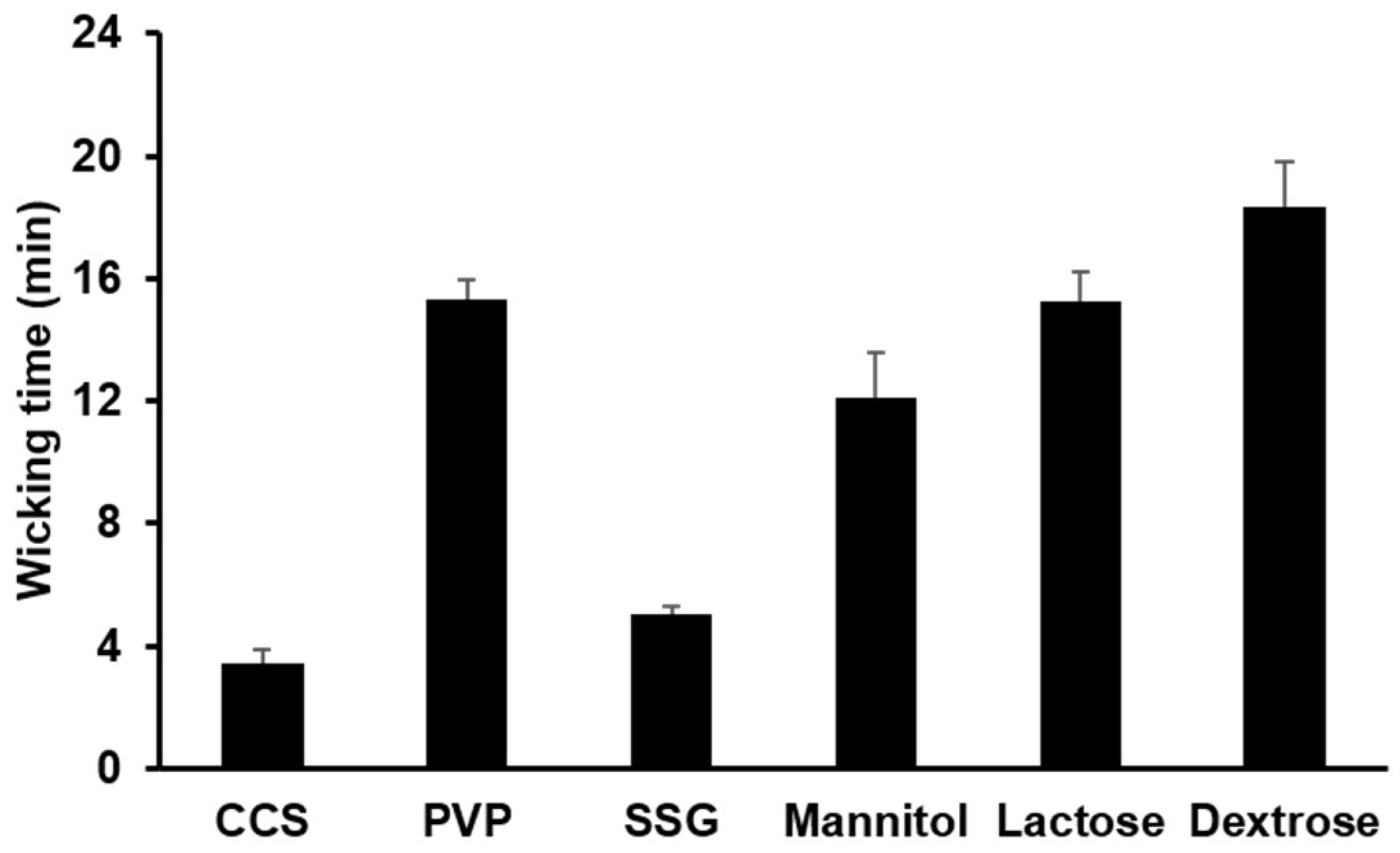
3.1.3. Water Uptake Ability of Hydrophilic Diluents and Disintegrants
3.1.4. The Impact of Different Hydrophilic Diluents/Disintegrants as Water Absorption Enhancer in PC-Based Dispersion Formulations on the Apparent Aqueous Solubility of CLC
3.2. Flowability of PC-Based Dispersion Formulations Powderized with Neusilin® US2
3.3. In Vitro Dissolution Study
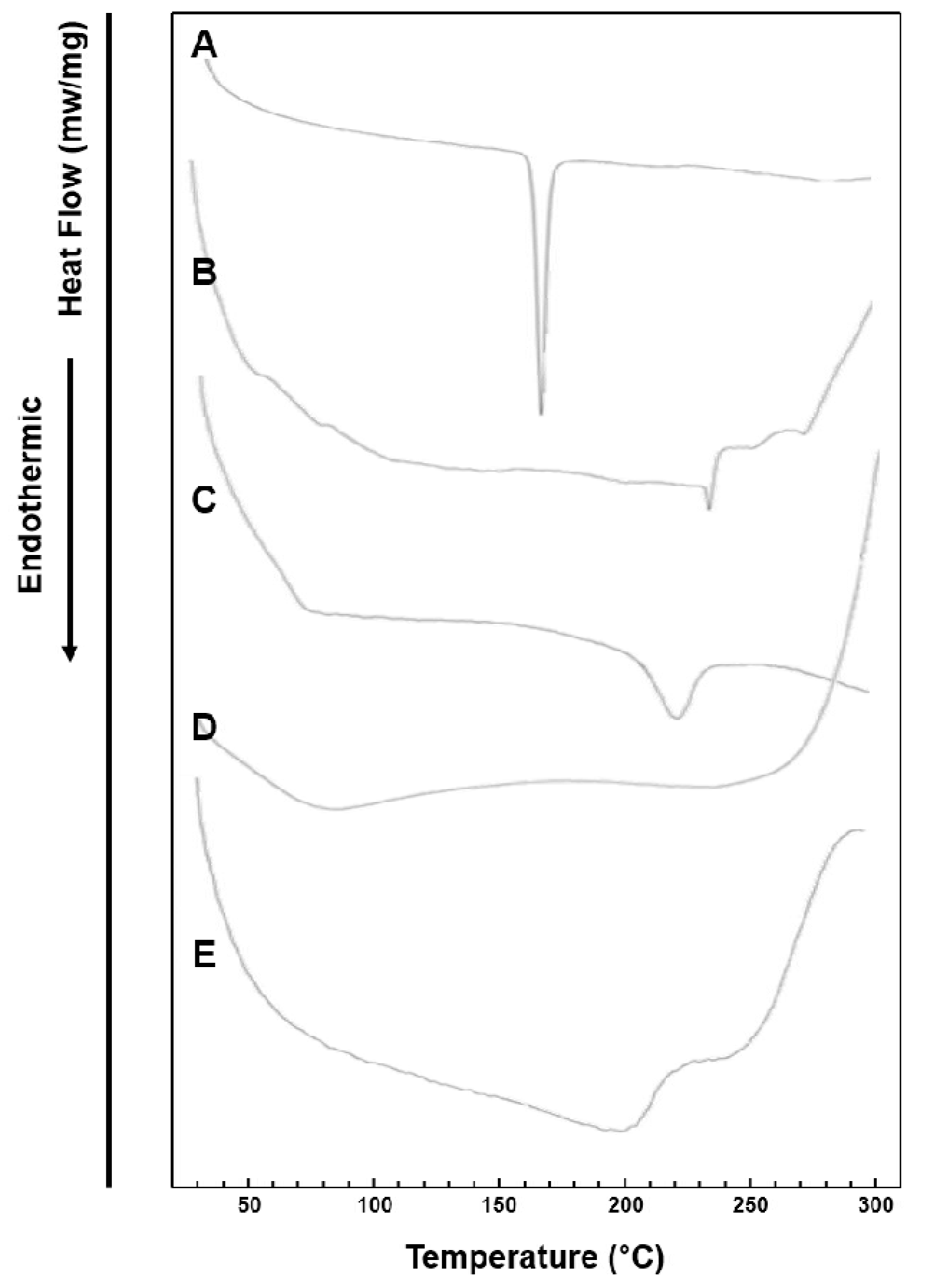
3.4. DSC Study
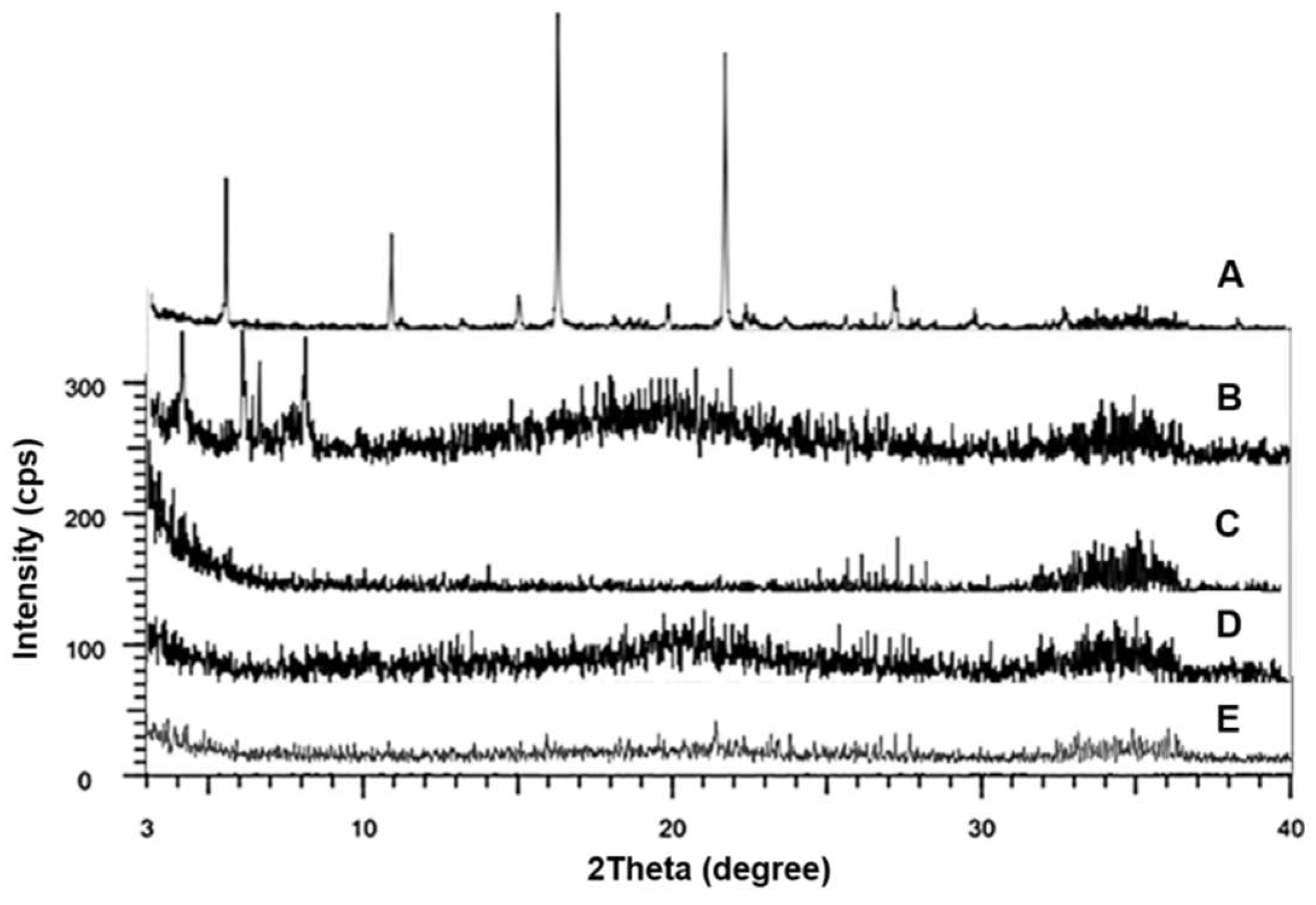
3.5. XRD Analysis
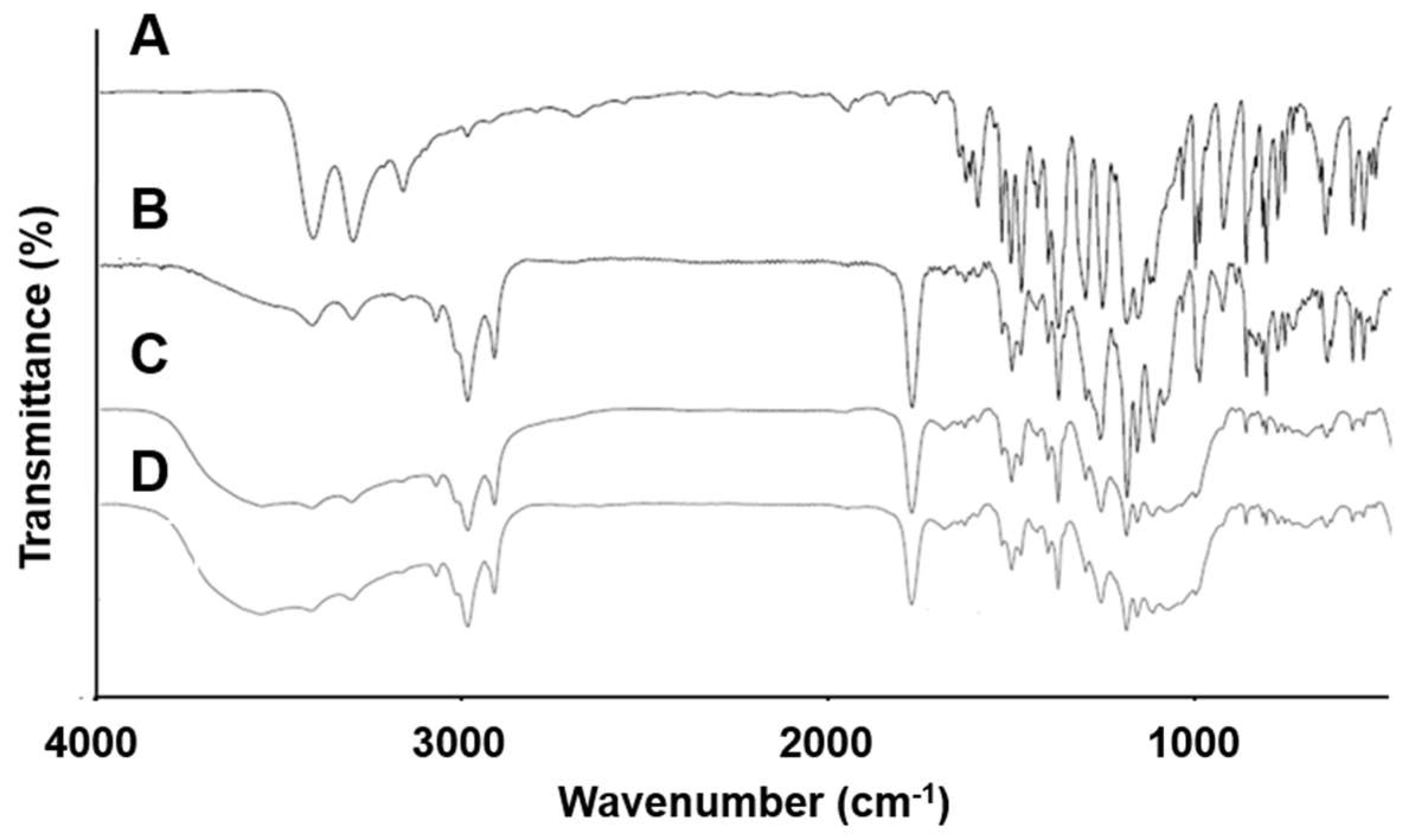
3.6. FT-IR Spectroscopy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lee, J.H.; Kim, M.J.; Yoon, H.; Shim, C.R.; Ko, H.A.; Cho, S.A.; Lee, D.; Khang, G. Enhanced dissolution rate of celecoxib using pvp and/or hpmc-based solid dispersions prepared by spray drying method. J. Pharm. Investig. 2013, 43, 205–213. [Google Scholar] [CrossRef]
- Punitha, S.; Karthikeyan, D.; Devi, P.; Vedha Hari, B. Enhancement of solubility and dissolution of celecoxib by solid dispersion technique. J. Pharm. Sci. Technol. 2009, 1, 63–68. [Google Scholar]
- Dixit, M.; Kini, A.G.; Kulkarni, P.K. Enhancing solubility and dissolution of celecoxib by spray drying using pluronic f 127. Indian J. Pharm. Educ. 2011, 45, 346–352. [Google Scholar]
- Dolenc, A.; Kristl, J.; Baumgartner, S.; Planinsek, O. Advantages of celecoxib nanosuspension formulation and transformation into tablets. Int. J. Pharm. 2009, 376, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Dhumal, R.; Shimpi, S.; Paradkar, A. Development of spray-dried co-precipitate of amorphous celecoxib containing storage and compression stabilizers. Acta Pharm. 2007, 57, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Punitha, S.; Vedha Hari, B.; Karthikeyan, D. Enhancement of celecoxib solubility by solid dispersion using mannitol. Int. J. Pharm. Pharm. Sci. 2010, 2, 109–111. [Google Scholar]
- Dahim, M.; Gustafsson, J.; Puisieux, F.; Ollivon, M. Solubilization of phospholipid/triacylglycerol aggregates by non-ionic surfactants. Chem. Phys. Lipids 1998, 97, 1–14. [Google Scholar] [CrossRef]
- Mirza, S.; Miroshnyk, I.; Habib, M.J.; Brausch, J.F.; Hussain, M.D. Enhanced dissolution and oral bioavailability of piroxicam formulations: Modulating effect of phospholipids. Pharmaceutics 2010, 2, 339–350. [Google Scholar] [CrossRef]
- Milovic, M.; Djuris, J.; Djekic, L.; Vasiljevic, D.; Ibric, S. Characterization and evaluation of solid self-microemulsifying drug delivery systems with porous carriers as systems for improved carbamazepine release. Int. J. Pharm. 2012, 436, 58–65. [Google Scholar] [CrossRef]
- Dhirendra, K.; Lewis, S.; Udupa, N.; Atin, K. Solid dispersions: A review. Pak. J. Pharm. Sci. 2009, 22, 234–246. [Google Scholar]
- Huang, Y.; Dai, W.-G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm. Sin. B 2014, 4, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Chen, Y.; Deng, J.; Jia, X.B.; Zhou, J.P.; Lv, H.X. Solid dispersion of berberine-phospholipid complex/tpgs 1000/sio2: Preparation, characterization and in vivo studies. Int. J. Pharm. 2014, 465, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Kakumanu, V.K.; Bansal, A.K. Stability and solubility of celecoxib-pvp amorphous dispersions: A molecular perspective. Pharm. Res. 2004, 21, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.Y.; Jong, J.E.; Lee, J.H.; Yang, J.W.; Park, S.M.; Lim, D.; Khang, G. Characterization and improvement of dissolution rate of solid dispersion of celecoxib in pvp k30/eudragit epo. Polym.-Korea 2014, 38, 434–440. [Google Scholar] [CrossRef][Green Version]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.S.; Shalaev, E.; Smith, S. Physical stability of pharmaceutical formulations: Solid-state characterization of amorphous dispersions. TrAC-Trend Anal. Chem. 2013, 49, 137–144. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification—The correlation of in-vitro drug product dissolution and in-vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.L.; Zhang, T.; Wang, C.L.; Huang, Z.J.; Luo, X.; Deng, Y.H. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2015, 10, 81–98. [Google Scholar] [CrossRef]
- Van Hoogevest, P.; Wendel, A. The use of natural and synthetic phospholipids as pharmaceutical excipients. Eur. J. Lipid Sci. Technol. 2014, 116, 1088–1107. [Google Scholar] [CrossRef]
- Hussain, M.D.; Saxena, V.; Brausch, J.F.; Talukder, R.M. Ibuprofen-phospholipid solid dispersions: Improved dissolution and gastric tolerance. Int. J. Pharm. 2012, 422, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Hammad, M.A.; Muller, B.W. Increasing drug solubility by means of bile salt-phosphatidylcholine-based mixed micelles. Eur. J. Pharm. Biopharm. 1998, 46, 361–367. [Google Scholar] [CrossRef]
- Ickenstein, L.M.; Sandstrom, M.C.; Mayer, L.D.; Edwards, K. Effects of phospholipid hydrolysis on the aggregate structure in dppc/dspe-peg(2000) liposome preparations after gel to liquid crystalline phase transition. BBA-Biomembr. 2006, 1758, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Arai, H.; Uchino, K.; Iwasaki, K.; Shibata, N.; Takada, K. Effect of adsorbents on the absorption of lansoprazole with surfactant. Int. J. Pharm. 2005, 289, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Gumaste, S.G.; Pawlak, S.A.; Dalrymple, D.M.; Nider, C.J.; Trombetta, L.D.; Serajuddin, A.T.M. Development of solid sedds, iv: Effect of adsorbed lipid and surfactant on tableting properties and surface structures of different silicates. Pharm. Res. 2013, 30, 3170–3185. [Google Scholar] [CrossRef] [PubMed]
- Baboota, S.; Faiyaz, S.; Ahuja, A.; Ali, J.; Shafiq, S.; Ahmad, S. Development and validation of a stability-indicating hplc method for analysis of celecoxib (cxb) in bulk drug and microemulsion formulations. Acta Chromatogr. 2007, 18, 116–129. [Google Scholar]
- Soares, L.A.L.; Ortega, G.G.; Petrovick, P.R.; Schmidt, P.C. Dry granulation and compression of spray-dried plant extracts. AAPS PharmSciTech 2005, 6, E359–E366. [Google Scholar] [CrossRef]
- Santomaso, A.; Lazzaro, P.; Canu, P. Powder flowability and density ratios: The impact of granules packing. Chem. Eng. Sci. 2003, 58, 2857–2874. [Google Scholar] [CrossRef]
- Morgen, M.; Bloom, C.; Beyerinck, R.; Bello, A.; Song, W.; Wilkinson, K.; Steenwyk, R.; Shamblin, S. Polymeric nanoparticles for increased oral bioavailability and rapid absorption using celecoxib as a model of a low-solubility, high-permeability drug. Pharm. Res. 2012, 29, 427–440. [Google Scholar] [CrossRef]
- Fong, S.Y.K.; Ibisogly, A.; Bauer-Brandl, A. Solubility enhancement of bcs class ii drug by solid phospholipid dispersions: Spray drying versus freeze-drying. Int. J. Pharm. 2015, 496, 382–391. [Google Scholar] [CrossRef]
- Paulson, S.K.; Vaughn, M.B.; Jessen, S.M.; Lawal, Y.; Gresk, C.J.; Yan, B.; Maziasz, T.J.; Cook, C.S.; Karim, A. Pharmacokinetics of celecoxib after oral administration in dogs and humans: Effect of food and site of absorption. J. Pharmacol. Exp. Ther. 2001, 297, 638–645. [Google Scholar] [PubMed]
- Gumaste, S.G.; Dalrymple, D.M.; Serajuddin, A.T.M. Development of solid sedds, V: Compaction and drug release properties of tablets prepared by adsorbing lipid-based formulations onto neusilinA (R) us2. Pharm. Res. 2013, 30, 3186–3199. [Google Scholar] [CrossRef] [PubMed]
- The United States Pharmacopeial Convention. <1174> Powder Flow; US Pharmacopoeia: Rockville, MD, USA, 2012; Volume 18. [Google Scholar]
- Seedher, N.; Bhatia, S. Solubility enhancement of cox-2 inhibitors using various solvent systems. AAPS PharmSciTech 2003, 4, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. Dissolution testing of uncompressed solid dosage forms. In Handbook of Pharmaceutical Manufacturing Formulations; CRC Press: Boca Raton, FL, USA, 2016; pp. 219–225. [Google Scholar]
- Reddy, M.N.; Rehana, T.; Ramakrishna, S.; Chowdary, K.; Diwan, P.V. B-cyclodextrin complexes of celecoxib: Molecular-modeling, characterization, and dissolution studies. AAPS PharmSciTech 2004, 6, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Bansal, A.K. Ternary amorphous composites of celecoxib, poly(vinyl pyrrolidone) and meglumine with enhanced solubility. Pharmazie 2005, 60, 830–836. [Google Scholar] [PubMed]
- Kallakunta, V.R.; Bandari, S.; Jukanti, R.; Veerareddy, P.R. Oral self emulsifying powder of lercanidipine hydrochloride: Formulation and evaluation. Powder Technol. 2012, 221, 375–382. [Google Scholar] [CrossRef]
- Krupa, A.; Majda, D.; Jachowicz, R.; Mozgawa, W. Solid-state interaction of ibuprofen and neusilin us2. Thermochim. Acta 2010, 509, 12–17. [Google Scholar] [CrossRef]
- Homayouni, A.; Sadeghi, F.; Varshosaz, J.; Garekani, H.A.; Nokhodchi, A. Comparing various techniques to produce micro/nanoparticles for enhancing the dissolution of celecoxib containing pvp. Eur. J. Pharm. Biopharm. 2014, 88, 261–274. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | Celecoxib (mg) | PC (mg) | Neusilin® US2 (mg) | Disintegrants | Diluents | ||||
|---|---|---|---|---|---|---|---|---|---|
| CCS (mg) | PVP (mg) | SSG (mg) | Mannitol (mg) | Lactose (mg) | Dextrose (mg) | ||||
| F1 | 100 | 10 | - | - | - | - | - | - | - |
| F2 | 100 | 30 | - | - | - | - | - | - | - |
| F3 | 100 | 50 | - | - | - | - | - | - | - |
| F4 | 100 | 100 | - | - | - | - | - | - | - |
| F5 | 100 | 150 | - | - | - | - | - | - | - |
| F6 | 100 | 10 | 200 | - | - | - | - | - | - |
| F7 | 100 | 30 | 200 | - | - | - | - | - | - |
| F8 | 100 | 50 | 200 | - | - | - | - | - | - |
| F9 | 100 | 100 | 200 | - | - | - | - | - | - |
| F10 | 100 | 150 | 200 | - | - | - | - | - | - |
| F11 | 100 | 150 | 200 | 50 | - | - | - | - | - |
| F12 | 100 | 150 | 200 | - | 50 | - | - | - | - |
| F13 | 100 | 150 | 200 | - | - | 50 | - | - | - |
| F14 | 100 | 150 | 200 | - | - | - | 50 | - | - |
| F15 | 100 | 150 | 200 | - | - | - | - | 50 | - |
| F16 | 100 | 150 | 200 | - | - | - | - | - | 50 |
| Formulation Code | Angle of Repose (θ) | Flow Rate (g/s) |
|---|---|---|
| F6 | 31.15 ± 0.92 | 4.43 ± 0.22 |
| F7 | 31.65 ± 0.37 | 4.50 ± 0.15 |
| F8 | 32.01 ± 0.55 | 4.74 ± 0.19 |
| F9 | 33.59 ± 0.71 | 4.90 ± 0.20 |
| F10 | 34.94 ± 0.34 | 5.54 ± 0.28 |
| F11 | 34.37 ± 0.35 | 5.76 ± 0.31 |
| F12 | 34.93 ± 0.48 | 5.95 ± 0.30 |
| F13 | 34.11 ± 0.70 | 5.43 ± 0.27 |
| F14 | 34.08 ± 0.64 | 5.83 ± 0.34 |
| F15 | 34.59 ± 0.84 | 5.44 ± 0.29 |
| F16 | 34.25 ± 0.43 | 5.87 ± 0.13 |
| Wavenumber of Peaks (cm−1) | |||
|---|---|---|---|
| CLC | PC | ||
| S=O (cm−1) | NH2 (cm−1) | P=O (cm−1) | |
| CLC powder | 1165/1347 | 3232/3338 | 1253 |
| F5 | 1164/1347 | 3234/3339 | 1235 |
| F10 | 1165/1348 | 3235/3343 | 1232 |
| F11 | 1165/1348 | 3237/3349 | 1232 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, K.; Cho, J.M.; Lee, H.; Kim, E.K.; Kim, H.C.; Kim, H.; Lee, J. Enhancement of Aqueous Solubility and Dissolution of Celecoxib through Phosphatidylcholine-Based Dispersion Systems Solidified with Adsorbent Carriers. Pharmaceutics 2019, 11, 1. https://doi.org/10.3390/pharmaceutics11010001
Jo K, Cho JM, Lee H, Kim EK, Kim HC, Kim H, Lee J. Enhancement of Aqueous Solubility and Dissolution of Celecoxib through Phosphatidylcholine-Based Dispersion Systems Solidified with Adsorbent Carriers. Pharmaceutics. 2019; 11(1):1. https://doi.org/10.3390/pharmaceutics11010001
Chicago/Turabian StyleJo, Kanghee, Jae Min Cho, Hyunjoo Lee, Eun Kyung Kim, Hong Chul Kim, Hyeongmin Kim, and Jaehwi Lee. 2019. "Enhancement of Aqueous Solubility and Dissolution of Celecoxib through Phosphatidylcholine-Based Dispersion Systems Solidified with Adsorbent Carriers" Pharmaceutics 11, no. 1: 1. https://doi.org/10.3390/pharmaceutics11010001
APA StyleJo, K., Cho, J. M., Lee, H., Kim, E. K., Kim, H. C., Kim, H., & Lee, J. (2019). Enhancement of Aqueous Solubility and Dissolution of Celecoxib through Phosphatidylcholine-Based Dispersion Systems Solidified with Adsorbent Carriers. Pharmaceutics, 11(1), 1. https://doi.org/10.3390/pharmaceutics11010001