Overview of Antibody Drug Delivery


Abstract

{kind=link}
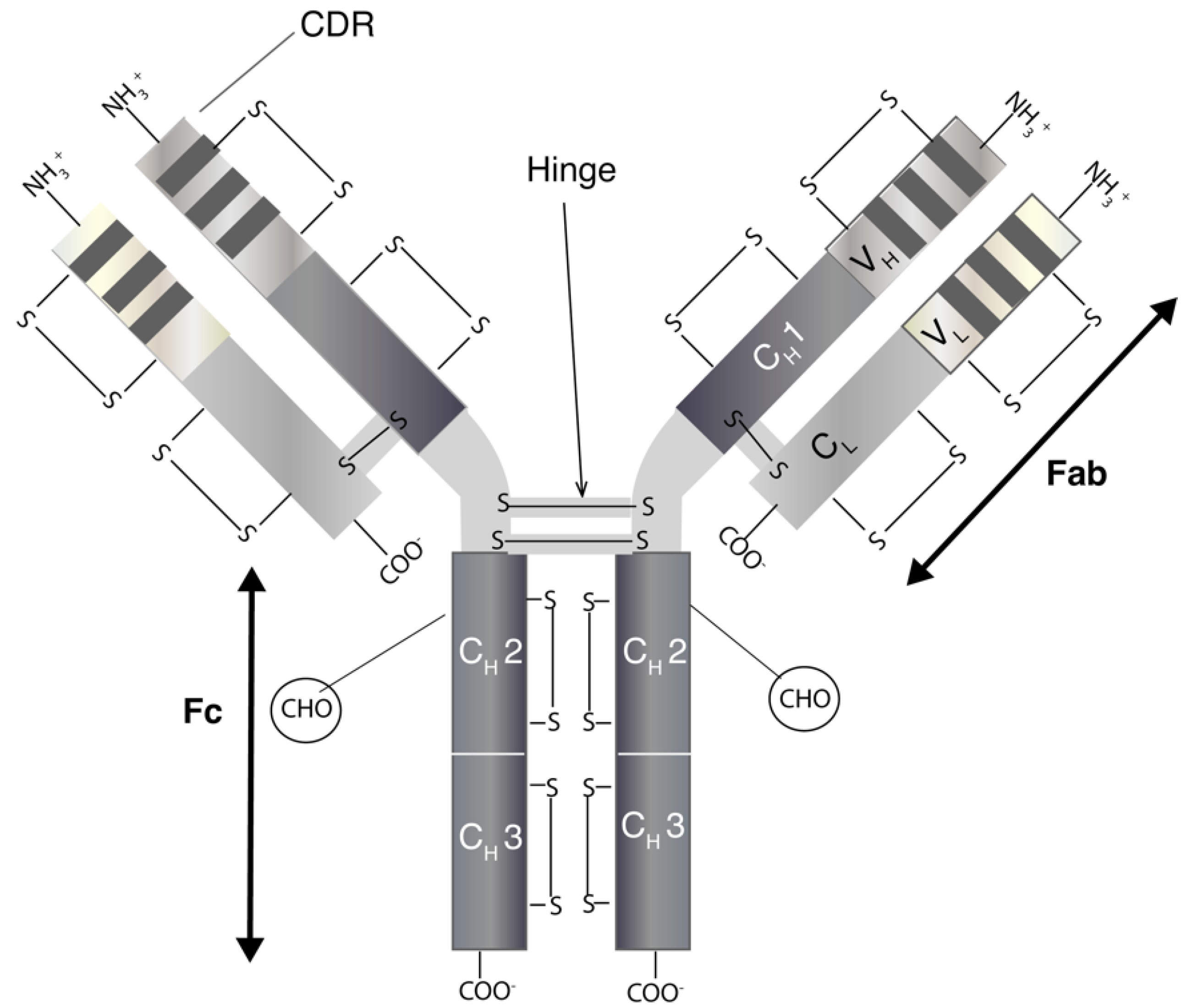{kind=link}
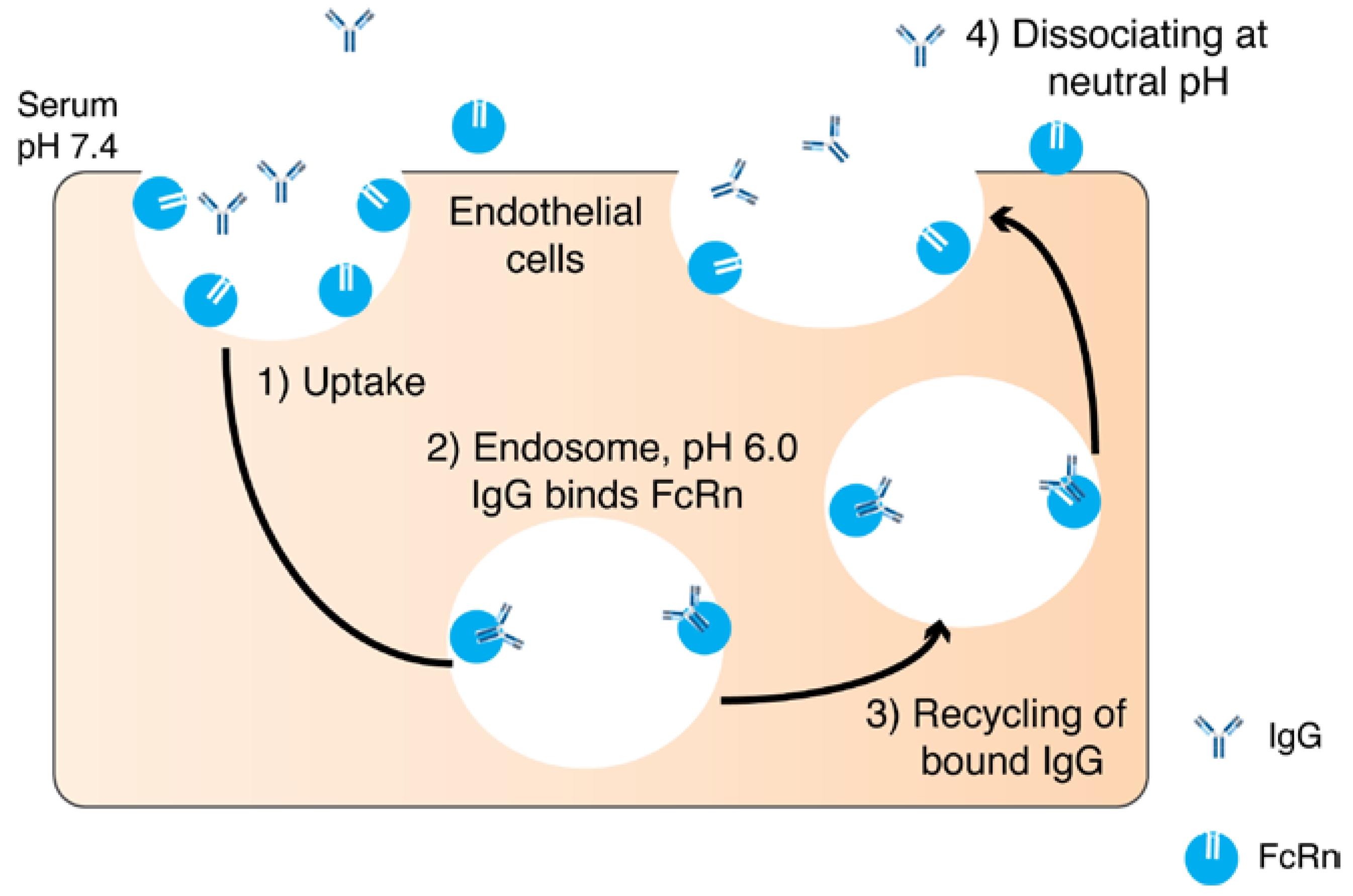{kind=link}
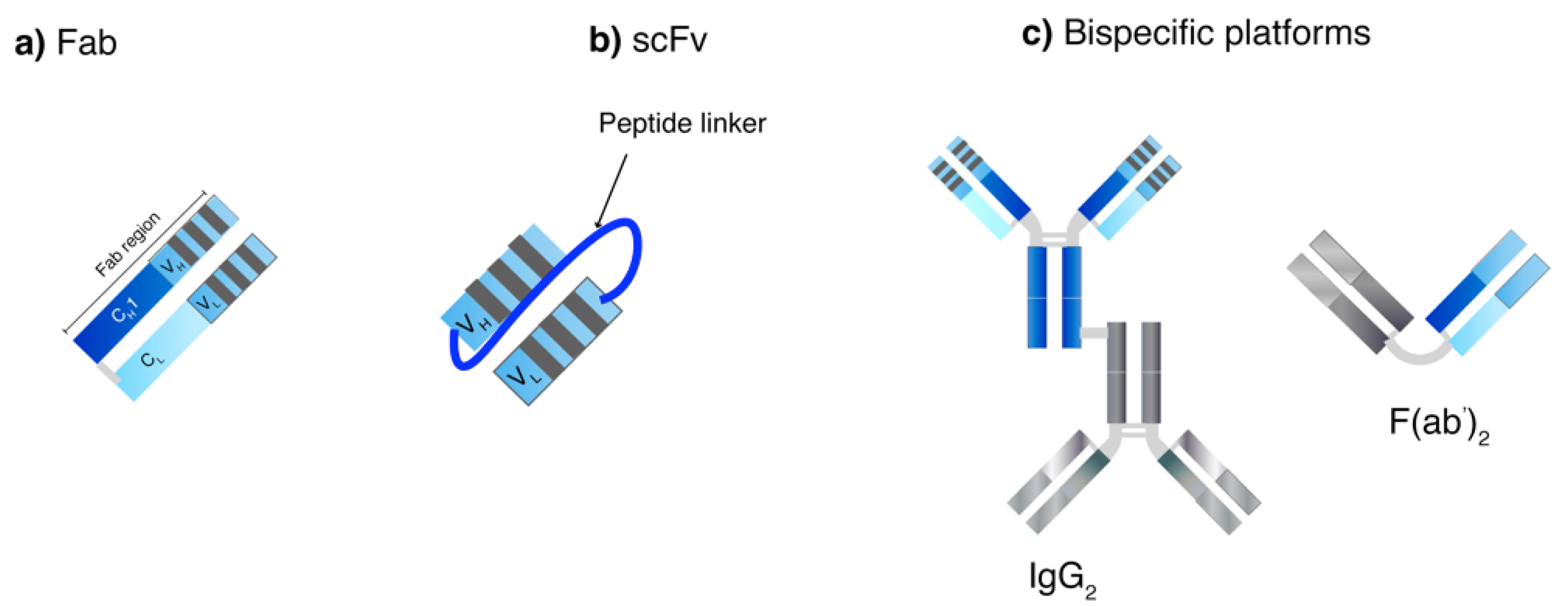{kind=link}
{kind=link}
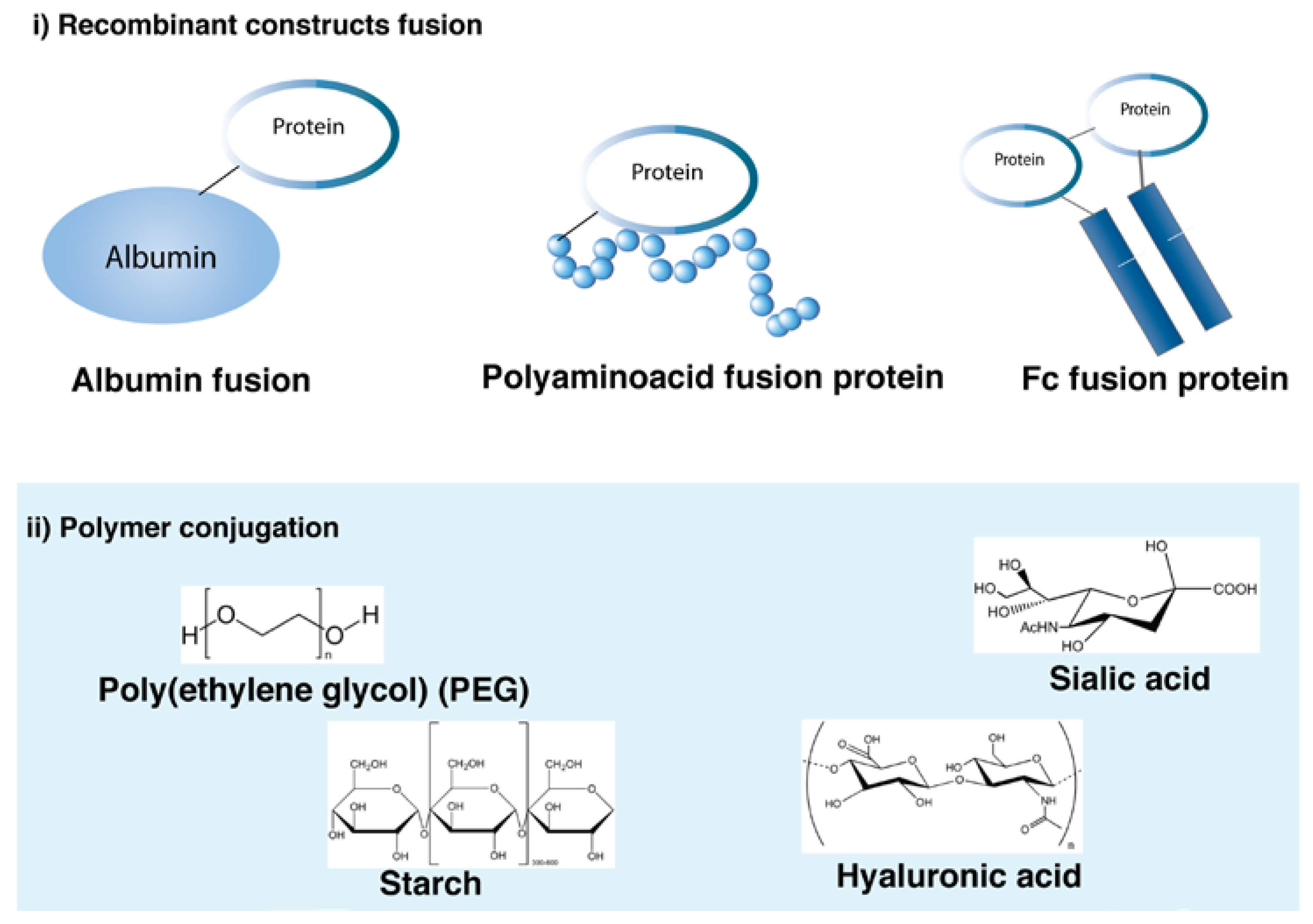{kind=link}
1. Introduction
2. General Limitations and Formulation Challenges
2.1. Pharmacokinetic Limitations
2.2. Formulation Challenges
3. Potential Strategies to Overcome Challenges in Antibody-Based Therapies
3.1. Use of Excipients to Stabilize Formulations
3.2. Production of Protein Scaffolds
3.3. mAb Formulations to Prolong the Duration of Action
3.3.1. Microparticulate Associated Formulations
3.3.2. Hydrogels and In Situ Forming Gels
3.3.3. Liposomes
3.4. Protein Modification to Increase Duration of Action
3.4.1. Albumin–Protein Fusions
3.4.2. Fc Fusion Proteins
3.4.3. Protein PEGylation
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Keizer, R.J.; Huitema, A.D.R.; Schellens, J.H.M.; Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 2010, 49, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.H. The history of monoclonal antibody development—Progress, remaining challenges and future innovations. Ann. Med. Surg. 2014, 3, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L. Ten years of Orthoclone OKT3 (muromonab-CD3): A review. J. Transpl. Coord. 1996, 6, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Wagner-Rousset, E.; Bussat, M.C.; Lokteff, M.; Klinguer-Hamour, C.; Haeuw, J.F.; Goetsch, L.; Wurch, T.; Dorsselaer, A.V.; Corvaïa, N. Trends in glycosylation, glycoanalysis and glycoengineering of therapeutic antibodies and Fc-fusion proteins. Curr. Pharm. Biotechnol. 2008, 9, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Mahmuda, A.; Bande, F.; Jameel, K.; Abdulhaleem, N.; Majid, R.A.; Hamat, R.A.; Abdullah, W.O.; Unyah, Z. Monoclonal antibodies: A review of therapeutic applications and future prospects. Trop. J. Pharm. Res. 2017, 16, 713. [Google Scholar] [CrossRef]
- Rodrigues, E.B.; Farah, M.E.; Maia, M.; Penha, F.M.; Regatieri, C.; Melo, G.B.; Pinheiro, M.M.; Zanetti, C.R. Therapeutic monoclonal antibodies in ophthalmology. Prog. Retin. Eye Res. 2009, 28, 117–144. [Google Scholar] [CrossRef] [PubMed]
- Wurch, T.; Pierré, A.; Depil, S. Novel protein scaffolds as emerging therapeutic proteins: From discovery to clinical proof-of-concept. Trends Biotechnol. 2012, 30, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Attaelmannan, M.; Levinson, S.S. Understanding and identifying monoclonal gammopathies. Clin. Chem. 2000, 46 Pt 2, 1230–1238. [Google Scholar] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Raju, T.S.; Scallon, B.J. Glycosylation in the Fc domain of IgG increases resistance to proteolytic cleavage by papain. Biochem. Biophys. Res. Commun. 2006, 341, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M.; Skerra, A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr. Opin. Chem. Biol. 2009, 13, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Dirks, N.L.; Meibohm, B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 2010, 49, 633–659. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Lum, L.G. “NextGen” Biologics: Bispecific Antibodies and Emerging Clinical Results. Expert Opin. Biol. Ther. 2016, 16, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.R. Separation and isolation of fractions of rabbit gamma-globulin containing the antibody and antigenic combining sites. Nature 1958, 182, 670–671. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gutshall, L.; Jiang, H.; Baker, A.; Beil, E.; Obmolova, G.; Carton, J.; Taudte, S.; Amegadzie, B. Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expr. Purif. 2009, 67, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Herrington-Symes, A.P.; Farys, M.; Khalili, H.; Brocchini, S. Antibody fragments: Prolonging circulation half-life special issue-antibody research. Adv. Biosci. Biotechnol. 2013, 4, 689–698. [Google Scholar] [CrossRef]
- Kourlas, H.; Schiller, D.S. Pegaptanib sodium for the treatment of neovascular age-related macular degeneration: A review. Clin. Ther. 2006, 28, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J.; Heier, J.S.; Hantsbarger, G.; Shams, N. Tolerability and Efficacy of Multiple Escalating Doses of Ranibizumab (Lucentis) for Neovascular Age-Related Macular Degeneration. Ophthalmology 2006, 113, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J.; Schwartz, S.D.; Blumenkranz, M.S.; Miller, J.; Haller, J.; Reimann, J.; Greene, W.; Shams, N. Maximum tolerated dose of a humanized anti-vascular endothelial growth factor antibody fragment for treating neovascular age-related macular degeneration. Ophthalmology 2005, 112, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Heier, J.S.; Antoszyk, A.N.; Pavan, P.R.; Leff, S.R.; Rosenfeld, P.J.; Ciulla, T.A.; Dreyer, R.F.; Gentile, R.C.; Sy, J.P.; Hantsbarger, G.; et al. Ranibizumab for Treatment of Neovascular Age-Related Macular Degeneration. A Phase I/II Multicenter, Controlled, Multidose Study. Ophthalmology 2006, 113. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L. Antibody fragments: Hope and hype. MAbs 2010, 2, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W. Pharmacokinetics, pharmacodynamics and pre-clinical characteristics of ophthalmic drugs that bind VEGF. Expert Rev. Clin. Pharmacol. 2014, 7, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Morrison, S.L. Effect of glycosylation on antibody function: Implications for genetic engineering. Trends Biotechnol. 1997, 15, 26–32. [Google Scholar] [CrossRef]
- Desjarlais, J.R.; Lazar, G.A. Modulation of antibody effector function. Exp. Cell Res. 2011, 317, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Wang, W.; Arakawa, T.; Ohtake, S. Developments and Challenges for mAb-Based Therapeutics. Antibodies 2013, 2, 452–500. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.R.; Morton, L.D.; Spindeldreher, S.; Kiessling, A.; Allenspach, R.; Hey, A.; Müller, P.; Frings, W.; Sims, J. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs 2010, 2, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Chaudhury, C.; Kim, J.; Bronson, C.L.; Wani, M.A.; Mohanty, S. Perspective—FcRn transports albumin: Relevance to immunology and medicine. Trends Immunol. 2006, 27, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.T.; Pehrson, R.; Tolmachev, V.; Daba, M.B.; Abrahmsén, L.; Ekblad, C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J. Biol. Chem. 2011, 286, 5234–5241. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Robinson, S.B.; Csaky, K.G. FcRn receptor-mediated pharmacokinetics of therapeutic IgG in the eye. Mol. Vis. 2009, 15, 2803–2812. [Google Scholar] [PubMed]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Qiao, S.W.; Yoshida, M.; Baker, K.; Lencer, W.I.; Blumberg, R.S. An FcRn-Dependent Role for Anti-flagellin Immunoglobulin G in Pathogenesis of Colitis in Mice. Gastroenterology 2009, 137, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, X.; Hernandez, L.D.; Lipari, P.; Flattery, A.; Chen, S.-C.; Kramer, S.; Polishook, J.D.; Racine, F.; Cape, H.; et al. Toxin-mediated paracellular transport of antitoxin antibodies facilitates protection against Clostridium difficile infection. Infect. Immun. 2015, 83, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.; Dickinson, B.L.; Blumberg, R.S.; Simister, N.E.; Lencer, W.I.; Walker, W.A. Distribution of the IgG Fc receptor, FcRn, in the human fetal intestine. Pediatr. Res. 2003, 53, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Pang, G.; Wang, Y.; Xie, J.; Chen, Q.; Hu, Z. Human FcRn can mediate the transport across intestinal mucosal barrier and prolong the half-life of rabbit IgG In Vivo. Braz. Arch. Biol. Technol. 2015, 58, 387–394. [Google Scholar] [CrossRef]
- Hornby, P.J.; Cooper, P.R.; Kliwinski, C.; Ragwan, E.; Mabus, J.R.; Harman, B.; Thompson, S.; Kauffman, A.L.; Yan, Z.; Tam, S.H.; et al. Human and non-human primate intestinal FcRn expression and immunoglobulin G transcytosis. Pharm. Res. 2014, 31, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, A.; Lozza, L.; Reece, S.T.; Perdomo, C.; Zedler, U.; Hahnke, K.; Oberbeck-Mueller, D.; Dorhoi, A.; Kaufmann, S.H.E. Neonatal Fc Receptor Regulation of Lung Immunoglobulin and CD103+ Dendritic Cells Confers Transient Susceptibility to Tuberculosis. Infect. Immun. 2016, 84, 2914–2921. [Google Scholar] [CrossRef] [PubMed]
- Bitonti, A.J.; Dumont, J.A.; Low, S.C.; Peters, R.T.; Kropp, K.E.; Palombella, V.J.; Stattel, J.M.; Lu, Y.; Tan, C.A.; Song, J.J.; et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 9763–9768. [Google Scholar] [CrossRef] [PubMed]
- Shatz, W.; Hass, P.E.; Mathieu, M.; Kim, H.S.; Leach, K.; Zhou, M.; Crawford, Y.; Shen, A.; Wang, K.; Chang, D.P.; et al. Contribution of Antibody Hydrodynamic Size to Vitreal Clearance Revealed through Rabbit Studies Using a Species-Matched Fab. Mol. Pharm. 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Gadkar, K.; Pastuskovas, C.V.; Le Couter, J.E.; Elliott, J.M.; Zhang, J.; Lee, C.V.; Sanowar, S.; Fuh, G.; Kim, H.S.; Lombana, T.N.; et al. Design and pharmacokinetic characterization of novel antibody formats for ocular therapeutics. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5390–5400. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.R.; Racher, A.J. Antibody production. Adv. Drug Deliv. Rev. 2006, 58, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Steinmeyer, D.E.; McCormick, E.L. The art of antibody process development. Drug Discov. Today 2008, 13, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Roque, A.C.A.; Lowe, C.R.; Taipa, M.Â. Antibodies and genetically engineered related molecules: Production and purification. Biotechnol. Prog. 2004, 20, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Gronemeyer, P.; Ditz, R.; Strube, J. Trends in Upstream and Downstream Process Development for Antibody Manufacturing. Bioengineering 2014, 1, 188–212. [Google Scholar] [CrossRef] [PubMed]
- Epstein, N.; Epstein, M. The hybridoma technology: I. Production of monoclonal antibodies. Adv. Biotechnol. Processes 1986, 6, 179–218. [Google Scholar] [PubMed]
- Little, M.; Kipriyanov, S.M.; Le Gall, F.; Moldenhauer, G. Of mice and men: Hybridoma and recombinant antibodies. Immunol. Today 2000, 21, 364–370. [Google Scholar] [CrossRef]
- Tomita, M.; Tsumoto, K. Hybridoma technologies for antibody production. Immunotherapy 2011, 3, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.R.; McCafferty, J.; Dübel, S.; Bradbury, A.R.M.; Sidhu, S.S. Recombinant Antibodies and In Vitro Selection Technologies. In Antibody Methods and Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 11–32. [Google Scholar]
- Kuhn, P.; Fühner, V.; Unkauf, T.; Moreira, G.M.S.G.; Frenzel, A.; Miethe, S.; Hust, M. Recombinant antibodies for diagnostics and therapy against pathogens and toxins generated by phage display. Proteom. Clin. Appl. 2016, 10, 922–948. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. MAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.R.M.; Sidhu, S.; Dübel, S.; McCafferty, J. Beyond natural antibodies: The power of in vitro display technologies. Nat. Biotechnol. 2011, 29, 245–254. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Glover, D.R. Engineering therapeutic proteins. Curr. Opin. Struct. Biol. 2000, 10, 417–420. [Google Scholar] [CrossRef]
- Constantinou, A.; Epenetos, A.A.; Hreczuk-Hirst, D.; Jain, S.; Wright, M.; Chester, K.A.; Deonarain, M.P. Site-specific polysialylation of an antitumor single-chain Fv fragment. Bioconjug. Chem. 2009, 20, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, M. Generating high-avidity human Mabs in mice. Nat. Biotechnol. 1996, 14, 826. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.J.; Souto, E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv. Drug Deliv. Rev. 2007, 59, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Harding, S.P.; Rogers, C.A.; Downes, S.M.; Lotery, A.J.; Wordsworth, S.; Reeves, B.C. Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: One-year findings from the IVAN randomized trial. Ophthalmology 2012, 119, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Fine, S.L.; Ying, G.S.; Jaffe, G.J.; Grunwald, J.E.; Toth, C.; Redford, M.; Ferris, F.L. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: Two-year results. Ophthalmology 2012, 119, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Moisseiev, E.; Waisbourd, M.; Ben-Artsi, E.; Levinger, E.; Barak, A.; Daniels, T.; Csaky, K.; Loewenstein, A.; Barequet, I.S. Pharmacokinetics of bevacizumab after topical and intravitreal administration in human eyes. Graefe Arch. Clin. Exp. Ophthalmol. 2014, 252, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.H.; Krohne, T.U.; Holz, F.G. Intraocular pharmacokinetics after a single intravitreal injection of 1.5 mg versus 3.0 mg of bevacizumab in humans. Retina 2011, 31, 1877–1884. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ziemssen, F.; Henke-Fahle, S.; Tatar, O.; Szurman, P.; Aisenbrey, S.; Schneiderhan-Marra, N.; Xu, X.; Grisanti, S. Vitreous levels of bevacizumab and vascular endothelial growth factor-A in patients with choroidal neovascularization. Ophthalmology 2008, 115, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.M.; Wong, S.J.; Hammad, A.M.; Falk, N.S.; O’Malley, M.R.; Khan, S. Vitreous levels of unbound bevacizumab and unbound vascular endothelial growth factor in two patients. Retina 2006, 26, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Jager, R.D.; Aiello, L.P.; Patel, S.C.; Cunningham, E.T. Risks of intravitreous injection: A comprehensive review. Retina 2004, 24, 676–698. [Google Scholar] [CrossRef] [PubMed]
- Dickmann, L. Ocular therapeutics: Drug delivery and pharmacology. Mol. Pharm. 2016, 13, 2875–2876. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, H.; Wirth, T.; Schenkwein, D.; Räty, J.K.; Ylä-Herttuala, S. Challenges in monoclonal antibody-based therapies. Ann. Med. 2009, 41, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Shi, S. Biologics: An update and challenge of their pharmacokinetics. Curr. Drug Metab. 2014, 15, 271–290. [Google Scholar] [CrossRef] [PubMed]
- Takakura, Y.; Mahato, R.I.; Nishikawa, M.; Hashida, M. Control of pharmacokinetic profiles of drug-macromolecule conjugates. Adv. Drug Deliv. Rev. 1996, 19, 377–399. [Google Scholar] [CrossRef]
- Supersaxo, A.; Hein, W.R.; Steffen, H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm. Res. 1990, 7, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.F.; Bhansali, S.G.; Morris, M.E. Mechanistic Determinants of Biotherapeutics Absorption Following SC Administration. AAPS J. 2012, 14, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A. Early development of therapeutic biologics—Pharmacokinetics. Curr. Drug Metab. 2006, 7, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Barar, J.; Javadzadeh, A.R.; Omidi, Y. Ocular novel drug delivery: Impacts of membranes and barriers. Expert Opin. Drug Deliv. 2008, 5, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Shpilberg, O.; Jackisch, C. Subcutaneous administration of rituximab (MabThera) and trastuzumab (Herceptin) using hyaluronidase. Br. J. Cancer 2013, 109, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Leveque, D. Subcutaneous administration of anticancer agents. Anticancer Res. 2014, 34, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.I. Recombinant human hyaluronidase (rHuPH20): An enabling platform for subcutaneous drug and fluid administration. Expert Opin. Drug Deliv. 2007, 4, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Pereira De Sousa, I.; Bernkop-Schnürch, A. Pre-systemic metabolism of orally administered drugs and strategies to overcome it. J. Control. Release 2014, 192, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.; Shores, E.; Wagner, C.; Mire-Sluis, A. Scientific and regulatory considerations on the immunogenicity of biologics. Trends Biotechnol. 2006, 24, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.; Lockwood, A.; Brocchini, S.; Khaw, P.T. The PK-Eye: A Novel In Vitro Ocular Flow Model for Use in Preclinical Drug Development. J. Pharm. Sci. 2015, 104, 3330–3342. [Google Scholar] [CrossRef] [PubMed]
- Brinch, K.S.; Frimodt-Møller, N.; Høiby, N.; Kristensen, H.-H. Influence of antidrug antibodies on plectasin efficacy and pharmacokinetics. Antimicrob. Agents Chemother. 2009, 53, 4794–4800. [Google Scholar] [CrossRef] [PubMed]
- Brinks, V.; Jiskoot, W.; Schellekens, H. Immunogenicity of therapeutic proteins: The use of animal models. Pharm. Res. 2011, 28, 2379–2385. [Google Scholar] [CrossRef] [PubMed]
- Van Beers, M.M.C.; Sauerborn, M.; Gilli, F.; Brinks, V.; Schellekens, H.; Jiskoot, W. Aggregated recombinant human interferon Beta induces antibodies but no memory in immune-tolerant transgenic mice. Pharm. Res. 2010, 27, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, M.A.; Tseng, C.M.; Roskos, L.K. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov. Today 2006, 11, 81–88. [Google Scholar] [CrossRef]
- Wang, B.; Lau, Y.Y.; Liang, M.; Vainshtein, I.; Zusmanovich, M.; Lu, H.; Magrini, F.; Sleeman, M.; Roskos, L. Mechanistic modeling of antigen sink effect for mavrilimumab following intravenous administration in patients with rheumatoid arthritis. J. Clin. Pharmacol. 2012, 52, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- McKeage, K.; Perry, C.M. Trastuzumab: A review of its use in the treatment of metastatic breast cancer overexpressing HER2. Drugs 2002, 62, 209–243. [Google Scholar] [CrossRef] [PubMed]
- Baert, F.; Noman, M.; Vermeire, S.; Van Assche, G.; D’Haens, G.; Carbonez, A.; Rutgeerts, P. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N. Engl. J. Med. 2003, 348, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Therapeutic protein aggregation: Mechanisms, design, and control. Trends Biotechnol. 2014, 32, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Mahler, H.C.; Friess, W.; Grauschopf, U.; Kiese, S. Protein aggregation: Pathways, induction factors and analysis. J. Pharm. Sci. 2009, 98, 2909–2934. [Google Scholar] [CrossRef] [PubMed]
- Philo, J.S.; Arakawa, T. Mechanisms of protein aggregation. Curr. Pharm. Biotechnol. 2009, 10, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Neergaard, M.S.; Nielsen, A.D.; Parshad, H.; De Weert, M.V. Stability of Monoclonal Antibodies at High-Concentration: Head-to-Head Comparison of the IgG1 and IgG4 Subclass. J. Pharm. Sci. 2014, 103, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Frokjaer, S.; Otzen, D.E. Protein drug stability: A formulation challenge. Nat. Rev. Drug Discov. 2005, 4, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef] [PubMed]
- Chavez, B.K.; Agarabi, C.D.; Read, E.K.; Boyne, M.T., II; Khan, M.A.; Brorson, K.A. Improved Stability of a Model IgG3 by DoE-Based Evaluation of Buffer Formulations. Biomed. Res. Int. 2016, 2016, 2074149. [Google Scholar] [CrossRef] [PubMed]
- Pikal-Cleland, K.A.; Cleland, J.L.; Anchordoquy, T.J.; Carpenter, J.F. Effect of glycine on pH changes and protein stability during freeze-thawing in phosphate buffer systems. J. Pharm. Sci. 2002, 91, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Henderson, I.; Nayar, R.; Randolph, T.W.; Carpenter, J.F. Effect of pH on stability of recombinant botulinum serotype A vaccine in aqueous solution and during storage of freeze-dried formulations. J. Pharm. Sci. 2008, 97, 5132–5146. [Google Scholar] [CrossRef] [PubMed]
- Angkawinitwong, U.; Sharma, G.; Brocchini, S. Solid-state protein formulation. Ther. Deliv. 2015, 6, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Farrell, R.A.; Marta, M.; Gaeguta, A.J.; Souslova, V.; Giovannoni, G.; Creeke, P.I. Development of resistance to biologic therapies with reference to IFN-β. Rheumatology 2012, 51, 590–599. [Google Scholar] [CrossRef] [PubMed]
- McKoy, J.M.; Stonecash, R.E.; Cournoyer, D.; Rossert, J.; Nissenson, A.R.; Raisch, D.W.; Casadevall, N.; Bennett, C.L. Epoetin-associated pure red cell aplasia: Past, present, and future considerations. Transfusion 2008, 48, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Ratanji, K.D.; Derrick, J.P.; Dearman, R.J.; Kimber, I. Immunogenicity of therapeutic proteins: Influence of aggregation. J. Immunotoxicol. 2014, 11, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Arriola-Villalobos, P.; Martinez-de-la-Casa, J.M.; Diaz-Valle, D.; Morales-Fernandez, L.; Fernandez-Perez, C.; Garcia-Feijoo, J. Glaukos iStent inject® Trabecular Micro-Bypass Implantation Associated with Cataract Surgery in Patients with Coexisting Cataract and Open-Angle Glaucoma or Ocular Hypertension: A Long-Term Study. J. Ophthalmol. 2016, 2016, 1056573. [Google Scholar] [CrossRef] [PubMed]
- Kossovsky, N.; Heggers, J.P.; Robson, M.C. Experimental demonstration of the immunogenicity of silicone–Protein complexes. J. Biomed. Mater. Res. 1987, 21, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Thirumangalathu, R.; Krishnan, S.; Ricci, M.S.; Brems, D.N.; Randolph, T.W.; Carpenter, J.F. Silicone oil- and agitation-induced aggregation of a monoclonal antibody in aqueous solution. J. Pharm. Sci. 2009, 98, 3167–3181. [Google Scholar] [CrossRef] [PubMed]
- Mintz, C.S.; Crea, R. Protein Scaffolds. Bioprocess. Int. 2013, 11, 40–48. [Google Scholar]
- Carter, P.J. Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 2011, 317, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Akash, M.S.H.; Rehman, K.; Tariq, M.; Chen, S. Development of therapeutic proteins: Advances and challenges. Turk. J. Biol. 2015, 39, 343–358. [Google Scholar] [CrossRef]
- Bhambhani, A.; Kissmann, J.M.; Joshi, S.B.; Volkin, D.B.; Kashi, R.S.; Middaugh, C.R. Formulation design and high-throughput excipient selection based on structural integrity and conformational stability of dilute and highly concentrated IgG1 monoclonal antibody solutions. J. Pharm Sci. 2012, 101, 1120–1135. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, K.; Sonali, N.; Moreno, M.; Nirmal, J.; Fernandez, A.A.; Venkatraman, S.; Agrawal, R. Protein delivery to the back of the eye: Barriers, carriers and stability of anti-VEGF proteins. Drug Discov. Today 2017, 22, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Westermaier, Y.; Veurink, M.; Riis-Johannessen, T.; Guinchard, S.; Gurny, R.; Scapozza, L. Identification of aggregation breakers for bevacizumab (Avastin®) self-association through similarity searching and interaction studies. Eur. J. Pharm. Biopharm. 2013, 85, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Ohshita, N.; Hirotsu, T.; Yamashita, Y.; Motoyama, K.; Koyama, S.; Iibuchi, R.; Uchida, T.; Mieda, S.; Handa, K.; et al. Stabilizing Effects for Antibody Formulations and Safety Profiles of Cyclodextrin Polypseudorotaxane Hydrogels. J. Pharm. Sci. 2017, 106, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Falconer, R.J.; Chan, C.; Hughes, K.; Munro, T.P. Stabilization of a monoclonal antibody during purification and formulation by addition of basic amino acid excipients. J. Chem. Technol. Biotechnol. 2011, 86, 942–948. [Google Scholar] [CrossRef]
- North, R.T.; Harvey, V.J.; Cox, L.C.; Ryan, S.N. Medical resource utilization for administration of trastuzumab in a new zealand oncology outpatient setting: A time and motion study. Clin. Outcomes Res. 2015, 7, 423–430. [Google Scholar] [CrossRef]
- Van der Kant, R.; Karow-Zwick, A.R.; Van Durme, J.; Blech, M.; Gallardo, R.; Seeliger, D.; Aßfalg, K.; Baatsen, P.; Compernolle, G.; Gils, A.; et al. Prediction and Reduction of the Aggregation of Monoclonal Antibodies. J. Mol. Biol. 2017, 429, 1244–1261. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, N.; Xiong, J.; Pace, S.E.; Kumar, V.; Middaugh, C.R.; Joshi, S.B.; Volkin, D.B. A Formulation Development Approach to Identify and Select Stable Ultra e High-Concentration Monoclonal Antibody Formulations with Reduced Viscosities. J. Pharm. Sci. 2017, 106, 3230–3241. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, S.D.; Walsh, R.B. Display scaffolds: Protein engineering for novel therapeutics. Curr. Opin. Pharmacol. 2008, 8, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Dostalek, M.; Gardner, I.; Gurbaxani, B.M.; Rose, R.H.; Chetty, M. Pharmacokinetics, Pharmacodynamics and Physiologically-Based Pharmacokinetic Modelling of Monoclonal Antibodies. Clin. Pharmacokinet. 2013, 52. [Google Scholar] [CrossRef] [PubMed]
- Pisal, D.S.; Kosloski, M.P.; Balu-Iyer, S.V. Delivery of therapeutic proteins. J. Pharm. Sci. 2010, 99, 2557–2575. [Google Scholar] [CrossRef] [PubMed]
- White, L.J.; Kirby, G.T.S.; Cox, H.C.; Qodratnama, R.; Qutachi, O.; Rose, F.R.A.J.; Shakesheff, K.M. Accelerating protein release from microparticles for regenerative medicine applications. Mater. Sci. Eng. C 2013, 33, 2578–2583. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.D. Analysis of initial burst in PLGA microparticles. Expert Opin. Drug Deliv. 2008, 5, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Angkawinitwong, U.; Awwad, S.; Khaw, P.T.; Brocchini, S.; Williams, G.R. Electrospun formulations of bevacizumab for sustained release in the eye. Acta Biomater. 2017, 64, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Adamson, P.; Wilde, T.; Dobrzynski, E.; Sychterz, C.; Polsky, R.; Kurali, E.; Haworth, R.; Tang, C.M.; Korczynska, J.; Cook, F.; et al. Single ocular injection of a sustained-release anti -VEGF delivers 6 months pharmacokinetics and ef fi cacy in a primate laser CNV model. J. Control. Release 2016, 244, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Foong, K.S.; Patel, R.; Forbes, A.; Day, R.M. Anti-tumor necrosis factor-alpha-loaded microspheres as a prospective novel treatment for Crohn’s disease fistulae. Tissue Eng. 2010, 16, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lau, L.C.M.; Lo, A.C.-Y.; Chau, Y. Injectable Chemically Crosslinked Hydrogel for the Controlled Release of Bevacizumab in Vitreous: A 6-Month In Vivo Study. Transl. Vis. Sci. Technol. 2015, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.; Al-Shohani, A.; Khaw, P.T.; Brocchini, S. Comparative Study of In Situ Loaded Antibody and PEG-Fab NIPAAM Gels. Macromol. Biosci. 2018, 18, 1700255. [Google Scholar] [CrossRef] [PubMed]
- Jatav, V.S.; Singh, H.; Singh, S.K. Recent Trends on Hydrogel in Human Body. Int. J. Res. Pharm. Biomed. Sci. 2011, 2, 442–447. [Google Scholar]
- Madan, M.; Bajaj, A.; Lewis, S.; Udupa, N.; Baig, J.A. In situ forming polymeric drug delivery systems. Indian J. Pharm. Sci. 2009, 71, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Rupenthal, I.D. Injectable implants for the sustained release of protein and peptide drugs. Drug Discov. Today 2013, 18, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Vaishya, R.D.; Mandal, A.; Patel, S.; Mitra, A.K. Extended release microparticle-in-gel formulation of octreotide: Effect of polymer type on acylation of peptide during in vitro release. Int. J. Pharm. 2015, 496, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Vaishya, R.; Khurana, V.; Patel, S.; Mitra, A.K. Long-term delivery of protein therapeutics. Expert Opin. Drug Deliv. 2015, 12, 415–440. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, L.E.; Ron, E.S. Temperature-responsive gels and thermogelling polymer matrices for protein and peptide delivery. Adv. Drug Deliv. Rev. 1998, 31, 197–221. [Google Scholar] [CrossRef]
- Drapala, P.W.; Brey, E.M.; Mieler, W.F.; Venerus, D.C.; Kang Derwent, J.J.; Pérez-Luna, V.H. Role of Thermo-responsiveness and Poly(ethylene glycol) Diacrylate Cross-link Density on Protein Release from Poly(N-isopropylacrylamide) Hydrogels. J. Biomater. Sci. Polym. Ed. 2011, 22, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Kang Derwent, J.J.; Mieler, W.F. Thermoresponsive hydrogels as a new ocular drug delivery platform to the posterior segment of the eye. Trans. Am. Ophthalmol. Soc. 2008, 106, 206–213. [Google Scholar] [PubMed]
- Egbu, R.; Brocchini, S.; Khaw, P.T.; Awwad, S. Antibody loaded collapsible hyaluronic acid hydrogels for intraocular delivery. Eur. J. Pharm. Biopharm. 2018, 124, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Conde, B.R.; Brewer, E.; Lowman, A.; Peppas, N.A. Complexation Hydrogels as Oral Delivery Vehicles of Therapeutic Antibodies: An in Vitro and ex Vivo Evaluation of Antibody Stability and Bioactivity. Ind. Eng. Chem. Res. 2015, 54, 10197–10205. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery—Liposomes versus lipid nanoparticles. Int. J. Nanomed. 2007, 2, 595–607. [Google Scholar]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Patil, Y.P.; Jadhav, S. Novel methods for liposome preparation. Chem. Phys. Lipids 2014, 177, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Narang, A.S.; Chang, R.K.; Hussain, M.A. Pharmaceutical development and regulatory considerations for nanoparticles and nanoparticulate drug delivery systems. J. Pharm. Sci. 2013, 102, 3867–3882. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Szoka, F.C. Chemical approaches to triggerable lipid vesicles for drug and gene delivery. Acc. Chem. Res. 2003, 36, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Briuglia, M.L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Vllasaliu, D.; Fowler, R.; Stolnik, S. PEGylated nanomedicines: Recent progress and remaining concerns. Expert Opin. Drug Deliv. 2014, 11, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.; Griffin, L.; Compson, J.E.; Jairaj, M.; Baker, T.; Ceska, T.; West, S.; Zaccheo, O.; Davé, E.; Lawson, A.D.; et al. Extending the half-life of a fab fragment through generation of a humanized anti-human serum albumin Fv domain: An investigation into the correlation between affinity and serum half-life. MAbs 2016, 8, 1336–1346. [Google Scholar] [CrossRef] [PubMed]
- O’Connor-Semmes, R.L.; Lin, J.; Hodge, R.J.; Andrews, S.; Chism, J.; Choudhury, A.; Nunez, D.J. GSK2374697, a Novel Albumin-Binding Domain Antibody (AlbudAb), Extends Systemic Exposure of Exendin-4: First Study in Humans—PK/PD and Safety. Clin. Pharmacol. Ther. 2014, 96, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.T.; Wilson, H.D.; Asano, S.; Nilchan, N.; Fuller, R.P.; Roush, W.R.; Rader, C.; Barbas, C.F. Human Serum Albumin Domain I Fusion Protein for Antibody Conjugation. Bioconjug. Chem. 2016, 27, 2271–2275. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Karle, A.; Meißburger, B.; Höfig, I.; Stork, R.; Kontermann, R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007, 282, 12650–12660. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Meng, F.; Jin, Q.; Sun, C.; Li, Y.; Li, H.; Jin, S. Fusion protein of single-chain variable domain fragments for treatment of myasthenia gravis. Neural Regen. Res. 2014, 9, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.T.; Cameron, J.; Plumridge, A.; Evans, L.; Sleep, D.; Sandlie, I. Single-chain variable fragment albumin fusions bind the neonatal Fc receptor (FcRn) in a species-dependent manner: Implications for in vivo half-life evaluation of albumin fusion therapeutics. J. Biol. Chem. 2013, 288, 24277–24285. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. EMBO Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W. Aflibercept (VEGF Trap-eye): The newest anti-VEGF drug. Br. J. Ophthalmol. 2012, 96, 1157–1158. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-F.; Lockhart, A.C. Aflibercept in the treatment of metastatic colorectal cancer. Clin. Med. Insights Oncol. 2012, 6, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Celik, N.; Scheuerle, A.; Auffarth, G.U.; Kopitz, J.; Dithmar, S. Intraocular pharmacokinetics of aflibercept and vascular endothelial growth factor-A. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5574–5578. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Kakinoki, M.; Sawada, T.; Wang, X.; Ohji, M. Ranibizumab and aflibercept: Intraocular pharmacokinetics and their effects on aqueous VEGF level in vitrectomized and nonvitrectomized macaque eyes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6501–6505. [Google Scholar] [CrossRef] [PubMed]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.-F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Albini, T.A.; Moshfeghi, A.A. Ziv-Aflibercept as a Possible Alternative to Aflibercept. Retin. Today, July 2014, pp. 67–68. Available online: http://retinatoday.com/2014/08/ziv-aflibercept-as-a-possible-alternative-to-aflibercept/ (accessed on 4 July 2018).
- Marmor, M.F. Retinal detachment from hyperosmotic intravitreal injection. Investig. Ophthalmol. Vis. Sci. 1979, 18, 1237–1244. [Google Scholar]
- De Oliveira Dias, J.R.; Badaró, E.; Novais, E.A.; Colicchio, D.; Chiarantin, G.M.D.; Matioli, M.M.; Verna, C.; Penha, F.M.; Barros, N.M.T.; Meyer, C.H.; et al. Preclinical Investigations of Intravitreal Ziv-Aflibercept. Ophthalmic Surg. Lasers Imaging Retin. 2014, 45, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Bailon, P.; Won, C. PEG-modified biopharmaceuticals. Expert Opin. Drug Deliv. 2009, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.R. Commercial challenges of protein drug delivery. Expert Opin. Drug Deliv. 2005, 2, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Didier, E.; Harris, P.; Siegel, N. PEGylated proteins: Evaluation of their safety in the absence of definitive metabolism studies. Drug Metab. Dispos. 2007, 35, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Ivens, I.A.; Baumann, A.; McDonald, T.A.; Humphries, T.J.; Michaels, L.A.; Mathew, P. PEGylated therapeutic proteins for haemophilia treatment: A review for haemophilia caregivers. Haemophilia 2013, 19, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Garg, N.K.; Lunde, E.; Han, K.Y.; Jain, S.; Azar, D.T. Corneal Neovascularization: An Anti-VEGF Therapy Review. Surv. Ophthalmol. 2012, 57, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Basile, A.S.; Hutmacher, M.; Nickens, D. Population Pharmacokinetics of Pegaptanib in Patients with Neovascular, Age-Related Macular Degeneration. J. Clin. Pharmacol. 2012, 52, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Goel, N.; Stephens, S. Certolizumab pegol. MAbs 2010, 2, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Bendele, A.; Seely, J.; Richey, C.; Sennello, G.; Shopp, G. Short communication: Renal tubular vacuolation in animals treated with polyethylene-glycol-conjugated proteins. Toxicol. Sci. 1998, 42, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Hennink, W.E.; Brinks, V. The immunogenicity of polyethylene glycol: Facts and fiction. Pharm. Res. 2013, 30, 1729–1734. [Google Scholar] [CrossRef] [PubMed]
- Saifer, M.G.P.; Williams, L.D.; Sobczyk, M.A.; Michaels, S.J.; Sherman, M.R. Selectivity of binding of PEGs and PEG-like oligomers to anti-PEG antibodies induced by methoxyPEG-proteins. Mol. Immunol. 2014, 57, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Garay, R.P.; El-Gewely, R.; Armstrong, J.K.; Garratty, G.; Richette, P. Antibodies against polyethylene glycol in healthy subjects and in patients treated with PEG-conjugated agents. Expert Opin. Drug Deliv. 2012, 9, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Piedmonte, D.M.; Treuheit, M.J. Formulation of Neulasta (pegfilgrastim). Adv. Drug Deliv. Rev. 2008, 60, 50–58. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awwad, S.; Angkawinitwong, U. Overview of Antibody Drug Delivery. Pharmaceutics 2018, 10, 83. https://doi.org/10.3390/pharmaceutics10030083
Awwad S, Angkawinitwong U. Overview of Antibody Drug Delivery. Pharmaceutics. 2018; 10(3):83. https://doi.org/10.3390/pharmaceutics10030083
Chicago/Turabian StyleAwwad, Sahar, and Ukrit Angkawinitwong. 2018. "Overview of Antibody Drug Delivery" Pharmaceutics 10, no. 3: 83. https://doi.org/10.3390/pharmaceutics10030083
APA StyleAwwad, S., & Angkawinitwong, U. (2018). Overview of Antibody Drug Delivery. Pharmaceutics, 10(3), 83. https://doi.org/10.3390/pharmaceutics10030083