Myeloma Bone Disease: Update on Pathogenesis and Novel Treatment Strategies



Abstract
1. Introduction
2. Pathogenesis of Osteolytic Bone Disease in MM
2.1. Physiologic Bone Remodeling
2.2. The Bone Niche in MM
2.3. Myeloma-Associated Bone Disease
2.3.1. Signaling Pathways Stimulating OC Activity in MM
RANK/RANKL Pathway
Notch/Jagged Pathway
Chemokines
Interleukins
2.3.2. Signaling Pathways Suppressing OB Activity in MM
Canonical and Non-Canonical Wnt Pathways
Activin A
Chemokines and Interleukins
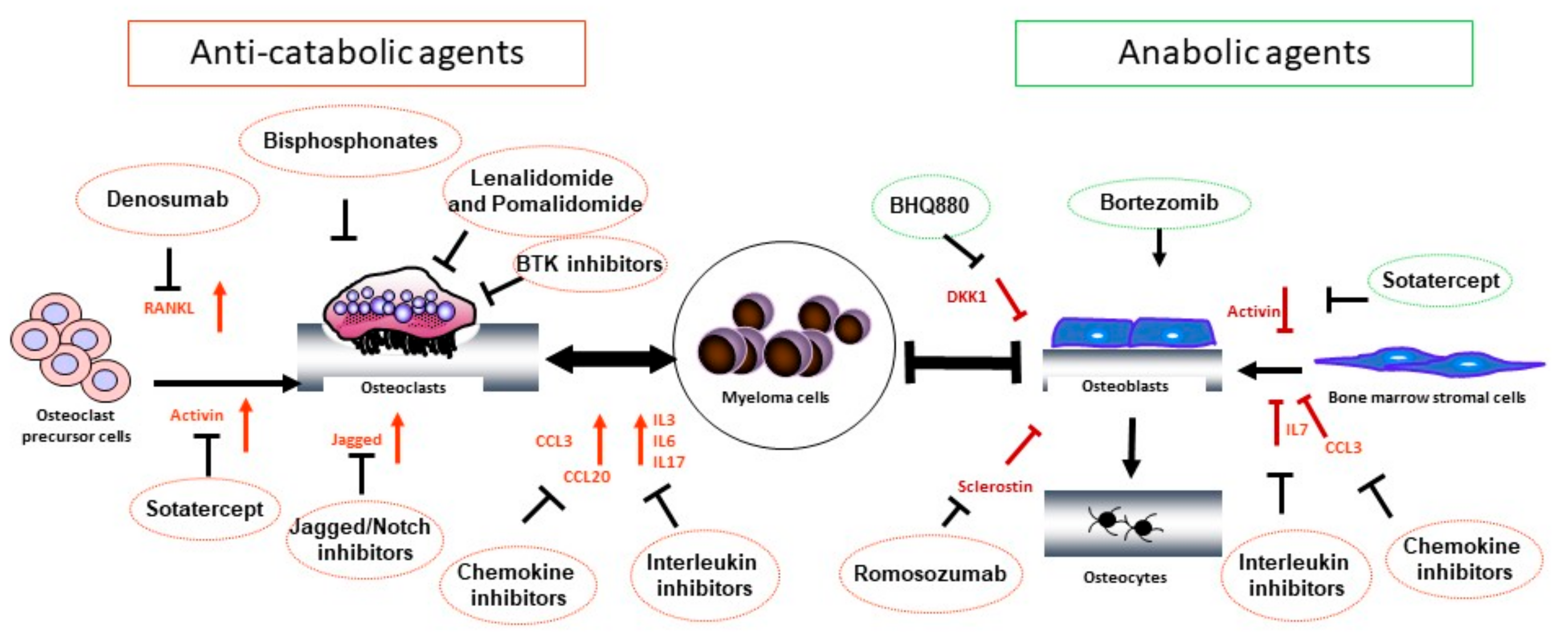
3. Treatment of Osteolytic Bone Disease in MM
3.1. Bisphosphonates
3.2. Denosumab
3.3. Anti-Tumor Therapies with Bone-Modifying Effects
3.3.1. Proteasome Inhibitors
3.3.2. Immunomodulatory Agents
3.4. Wnt Pathway Modulators
3.5. Promising Agents in Preclinical and Early Clinical Development
4. Concluding Remarks
Funding
Conflicts of Interest
References
- Moreau, P.; San Miguel, J.; Sonneveld, P.; Mateos, M.V.; Zamagni, E.; Avet-Loiseau, H.; Hajek, R.; Dimopoulos, M.A.; Ludwig, H.; Einsele, H.; et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple Myeloma: Diagnosis and Treatment. Mayo Clin. Proc. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Finkelstein, J.; Shullenberger, C.C.; Alexanian, R. Bone healing in multiple myeloma with melphalan chemotherapy. Ann. Intern. Med. 1972, 76, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.; Weisel, K.; Grandjean, C.; Oehrlein, K.; Zago, M.; Spira, D.; Horger, M. Increasing bone sclerosis during bortezomib therapy in multiple myeloma patients: Results of a reduced-dose whole-body MDCT study. Am. J. Roentgenol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hinge, M.; Andersen, K.T.; Lund, T.; Jørgensen, H.B.; Holdgaard, P.C.; Ormstrup, T.E.; Østergaard, L.L.; Plesner, T. Bone healing in multiple myeloma: A prospective evaluation of the impact of first-line anti-myeloma treatment. Haematologica 2016, 101, e419–e422. [Google Scholar] [CrossRef] [PubMed]
- Silbermann, R.; Roodman, G.D. Myeloma bone disease: Pathophysiology and management. J. Bone Oncol. 2013, 2, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Kastritis, E.; Zomas, A.; Terpos, E.; Katodritou, E.; Symeonidis, A.; Delimpasi, S.; Pouli, A.; Vassilakopoulos, T.P.; Michalis, E.; et al. Hypercalcemia remains an adverse prognostic factor for newly diagnosed multiple myeloma patients in the era of novel antimyeloma therapies. Eur. J. Haematol. 2017, 99, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Sonmez, M.; Akagun, T.; Topbas, M.; Cobanoglu, U.; Sonmez, B.; Yilmaz, M.; Ovali, E.; Omay, S.B. Effect of pathologic fractures on survival in multiple myeloma patients: A case control study. J. Exp. Clin. Cancer Res. 2008, 27. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Mechanisms of bone metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Pianko, M.J.; Terpos, E.; Roodman, G.D.; Divgi, C.R.; Zweegman, S.; Hillengass, J.; Lentzsch, S. Whole-body low-dose computed tomography and advanced imaging techniques for multiple myeloma bone disease. Clin. Cancer Res. 2014, 20, 5888–5897. [Google Scholar] [CrossRef] [PubMed]
- Hillengass, J.; Moulopoulos, L.A.; Delorme, S.; Koutoulidis, V.; Mosebach, J.; Hielscher, T.; Drake, M.; Rajkumar, S.V.; Oestergaard, B.; Abildgaard, N.; et al. Whole-body computed tomography versus conventional skeletal survey in patients with multiple myeloma: A study of the International Myeloma Working Group. Blood Cancer J. 2017, 7, e599. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Terpos, E.; Nanni, C.; Moreau, P.; Lentzsch, S.; Zweegman, S.; Hillengass, J.; Engelhardt, M.; Usmani, S.Z.; Vesole, D.H.; et al. Role of 18 F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders: A consensus statement by the International Myeloma Working Group. Lancet Oncol. 2017, 18, e206–e217. [Google Scholar] [CrossRef]
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; García-Sanz, R.; Durie, B.; Legieć, W.; Krejčí, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381. [Google Scholar] [CrossRef]
- Vallet, S.; Smith, M.R.; Raje, N. Novel bone-targeted strategies in oncology. Clin. Cancer Res. 2010, 16, 4084–4093. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, J.A.; Glimcher, M.J.; Cooper, R.R.; Recker, R. Bone biology. Part I: Structure, blood supply, cells, matrix, and mineralization. J. Bone Jt. Surg. Ser. A 1995, 45, 371–386. [Google Scholar]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Huang, W. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front. Biosci. 2007, 1, 3068–3092. [Google Scholar] [CrossRef]
- Harada, S.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef] [PubMed]
- Christenson, R.H. Biochemical markers of bone metabolism: An overview. Clin. Biochem. 1997, 30, 573–593. [Google Scholar] [CrossRef]
- Arnulf, B.; Lecourt, S.; Soulier, J.; Ternaux, B.; Lacassagne, M.N.; Crinquette, A.; Dessoly, J.; Sciaini, A.K.; Benbunan, M.; Chomienne, C.; et al. Phenotypic and functional characterization of bone marrow mesenchymal stem cells derived from patients with multiple myeloma. Leukemia 2007, 21, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Umezu, T.; Tadokoro, H.; Azuma, K.; Yoshizawa, S.; Ohyashiki, K.; Ohyashiki, J.H. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014, 124, 3748–3757. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Zimmerhackl, A.; Fulciniti, M.; Tonon, G.; Hainz, U.; Tai, Y.-T.; Vallet, S.; Halama, N.; Jäger, D.; Olson, D.L.; et al. The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: Therapeutic implications. Br. J. Haematol. 2011, 155, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Bashari, M.H.; Morelli, E.; Tonon, G.; Malvestiti, S.; Vallet, S.; Jarahian, M.; Seckinger, A.; Hose, D.; Bakiri, L.; et al. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 2017, 31, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Hiura, K.; Wilde, J.; Shioyasono, A.; Moriyama, K.; Hashimoto, T.; Kido, S.; Oshima, T.; Shibata, H.; Ozaki, S.; et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: A vicious cycle between bone destruction and myeloma expansion. Blood 2004, 104, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pennisi, A.; Yaccoby, S. Role of decorin in the antimyeloma effects of osteoblasts. Blood 2008, 112, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Yaccoby, S.; Wezeman, M.J.; Zangari, M.; Walker, R.; Cottler-Fox, M.; Gaddy, D.; Ling, W.; Saha, R.; Barlogie, B.; Tricot, G.; et al. Inhibitory effects of osteoblasts and increased bone formation on myeloma in novel culture systems and a myelomatous mouse model. Haematologica 2006, 91, 192–199. [Google Scholar] [PubMed]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Khoo, W.H.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8993. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Trotter, T.N.; Gibson, J.T.; Sherpa, T.L.; Gowda, P.S.; Peker, D.; Yang, Y. Adipocyte-Lineage Cells Support Growth and Dissemination of Multiple Myeloma in Bone. Am. J. Pathol. 2016, 186, 3054–3063. [Google Scholar] [CrossRef] [PubMed]
- Caers, J.; Deleu, S.; Belaid, Z.; De Raeve, H.; Van Valckenborgh, E.; De Bruyne, E.; DeFresne, M.P.; Van Riet, I.; Van Camp, B.; Vanderkerken, K. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia 2007, 21, 1580–1584. [Google Scholar] [CrossRef] [PubMed]
- Bullwinkle, E.M.; Parker, M.D.; Bonan, N.F.; Falkenberg, L.G.; Davison, S.P.; DeCicco-Skinner, K.L. Adipocytes contribute to the growth and progression of multiple myeloma: Unraveling obesity related differences in adipocyte signaling. Cancer Lett. 2016, 380, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Shi, Q.; Liu, Z.; He, J.; Liu, H.; Lin, P.; Cui, J.; Yang, J. Resistin induces multidrug resistance in myeloma by inhibiting cell death and upregulating ABC transporter expression. Haematologica 2017, 102, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Pratt, G.; Goodyear, O.; Moss, P. Immunodeficiency and immunotherapy in multiple myeloma. Br. J. Haematol. 2007, 138, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Favreau, M.; Menu, E.; Gaublomme, D.; Vanderkerken, K.; Faict, S.; Maes, K.; De Bruyne, E.; Govindarajan, S.; Drennan, M.; Van Calenbergh, S.; et al. Leptin receptor antagonism of iNKT cell function: A novel strategy to combat multiple myeloma. Leukemia 2017, 31, 2678–2685. [Google Scholar] [CrossRef] [PubMed]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.T.; Richardson, P.G.; et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.T.; Boehrer, A.; Sanchez, C.; Mercier, D.; Baier, C.; Le Treut, T.; Sébahoun, G. Differential expression of natural killer cell activating receptors in blood versus bone marrow in patients with monoclonal gammopathy. Immunology 2013, 139, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.V.; Brahmandam, M.; Carrasco, R.; Bandi, M.; Hideshima, T.; Bianchi, G.; Podar, K.; Tai, Y.T.; Mitsiades, C.; et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: A therapeutic target. Cancer Cell 2009, 16, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Cho, S.; Anderson, K. Osteoclast Immunosuppressive Effects in Multiple Myeloma: Role of Programmed Cell Death Ligand 1. Front. Immunol. 2018, 10, 1822. [Google Scholar] [CrossRef] [PubMed]
- Melton, L.J., 3rd; Rajkumar, S.V.; Khosla, S.; Achenbach, S.J.; Oberg, A.L.; Kyle, R.A. Fracture risk in monoclonal gammopathy of undetermined significance. J. Bone Miner. Res. 2004, 19, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Bataille, R.; Chappard, D.; Marcelli, C.; Dessauw, P.; Sany, J.; Baldet, P.; Alexandre, C. Mechanisms of bone destruction in multiple myeloma: The importance of an unbalanced process in determining the severity of lytic bone disease. J. Clin. Oncol. 1989, 7, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Bataille, R.; Chappard, D.; Basle, M.F. Quantifiable excess of bone resorption in monoclonal gammopathy is an early symptom of malignancy: A prospective study of 87 bone biopsies. Blood 1996, 87, 4762–4769. [Google Scholar] [PubMed]
- Vallet, S.; Hoyle, N.R.; Kyle, R.A.; Podar, K.; Pecherstorfer, M. A role for bone turnover markers β-CrossLaps (CTX) and amino-terminal propeptide of type I collagen (PINP) as potential indicators for disease progression from MGUS to multiple myeloma. Leuk. Lymphoma 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Dimopoulos, M.A.; Sezer, O.; Roodman, D.; Abildgaard, N.; Vescio, R.; Tosi, P.; Garcia-Sanz, R.; Davies, F.; Chanan-Khan, A.; et al. The use of biochemical markers of bone remodeling in multiple myeloma: A report of the International Myeloma Working Group. Leukemia 2010, 24, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; De Luca, A.; Amodio, N.; Manno, M.; Raccosta, S.; Taverna, S.; Bellavia, D.; Naselli, F.; Fontana, S.; Schillaci, O.; et al. Involvement of multiple myeloma cell-derived exosomes in osteoclast differentiation. Oncotarget 2015, 6, 13772–13789. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Ferretti, M.; Bolzoni, M.; Storti, P.; Lazzaretti, M.; Dalla Palma, B.; Bonomini, S.; Martella, E.; Agnelli, L.; Neri, A.; et al. Increased osteocyte death in multiple myeloma patients: Role in myeloma-induced osteoclast formation. Leukemia 2012, 26, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Colla, S.; Sala, R.; Moroni, M.; Lazzaretti, M.; La Monica, S.; Bonomini, S.; Hojden, M.; Sammarelli, G.; Barille, S.; et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: A potential role in multiple myeloma bone disease. Blood 2002, 100, 4615–4621. [Google Scholar] [CrossRef] [PubMed]
- Sezer, O.; Heider, U.; Zavrski, I.; Kuhne, C.A.; Hofbauer, L.C. RANK ligand and osteoprotegerin in myeloma bone disease. Blood 2003, 101, 2094–2098. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Previs, R.A.; Coleman, R.L.; Harris, A.L.; Sood, A.K. Molecular pathways: Translational and therapeutic implications of the Notch signaling pathway in cancer. Clin. Cancer Res. 2015, 21, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Mirandola, L.; Platonova, N.; Apicella, L.; Basile, A.; Figueroa, A.J.; Cobos, E.; Chiriva-Internati, M.; Chiaramonte, R. Notch-directed microenvironment reprogramming in myeloma: A single path to multiple outcomes. Leukemia 2013, 27, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Thümmler, K.; Mirandola, L.; Garavelli, S.; Todoerti, K.; Apicella, L.; Lazzari, E.; Lancellotti, M.; Platonova, N.; Akbar, M.; et al. Notch signaling drives multiple myeloma induced osteoclastogenesis. Oncotarget 2014, 5, 10393–10406. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Raje, N.; Ishitsuka, K.; Hideshima, T.; Podar, K.; Chhetri, S.; Pozzi, S.; Breitkreutz, I.; Kiziltepe, T.; Yasui, H.; et al. MLN3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood 2007, 110, 3744–3752. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Abe, M. Bone destruction in multiple myeloma. Ann. N. Y. Acad. Sci. 2006, 1068, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Kato, C.; Manno, M.; Ogaki, M.; Satou, T.; Itoh, T.; Kusunoki, T.; Tanimori, Y.; Fujiwara, K.; Matsuoka, H.; et al. Macrophage inflammatory protein-1alpha (MIP-1alpha) enhances a receptor activator of nuclear factor kappaB ligand (RANKL) expression in mouse bone marrow stromal cells and osteoblasts through MAPK and PI3K/Akt pathways. Mol. Cell. Biochem. 2007, 304, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Politou, M.; Szydlo, R.; Goldman, J.M.; Apperley, J.F.; Rahemtulla, A. Serum levels of macrophage inflammatory protein-1 alpha (MIP-1alpha) correlate with the extent of bone disease and survival in patients with multiple myeloma. Br. J. Haematol. 2003, 123, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Lisignoli, G.; Colla, S.; Lazzaretti, M.; Storti, P.; Mancini, C.; Bonomini, S.; Manferdini, C.; Codeluppi, K.; Facchini, A.; et al. CC-Chemokine ligand 20/macrophage inflammatory protein-3alpha and CC-chemokine receptor 6 are overexpressed in myeloma microenvironment related to osteolytic bone lesions. Cancer Res. 2008, 68, 6840–6850. [Google Scholar] [CrossRef] [PubMed]
- Dalla Palma, B.; Guasco, D.; Pedrazzoni, M.; Bolzoni, M.; Accardi, F.; Costa, F.; Sammarelli, G.; Craviotto, L.; De Filippo, M.; Ruffini, L.; et al. Osteolytic lesions, cytogenetic features and bone marrow levels of cytokines and chemokines in multiple myeloma patients: Role of chemokine (C-C motif) ligand 20. Leukemia 2016, 30, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Schlossman, R.; Richardson, P.; Anderson, K.C. The role of tumor necrosis factor α in the pathophysiology of human multiple myeloma: Therapeutic applications. Oncogene 2001, 20, 4519–4527. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Gilbert, L.; He, X.; Rubin, J.; Nanes, M.S. Transcriptional regulation of the osterix (Osx, Sp7) promoter by tumor necrosis factor identifies disparate effects of mitogen-activated protein kinase and NF kappa B pathways. J. Biol. Chem. 2006, 281, 6297–6306. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Katagiri, T.; Tamura, T.; Wada, S.; Findlay, D.M.; Martin, T.J.; Hirota, H.; Taga, T.; Kishimoto, T.; et al. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J. Exp. Med. 1995, 182, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Chung, H.Y.; Ehrlich, L.A.; Jelinek, D.F.; Callander, N.S.; Roodman, G.D.; Choi, S.J. IL-3 expression by myeloma cells increases both osteoclast formation and growth of myeloma cells. Blood 2004, 103, 2308–2315. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.A.; Chung, H.Y.; Ghobrial, I.; Choi, S.J.; Morandi, F.; Colla, S.; Rizzoli, V.; Roodman, G.D.; Giuliani, N. IL-3 is a potential inhibitor of osteoblast differentiation in multiple myeloma. Blood 2005, 106, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Silbermann, R.; Bolzoni, M.; Storti, P.; Guasco, D.; Bonomini, S.; Zhou, D.; Wu, J.; Anderson, J.L.; Windle, J.J.; Aversa, F.; et al. Bone marrow monocyte-/macrophage-derived activin A mediates the osteoclastogenic effect of IL-3 in multiple myeloma. Leukemia 2014, 28, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Noonan, K.; Marchionni, L.; Anderson, J.; Pardoll, D.; Roodman, G.D.; Borrello, I. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood 2010, 116, 3554–3563. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Yamamoto, I.; Iwato, K.; Tanaka, H.; Asaoku, H.; Tanabe, O.; Ishikawa, H.; Nobuyoshi, M.; Ohmoto, Y.; Hirai, Y.; et al. Interleukin-1 beta rather than lymphotoxin as the major bone resorbing activity in human multiple myeloma. Blood 1989, 73, 1646–1649. [Google Scholar] [PubMed]
- Hjertner, O.; Torgersen, M.L.; Seidel, C.; Hjorth-Hansen, H.; Waage, A.; Børset, M.; Sundan, A. Hepatocyte growth factor (HGF) induces interleukin-11 secretion from osteoblasts: A possible role for HGF in myeloma-associated osteolytic bone disease. Blood 1999, 94, 3883–3888. [Google Scholar] [PubMed]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Mao, J.; Belsky, J.; Mitzner, L.; Farhi, A.; Mitnick, M.A.; Wu, D.; Insogna, K.; Lifton, R.P. High Bone Density Due to a Mutation in LDL-Receptor–Related Protein 5. N. Engl. J. Med. 2002, 346, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Spaan, I.; Raymakers, R.A.; Van De Stolpe, A.; Peperzak, V. Wnt signaling in multiple myeloma: A central player in disease with therapeutic potential. J. Hematol. Oncol. 2018, 11, 67. [Google Scholar] [CrossRef] [PubMed]
- Politou, M.C.; Heath, D.J.; Rahemtulla, A.; Szydlo, R.; Anagnostopoulos, A.; Dimopoulos, M.A.; Croucher, P.I.; Terpos, E. Serum concentrations of Dickkopf-1 protein are increased in patients with multiple myeloma and reduced after autologous stem cell transplantation. Int. J. Cancer 2006, 119, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Eda, H.; Santo, L.; Wein, M.N.; Hu, D.Z.; Cirstea, D.D.; Nemani, N.; Tai, Y.T.; Raines, S.E.; Kuhstoss, S.A.; Munshi, N.C.; et al. Regulation of Sclerostin Expression in Multiple Myeloma by Dkk-1: A Potential Therapeutic Strategy for Myeloma Bone Disease. J. Bone Miner. Res. 2016, 31, 1225–1234. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Ramos Fuentes, F.J.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Sato, A.Y.; Bellido, T. Role and mechanism of action of sclerostin in bone. Bone 2017, 96, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, N.; Ye, L.; Kobayashi, T.; Mochida, Y.; Yamauchi, M.; Kronenberg, H.M.; Feng, J.Q.; Mishina, Y. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 2008, 135, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Condon, K.W.; Kuhstoss, S.A.; Plotkin, L.I.; Bellido, T.; Roodman, G.D. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017, 31, 2686–2694. [Google Scholar] [CrossRef] [PubMed]
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D., Jr. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N. Engl. J. Med. 2003, 349, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Gunn, W.G.; Conley, A.; Deininger, L.; Olson, S.D.; Prockop, D.J.; Gregory, C.A. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: A potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells 2006, 24, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Walker, B.A.; Brewer, D.; Gregory, W.M.; Ashcroft, J.; Ross, F.M.; Jackson, G.H.; Child, A.J.; Davies, F.E.; Morgan, G.J. A gene expression-based predictor for myeloma patients at high risk of developing bone disease on bisphosphonate treatment. Clin. Cancer Res. 2011, 17, 6347–6355. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Donofrio, G.; Storti, P.; Guasco, D.; Toscani, D.; Lazzaretti, M.; Bonomini, S.; Agnelli, L.; Capocefalo, A.; Dalla Palma, B.; et al. Myeloma cells inhibit non-canonical wnt co-receptor ror2 expression in human bone marrow osteoprogenitor cells: Effect of wnt5a/ror2 pathway activation on the osteogenic differentiation impairment induced by myeloma cells. Leukemia 2013, 27, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Mukherjee, S.; Vaghela, N.; Hideshima, T.; Fulciniti, M.; Pozzi, S.; Santo, L.; Cirstea, D.; Patel, K.; Sohani, A.R.; et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc. Natl. Acad. Sci. USA 2010, 107, 5124–5129. [Google Scholar] [CrossRef] [PubMed]
- Fuller, K.; Bayley, K.E.; Chambers, T.J. Activin A is an essential cofactor for osteoclast induction. Biochem. Biophys. Res. Commun. 2000, 268, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Colla, S.; Morandi, F.; Lazzaretti, M.; Sala, R.; Bonomini, S.; Grano, M.; Colucci, S.; Svaldi, M.; Rizzoli, V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005, 106, 2472–2483. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Pozzi, S.; Patel, K.; Vaghela, N.; Fulciniti, M.T.; Veiby, P.; Hideshima, T.; Santo, L.; Cirstea, D.; Scadden, D.T.; et al. A novel role for CCL3 (MIP-1α) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia 2011, 25, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Kassen, D.; Lath, D.; Lach, A.; Evans, H.; Chantry, A.; Rabin, N.; Croucher, P.; Yong, K.L. Myeloma impairs mature osteoblast function but causes early expansion of osteo-progenitors: Temporal changes in bone physiology and gene expression in the KMS12BM model. Br. J. Haematol. 2016, 172, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Ismaila, N.; Flynn, P.J.; Halabi, S.; Jagannath, S.; Ogaily, M.S.; Omel, J.; Raje, N.; Roodman, G.D.; Yee, G.C.; et al. Role of bone-modifying agents in multiple myeloma: American society of clinical oncology clinical practice guideline update. J. Clin. Oncol. 2018, 36, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Podar, K. New insights, recent advances, and current challenges in the biological treatment of multiple myeloma. Expert Opin. Biol. Ther. 2013, 13, S35–S53. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Anderson, K.C. Introduction: The evolving role of bisphosphonate therapy in multiple myeloma. Blood 2000, 96, 381–383. [Google Scholar] [PubMed]
- Dunford, J.E.; Kwaasi, A.A.; Rogers, M.J.; Barnett, B.L.; Ebetino, F.H.; Russell, R.G.; Oppermann, U.; Kavanagh, K.L. Structure-activity relationships among the nitrogen containing bisphosphonates in clinical use and other analogues: Time-dependent inhibition of human farnesyl pyrophosphate synthase. J. Med. Chem. 2008, 51, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Davies, F.E.; Gregory, W.M.; Cocks, K.; Bell, S.E.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.T.; Owen, R.G.; Feyler, S.; et al. First-line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC Myeloma IX): A randomised controlled trial. Lancet 2010, 376, 1989–1999. [Google Scholar] [CrossRef]
- Perazella, M.A.; Markowitz, G.S. Bisphosphonate nephrotoxicity. Kidney Int. 2008, 74, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Yee, G.C.; Somerfield, M.R.; Flynn, P.J.; Halabi, S.; Jagannath, S.; Orlowski, R.Z.; Roodman, D.G.; Twilde, P.; Anderson, K. American Society of Clinical Oncology 2007 clinical practice guideline update on the role of bisphosphonates in multiple myeloma. J. Clin. Oncol. 2007, 25, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Woo, S.-B.; Hande, K.; Yap, J.T.; Richardson, P.G.; Vallet, S.; Treister, N.; Hideshima, T.; Sheehy, N.; Chhetri, S.; et al. Clinical, radiographic, and biochemical characterization of multiple myeloma patients with osteonecrosis of the jaw. Clin. Cancer Res. 2008, 14, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Schilcher, J.; Aspenberg, P. Incidence of stress fractures of the femoral shaft in women treated with bisphosphonate. Acta Orthop. 2009, 80, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Black, D.M.; Kelly, M.P.; Genant, H.K.; Palermo, L.; Eastell, R.; Bucci-Rechtweg, C.; Cauley, J.; Leung, P.C.; Boonen, S.; Santora, A.; et al. Bisphosphonates and Fractures of the Subtrochanteric or Diaphyseal Femur. N. Engl. J. Med. 2010, 362, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.R. Bisphosphonate effects on bone turnover, microdamage, and mechanical properties: What we think we know and what we know that we don’t know. Bone 2011, 49, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, M.C.H.; Boskey, A.L. Atypical subtrochanteric femoral shaft fractures: Role for mechanics and bone quality. Arthritis Res. Ther. 2012, 14, 220. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Vescio, R.; Montgomery, C.W.; Badros, A.; Munshi, N.; Orlowski, R.; Hadala, J.T.; Warsi, G.; Argonza-Aviles, E.; Ericson, S.G.; et al. Bone marker-directed dosing of zoledronic acid for the prevention of skeletal complications in patients with multiple myeloma: Results of the Z-MARK study. Clin. Cancer Res. 2016, 22, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Kostenuik, P.J.; Nguyen, H.Q.; McCabe, J.; Warmington, K.S.; Kurahara, C.; Sun, N.; Chen, C.; Li, L.; Cattley, R.C.; Van, G.; et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J. Bone Miner. Res. 2009, 24, 182–195. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R.; Lewiecki, E.M.; Cohen, S.B.; Bolognese, M.A.; Woodson, G.C.; Moffett, A.H.; Peacock, M.; Miller, P.D.; Lederman, S.N.; Chesnut, C.H.; et al. Denosumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2006, 354, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Steger, G.G.; Figueroa, J.; Alvarado, C.; Solal-Celigny, P.; Body, J.J.; de Boer, R.; Berardi, R.; Gascon, P.; Tonkin, K.S.; et al. Randomized active-controlled phase II study of denosumab efficacy and safety in patients with breast cancer-related bone metastases. J. Clin. Oncol. 2007, 25, 4431–4437. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Horvath, N.; Spencer, A.; Taylor, K.; Vadhan-Raj, S.; Vescio, R.; Smith, J.; Qian, Y.; Yeh, H.; Jun, S. An open-label, phase 2 trial of denosumab in the treatment of relapsed or plateau-phase multiple myeloma. Am. J. Hematol. 2009, 84, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Henry, D.H.; Costa, L.; Goldwasser, F.; Hirsh, V.; Hungria, V.; Prausova, J.; Scagliotti, G.V.; Sleeboom, H.; Spencer, A.; Vadhan-Raj, S.; et al. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J. Clin. Oncol. 2011, 29, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Vadhan-Raj, S.; Willenbacher, W.; Terpos, E.; Hungria, V.; Spencer, A.; Alexeeva, Y.; Facon, T.; Stewart, A.K.; Feng, A.; et al. Evaluating results from the multiple myeloma patient subset treated with denosumab or zoledronic acid in a randomized phase 3 trial. Blood Cancer J. 2016, 6, e378. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Roodman, D.; Willenbacher, W.; Shimizu, K.; García-Sanz, R.; Durie, B.; Zhu, L.; Bhatta, S.; Raje, N. Comparison of denosumab with zoledronic acid for the treatment of bone disease in patients with newly diagnosed Multiple Myeloma; an international, randomized, double blind trial. EHA 2017, 102, S782. [Google Scholar]
- Roccaro, A.M.; Hideshima, T.; Richardson, P.G.; Russo, D.; Ribatti, D.; Vacca, A.; Dammacco, F.; Anderson, K.C. Bortezomib as an antitumor agent. Curr. Pharm. Biotechnol. 2006, 7, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Raje, N.; Schoonmaker, J.A.; Liu, J.C.; Hideshima, T.; Wein, M.N.; Jones, D.C.; Vallet, S.; Bouxsein, M.L.; Pozzi, S.; et al. Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. J. Clin. Investig. 2008, 118, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Von Metzler, I.; Krebbel, H.; Hecht, M.; Manz, R.A.; Fleissner, C.; Mieth, M.; Kaiser, M.; Jakob, C.; Sterz, J.; Kleeberg, L.; et al. Bortezomib inhibits human osteoclastogenesis. Leukemia 2007, 21, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Zangari, M.; Esseltine, D.; Lee, C.K.; Barlogie, B.; Elice, F.; Burns, M.J.; Kang, S.H.; Yaccoby, S.; Najarian, K.; Richardson, P.; et al. Response to bortezomib is associated to osteoblastic activation in patients with multiple myeloma. Br. J. Haematol. 2005, 131, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Lazzaretti, M.; Bonomini, S.; Crugnola, M.; Mancini, C.; Martella, E.; Ferrari, L.; Tabilio, A.; et al. The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood 2007, 110, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Lund, T.; Søe, K.; Abildgaard, N.; Garnero, P.; Pedersen, P.T.; Ormstrup, T.; Delaissé, J.M.; Plesner, T. First-line treatment with bortezomib rapidly stimulates both osteoblast activity and bone matrix deposition in patients with multiple myeloma, and stimulates osteoblast proliferation and differentiation in vitro. Eur. J. Haematol. 2010, 85, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Toscani, D.; Palumbo, C.; Dalla Palma, B.; Ferretti, M.; Bolzoni, M.; Marchica, V.; Sena, P.; Martella, E.; Mancini, C.; Ferri, V.; et al. The Proteasome Inhibitor Bortezomib Maintains Osteocyte Viability in Multiple Myeloma Patients by Reducing both Apoptosis and Autophagy: A New Function for Proteasome Inhibitors. J. Bone Miner. Res. 2016, 31, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Gries, M.; Kurihara, N.; Honjo, T.; Anderson, J.; Donnenberg, V.; Donnenberg, A.; Ghobrial, I.; Mapara, M.Y.; Stirling, D.; et al. Thalidomide derivative CC-4047 inhibits osteoclast formation by down-regulation of PU.1. Blood 2006, 107, 3098–3105. [Google Scholar] [CrossRef] [PubMed]
- Breitkreutz, I.; Raab, M.S.; Vallet, S.; Hideshima, T.; Raje, N.; Mitsiades, C.; Chauhan, D.; Okawa, Y.; Munshi, N.C.; Richardson, P.G.; et al. Lenalidomide inhibits osteoclastogenesis, survival factors and bone-remodeling markers in multiple myeloma. Leukemia 2008, 22, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Storti, P.; Bonomini, S.; Todoerti, K.; Guasco, D.; Toscani, D.; Agnelli, L.; Neri, A.; Rizzoli, V.; Giuliani, N. Immunomodulatory drugs lenalidomide and pomalidomide inhibit multiple myeloma-induced osteoclast formation and the RANKL/OPG ratio in the myeloma microenvironment targeting the expression of adhesion molecules. Exp. Hematol. 2012, 41, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Christoulas, D.; Gkotzamanidou, M.; Bratengeier, C.; Gavriatopoulou, M.; Migkou, M.; Papatheodorou, A.; Kastritis, E.; Woloszczuk, W.; Dimopoulos, M.A.C.E.T. Circulating Levels of the Wnt Inhibitors Dickkopf-1 and Sclerostin in Different Phases of Multiple Myeloma: Alterations Post-Therapy with Lenalidomide and Dexamethasone with or without Bortezomib. Blood 2010, 116, 2963. [Google Scholar]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.-T.; Chauhan, D.; et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Zheng, Y.; Zheng, C.; Wang, L.; Qin, H.; Hong, S.; Li, H.; Lu, Y.; He, J.; Yang, J.; et al. Active vaccination with Dickkopf-1 induces protective and therapeutic antitumor immunity in murine multiple myeloma. Blood 2012, 119, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, S.; Fulciniti, M.; Yan, H.; Vallet, S.; Eda, H.; Patel, K.; Santo, L.; Cirstea, D.; Hideshima, T.; Schirtzinge, L.; et al. In vivo and in vitro effects of a novel anti-Dkk1 neutralizing antibody in multiple myeloma. Bone 2013, 53, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Heath, D.J.; Chantry, A.D.; Buckle, C.H.; Coulton, L.; Shaughnessy, J.D.; Evans, H.R.; Snowden, J.A.; Stover, D.R.; Vanderkerken, K.; Croucher, P.I. Inhibiting Dickkopf-1 (Dkk1) Removes Suppression of Bone Formation and Prevents the Development of Osteolytic Bone Disease in Multiple Myeloma. J. Bone Miner. Res. 2008, 24, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Abonour, R.; Beck, J.T.; Bensinger, W.; Facon, T.; Stockerl-Goldstein, K.; Baz, R.; Siegel, D.S.; Neben, K.; Lonial, S.; Suvannasankha, A.; et al. Early Evidence of Anabolic Bone Activity of BHQ880, a Fully Human Anti-DKK1 Neutralizing Antibody: Results of a Phase 2 Study in Previously Untreated Patients with Smoldering Multiple Myeloma at Risk for Progression. Blood 2012, 120, 331. [Google Scholar]
- Iyer, S.P.; Beck, J.T.; Stewart, A.K.; Shah, J.; Kelly, K.R.; Isaacs, R.; Bilic, S.; Sen, S.; Munshi, N.C. A Phase IB multicentre dose-determination study of BHQ880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br. J. Haematol. 2014, 167, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.M.; Reagan, M.R.; Youlten, S.E.; Mohanty, S.T.; Seckinger, A.; Terry, R.L.; Pettitt, J.A.; Simic, M.K.; Cheng, T.L.; Morse, A.; et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017, 129, 3452–3464. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.M.; Delgado-Calle, J. Sclerostin: An Emerging Target for the Treatment of Cancer-Induced Bone Disease. Curr. Osteoporos. Rep. 2017, 15, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Vallet, S. Sotatercept, a soluble activin receptor type 2A IgG-Fc fusion protein for the treatment of anemia and bone loss. Curr. Opin. Mol. Ther. 2010, 12, 586–597. [Google Scholar] [PubMed]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Scullen, T.; Santo, L.; Vallet, S.; Fulciniti, M.; Eda, H.; Cirstea, D.; Patel, K.; Nemani, N.; Yee, A.; Mahindra, A.; et al. Lenalidomide in combination with an activin A-neutralizing antibody: Preclinical rationale for a novel anti-myeloma strategy. Leukemia 2013, 27, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 Inhibition Reverses Multiple Myeloma–Induced Epigenetic Suppression of Osteoblast Differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Deleu, S.; Lemaire, M.; Arts, J.; Menu, E.; Van Valckenborgh, E.; King, P.; Vande Broek, I.; De Raeve, H.; Van Camp, B.; Croucher, P.; et al. The effects of JNJ-26481585, a novel hydroxamate-based histone deacetylase inhibitor, on the development of multiple myeloma in the 5T2MM and 5T33MM murine models. Leukemia 2009, 23, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; De Veirman, K.; Evans, H.; Santini, G.C.; Vande Broek, I.; Leleu, X.; De Becker, A.; Van Camp, B.; Croucher, P.; Vanderkerken, K.; et al. Effect of the HDAC inhibitor vorinostat on the osteogenic differentiation of mesenchymal stem cells in vitro and bone formation in vivo. Acta Pharmacol. Sin. 2013, 34, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, L.; Apicella, L.; Colombo, M.; Yu, Y.; Berta, D.G.; Platonova, N.; Lazzari, E.; Lancellotti, M.; Bulfamante, G.; Cobos, E.; et al. Anti-Notch treatment prevents multiple myeloma cells localization to the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia 2013, 27, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, R.; Nickel, N.; Godau, J.; Willie, B.M.; Duda, G.N.; Schwarzer, R.; Cirovic, B.; Leutz, A.; Manz, R.; Bogen, B.; et al. Notch pathway inhibition controls myeloma bone disease in the murine MOPC315.BM model. Blood Cancer J. 2014, 4, e217. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Walls, M.; Qiu, M.; Ding, R.; Denlinger, R.H.; Wong, A.; Tsaparikos, K.; Jani, J.P.; Hosea, N.; Sands, M.; et al. Evaluation of Selective Gamma-Secretase Inhibitor PF-03084014 for Its Antitumor Efficacy and Gastrointestinal Safety to Guide Optimal Clinical Trial Design. Mol. Cancer Ther. 2010, 9, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J.; et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; De Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; LoRusso, P.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A phase I first-in-human study of enoticumab (REGN421), a fully human delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Anderson, K.C. CCR1 as a target for multiple myeloma. Expert Opin. Ther. Targets 2011, 15, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Menu, E.; De Leenheer, E.; De Raeve, H.; Coulton, L.; Imanishi, T.; Miyashita, K.; Van Valckenborgh, E.; Van Riet, I.; Van Camp, B.; Horuk, R.; et al. Role of CCR1 and CCR5 in homing and growth of multiple myeloma and in the development of osteolytic lesions: A study in the 5TMM model. Clin. Exp Metastasis 2006, 23, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Prabhala, R.H.; Fulciniti, M.; Pelluru, D.; Rashid, N.; Nigroiu, A.; Nanjappa, P.; Pai, C.; Lee, S.; Prabhala, N.S.; Bandi, R.L.; et al. Targeting IL-17A in multiple myeloma: A potential novel therapeutic approach in myeloma. Leukemia 2016, 30, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Fulciniti, M.; Hideshima, T.; Vermot-Desroches, C.; Pozzi, S.; Nanjappa, P.; Shen, Z.; Patel, N.; Smith, E.S.; Wang, W.; Prabhala, R.; et al. A high-affinity fully human anti-IL-6 mAb, 1339, for the treatment of multiple myeloma. Clin. Cancer Res. 2009, 15, 7144–7152. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Chang, B.Y.; Kong, S.Y.; Fulciniti, M.; Yang, G.; Calle, Y.; Hu, Y.; Lin, J.; Zhao, J.J.; Cagnetta, A.; et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood 2012, 120, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Hiasa, M.; Teramachi, J.; Oda, A.; Amachi, R.; Harada, T.; Nakamura, S.; Miki, H.; Fujii, S.; Kagawa, K.; Watanabe, K.; et al. Pim-2 kinase is an important target of treatment for tumor progression and bone loss in myeloma. Leukemia 2015, 29, 207–217. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Zoledronate | Denosumab | |
|---|---|---|
| Agent | Nitrogen-containing bisphosphonate | Fully human anti-RANKL IgG2 monoclonal antibody |
| Indications [97] | Patients treated for active myeloma with or without lytic lesions. | Myeloma patients with evidence of lytic bone lesions |
| Dosing schedule [97,109] | Renal-adapted, iv administration every 3–4 weeks or every 12 weeks | sc administration, every 4 weeks |
| Suggested duration of treatment [97] | Up to 2 years | No recommendations available |
| Median time to first SRE [14] | 24 months | 22.8 months |
| Median PFS [116] | 35.4 months | 46.1 months |
| Renal toxicity [14] | 17% | 10% |
| ONJ [14] | 3% | 4% |
| Hypocalcemia [14] | 12% | 17% |
| Monitoring |
|
|
| Molecular Target | Function | Therapeutic Relevance |
|---|---|---|
| Jagged/Notch pathway [35,61] |
| |
| CCL3 (MIP-1α) [62,63,64] |
| |
| CCL20 (MIP-3α) [66] |
| |
| IL-3 [72,73,74] |
| |
| IL-17 [75] |
|
|
| IL-7 [94] |
| |
| IL-6, IL-1β, IL-11 [71,76,77] |
|
|
| DKK1 [83,88] |
| |
| Sclerostin [23,86] |
|
|
| Activin A [92,93] |
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallet, S.; Filzmoser, J.-M.; Pecherstorfer, M.; Podar, K. Myeloma Bone Disease: Update on Pathogenesis and Novel Treatment Strategies. Pharmaceutics 2018, 10, 202. https://doi.org/10.3390/pharmaceutics10040202
Vallet S, Filzmoser J-M, Pecherstorfer M, Podar K. Myeloma Bone Disease: Update on Pathogenesis and Novel Treatment Strategies. Pharmaceutics. 2018; 10(4):202. https://doi.org/10.3390/pharmaceutics10040202
Chicago/Turabian StyleVallet, Sonia, Julia-Marie Filzmoser, Martin Pecherstorfer, and Klaus Podar. 2018. "Myeloma Bone Disease: Update on Pathogenesis and Novel Treatment Strategies" Pharmaceutics 10, no. 4: 202. https://doi.org/10.3390/pharmaceutics10040202
APA StyleVallet, S., Filzmoser, J.-M., Pecherstorfer, M., & Podar, K. (2018). Myeloma Bone Disease: Update on Pathogenesis and Novel Treatment Strategies. Pharmaceutics, 10(4), 202. https://doi.org/10.3390/pharmaceutics10040202