
Serotype-Specific Killing of Large Cell Carcinoma Cells by Reovirus



Abstract
:1. Introduction
2. Results
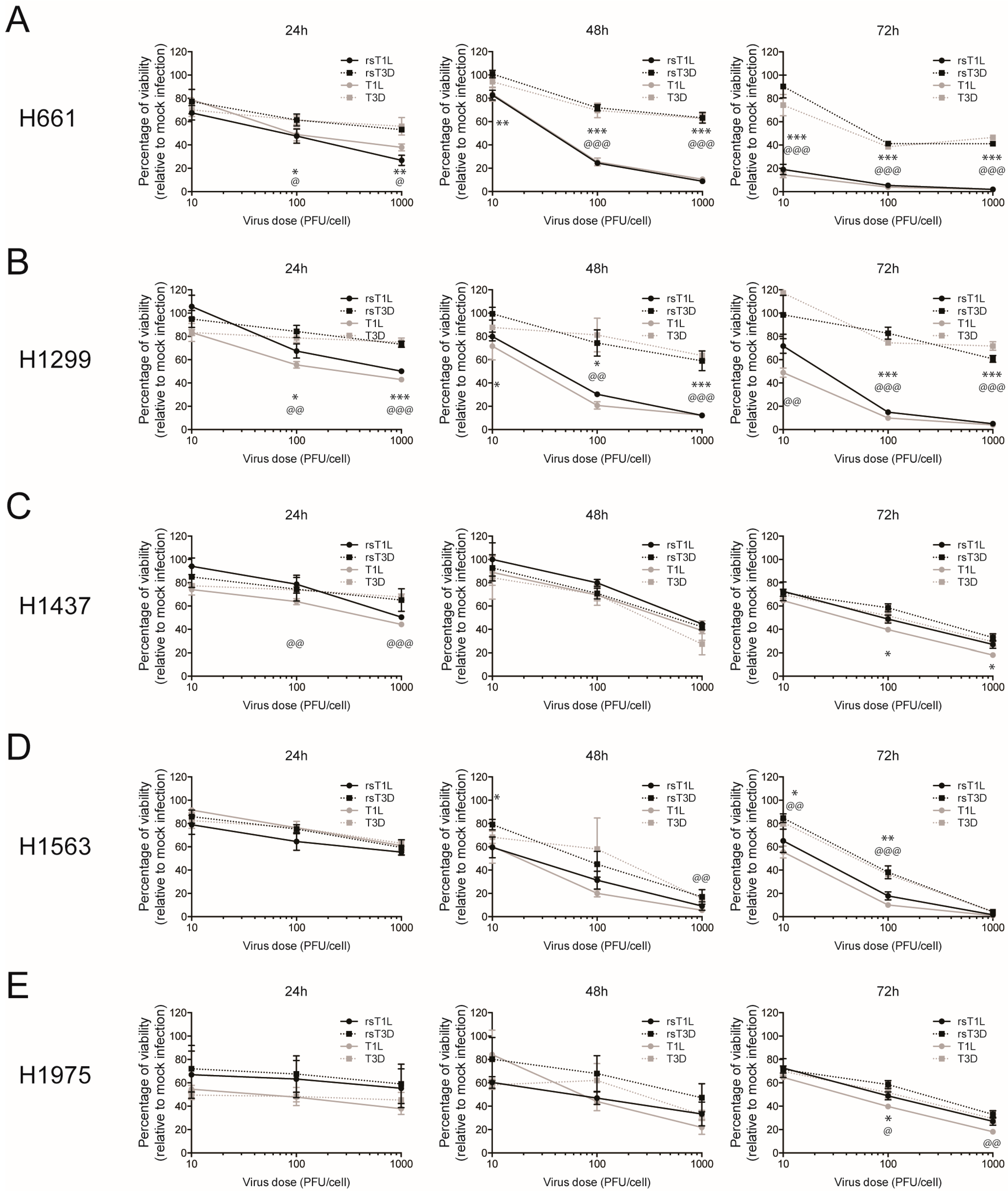
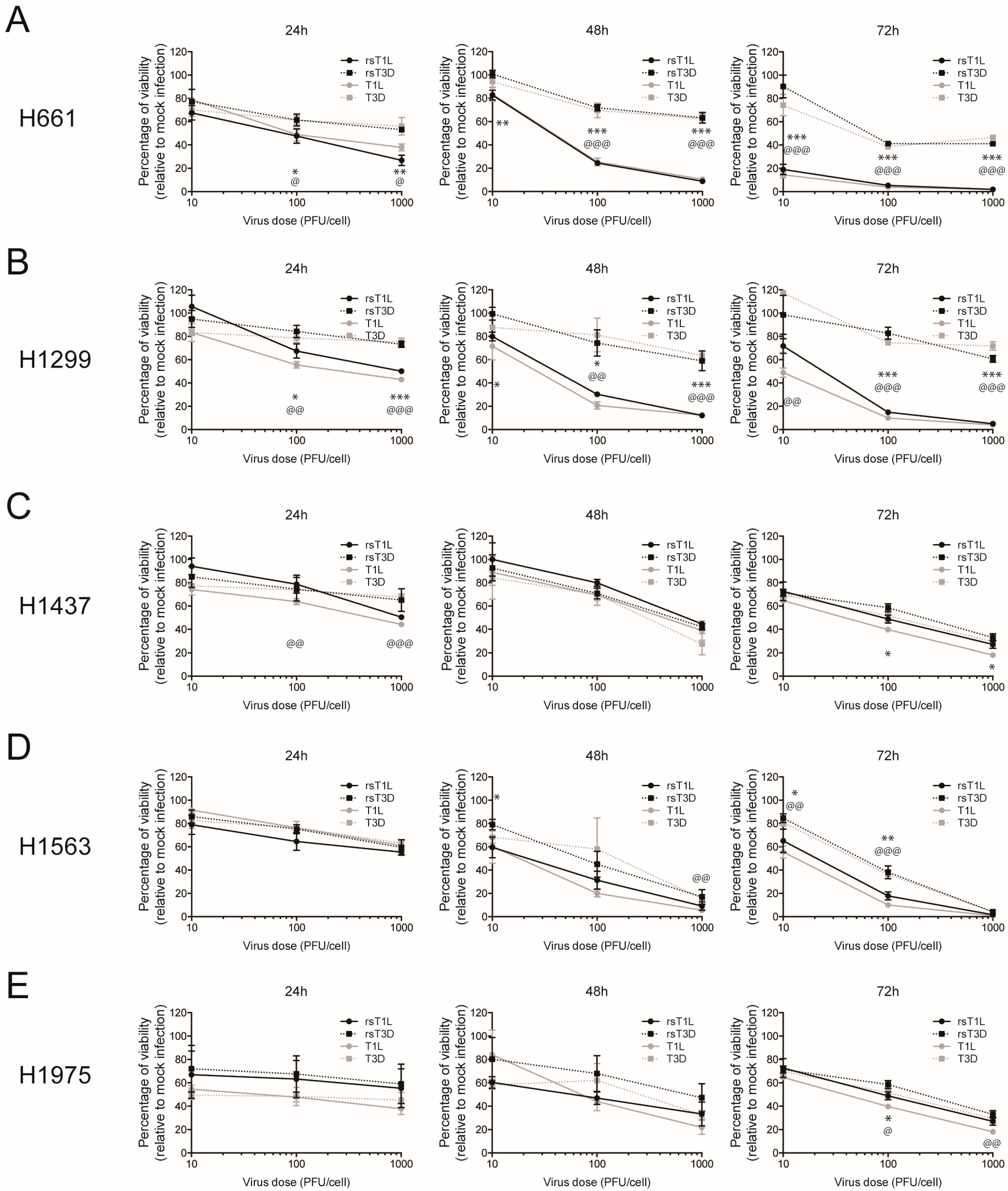
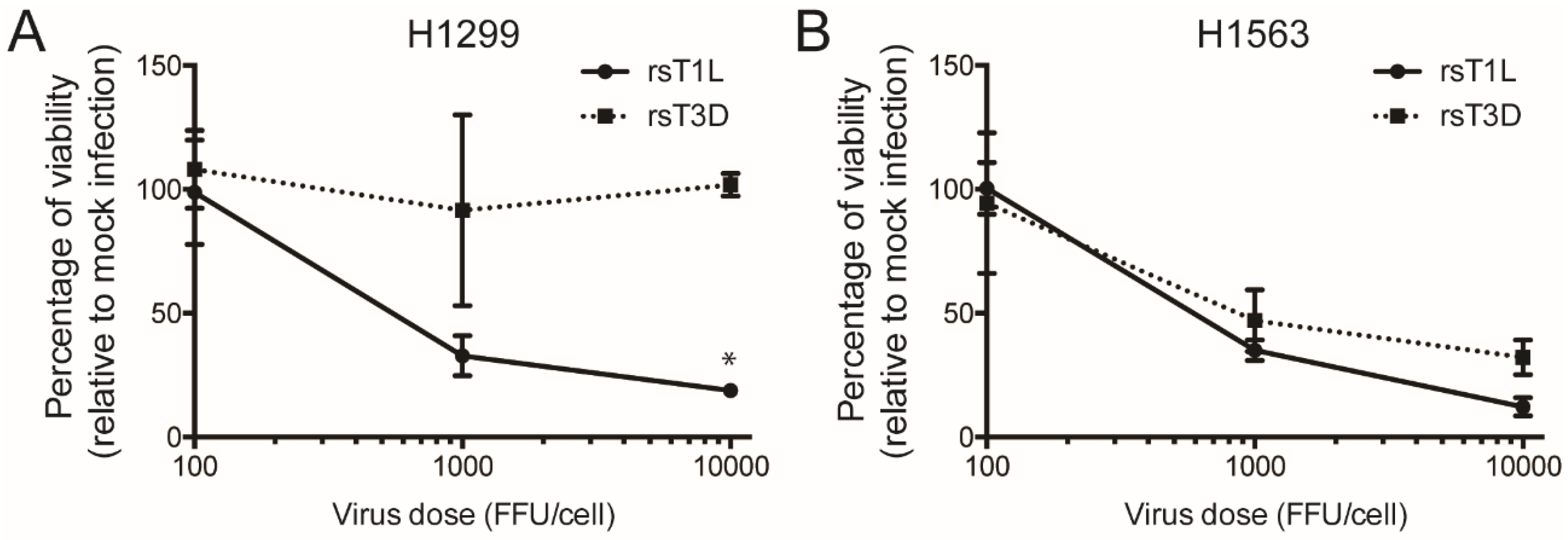
2.1. Recombinant Reovirus Strains rsT1L and rsT3D Differ in the Capacity to Kill Large Cell Carcinoma Cell Lines
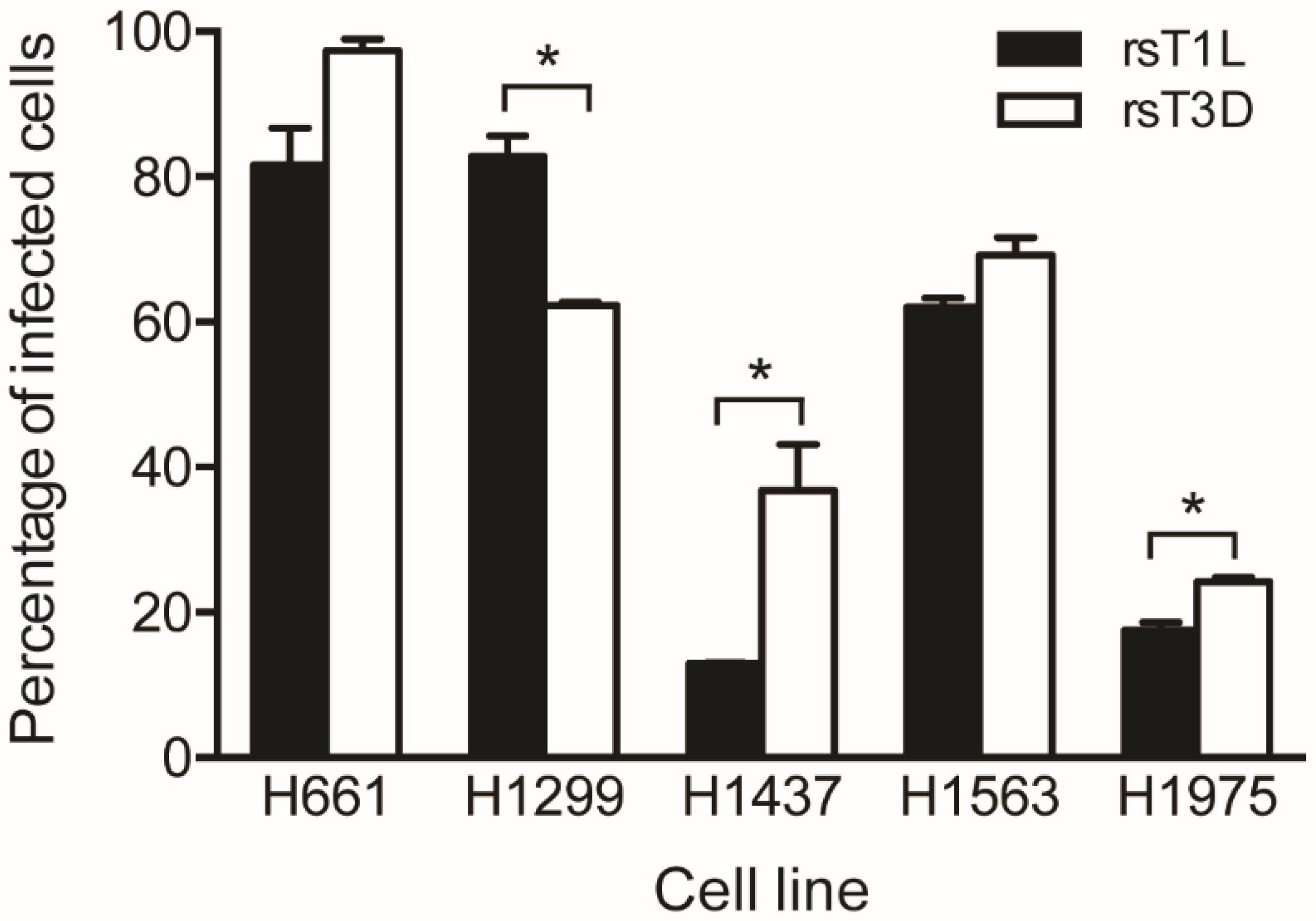
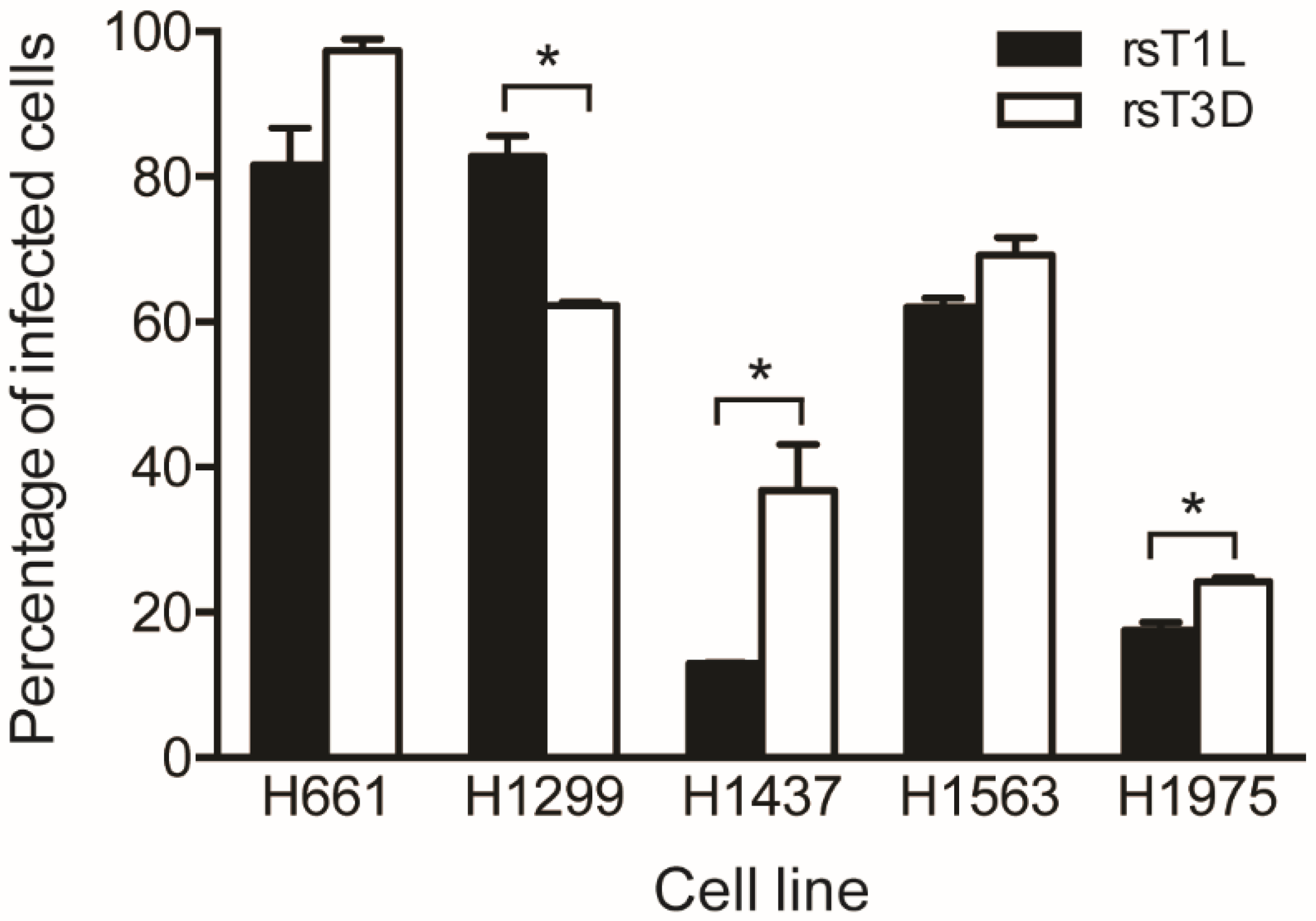
2.2. Reovirus Infectivity in NSCLC Cell Lines
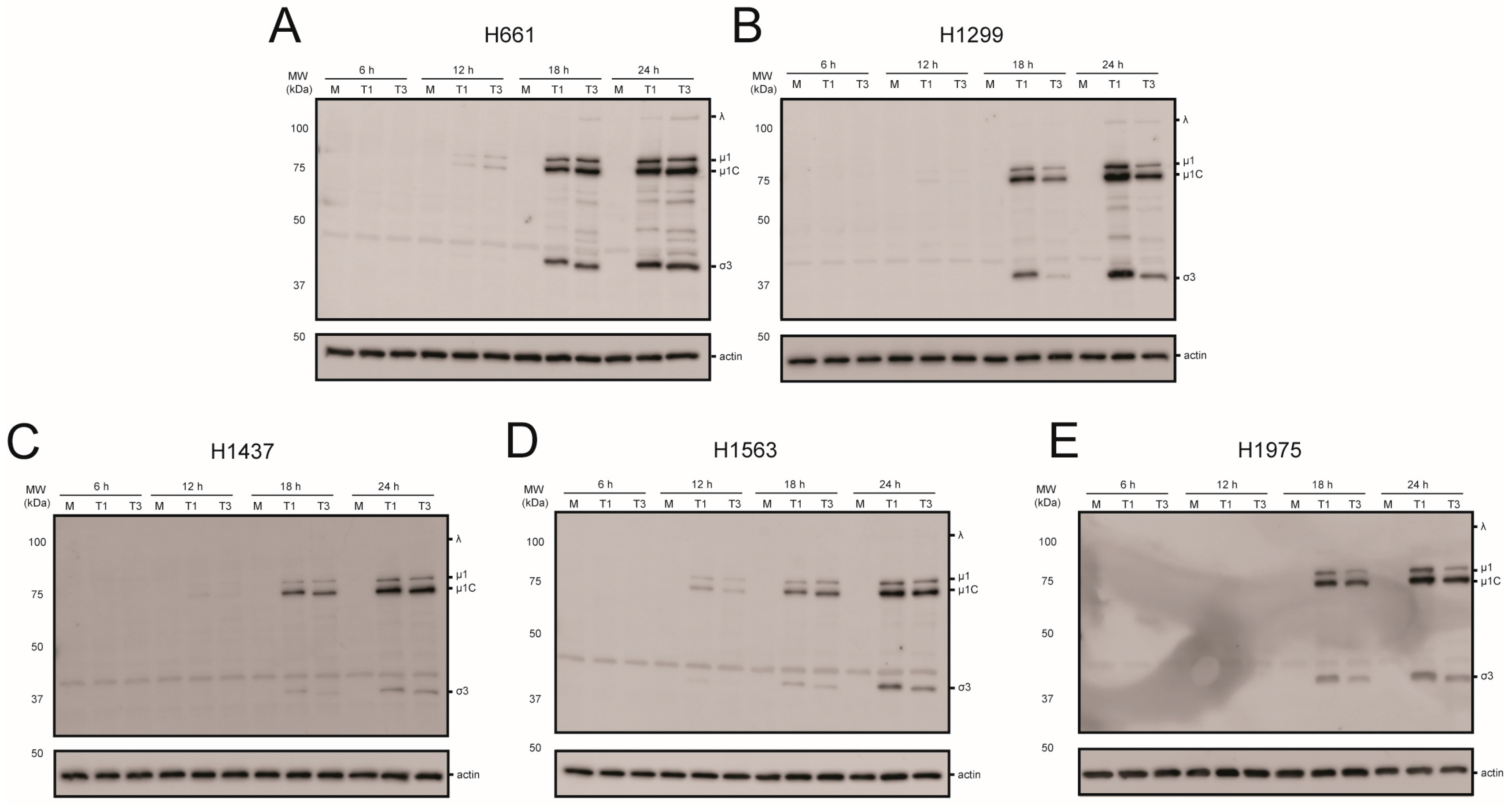
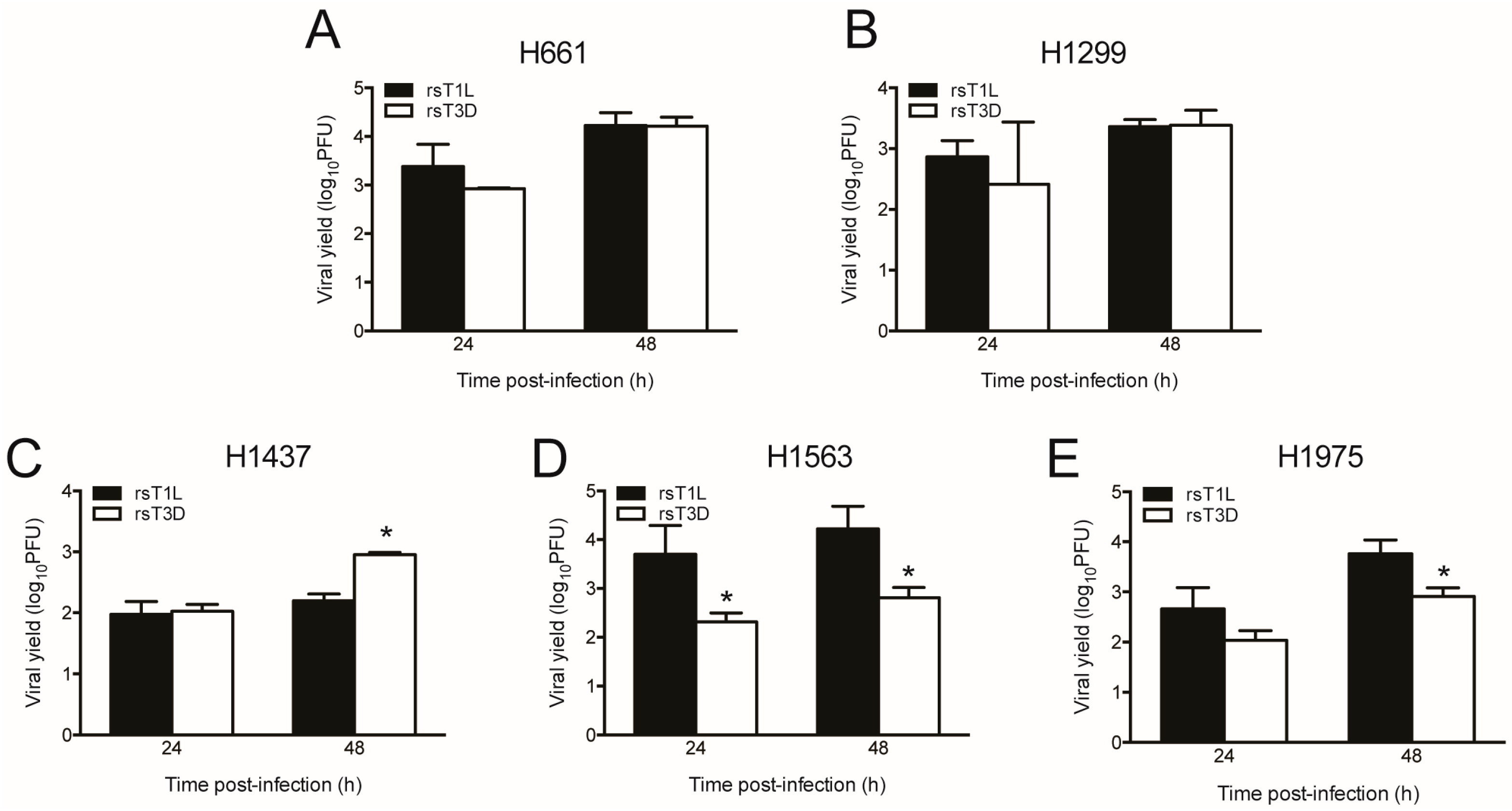
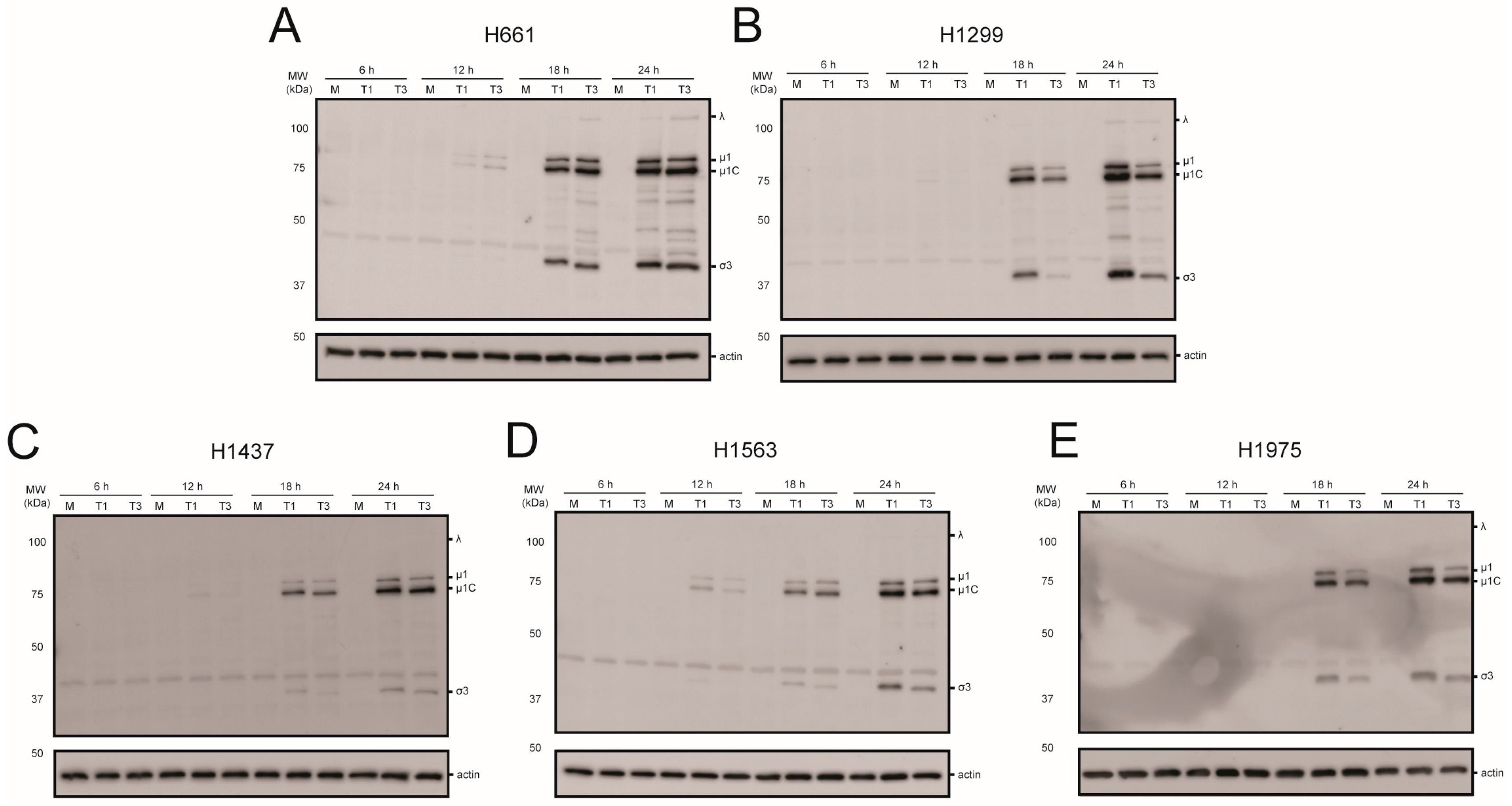
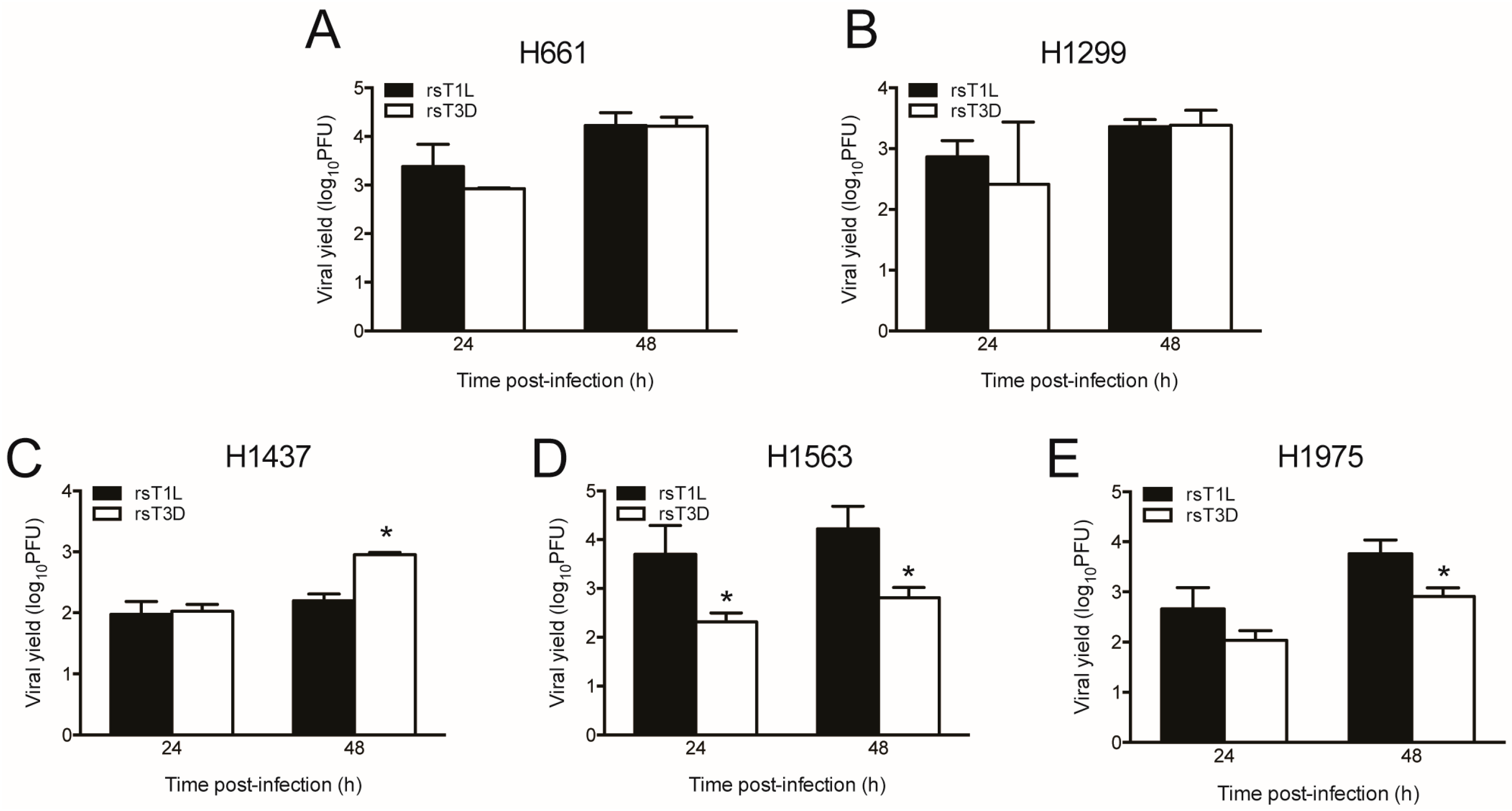
2.3. rsT1L and rsT3D Gene Expression and Replication in NSCLC Cell Lines
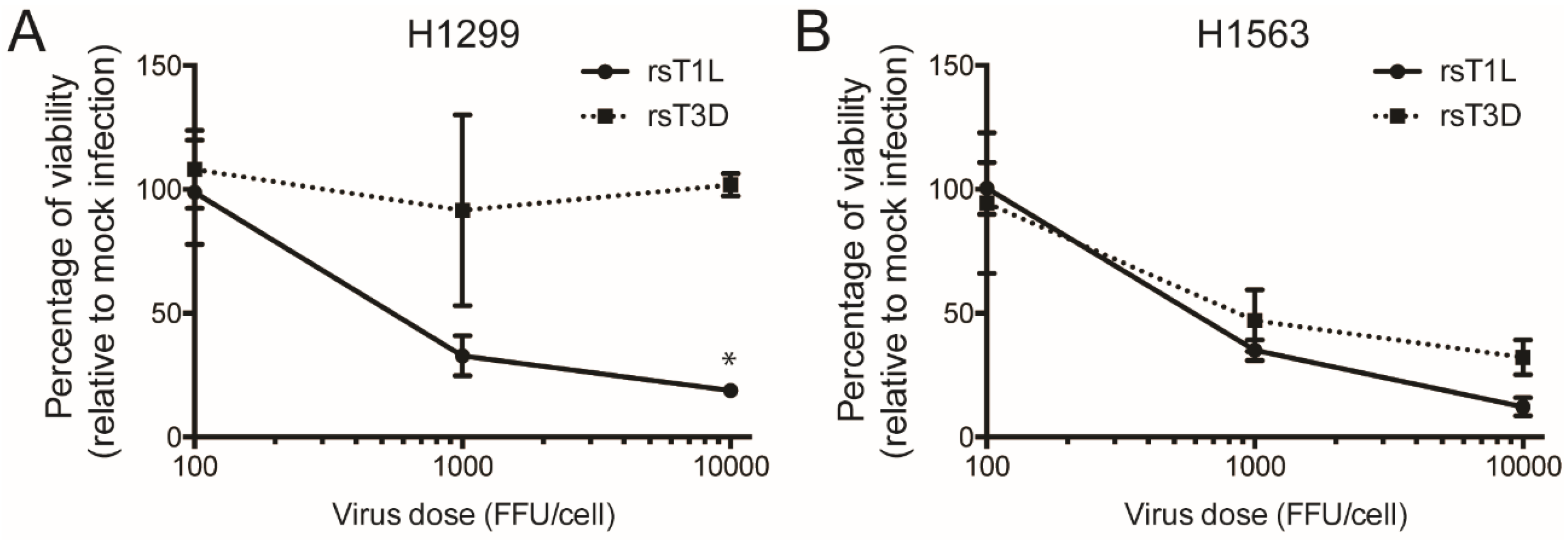
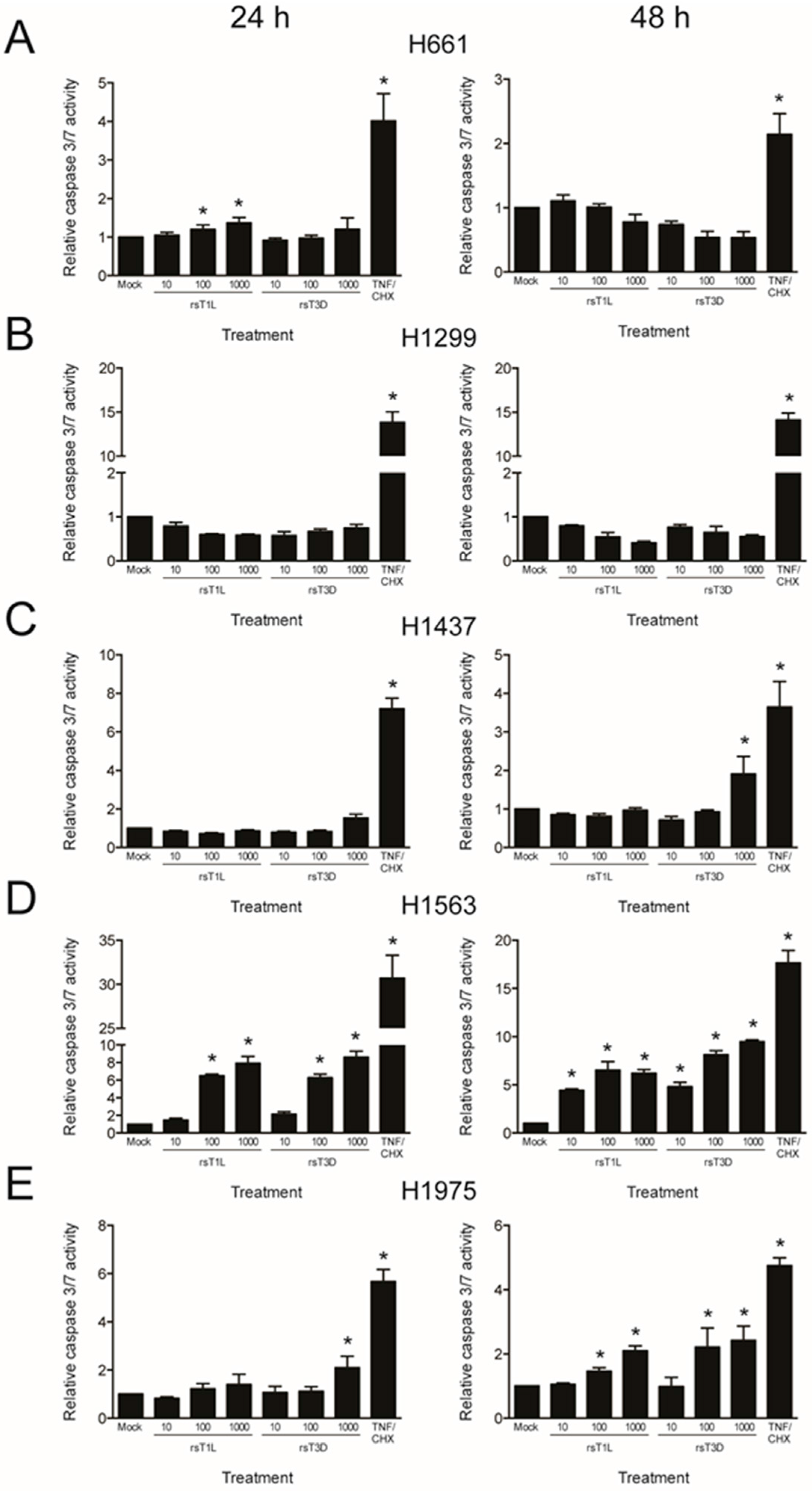
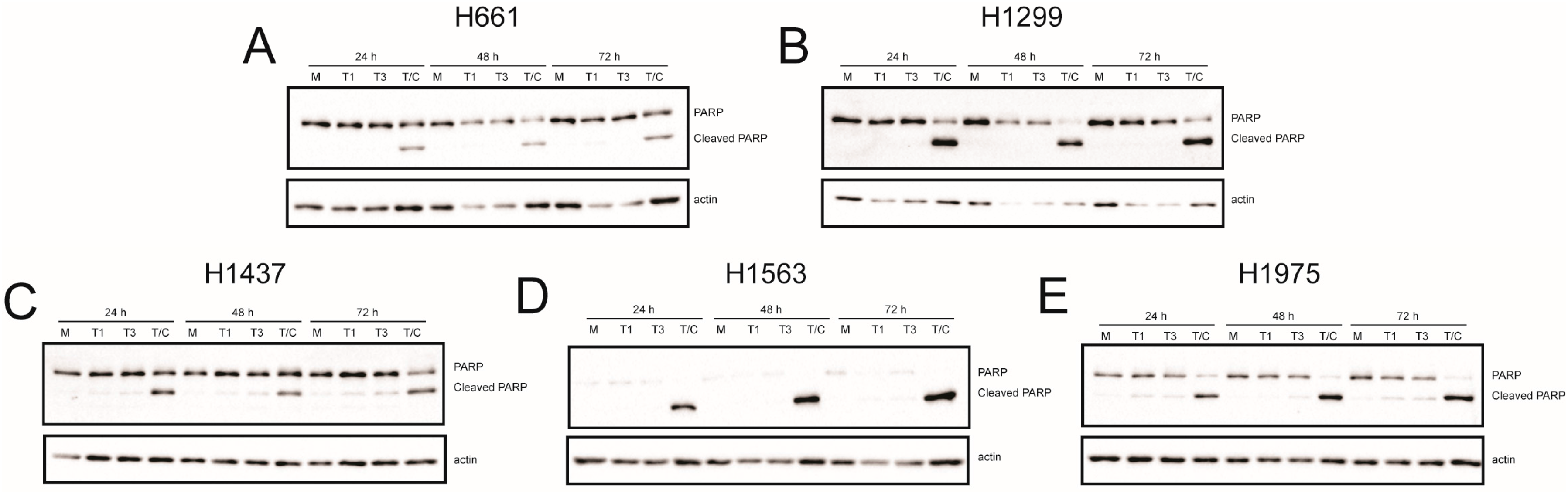
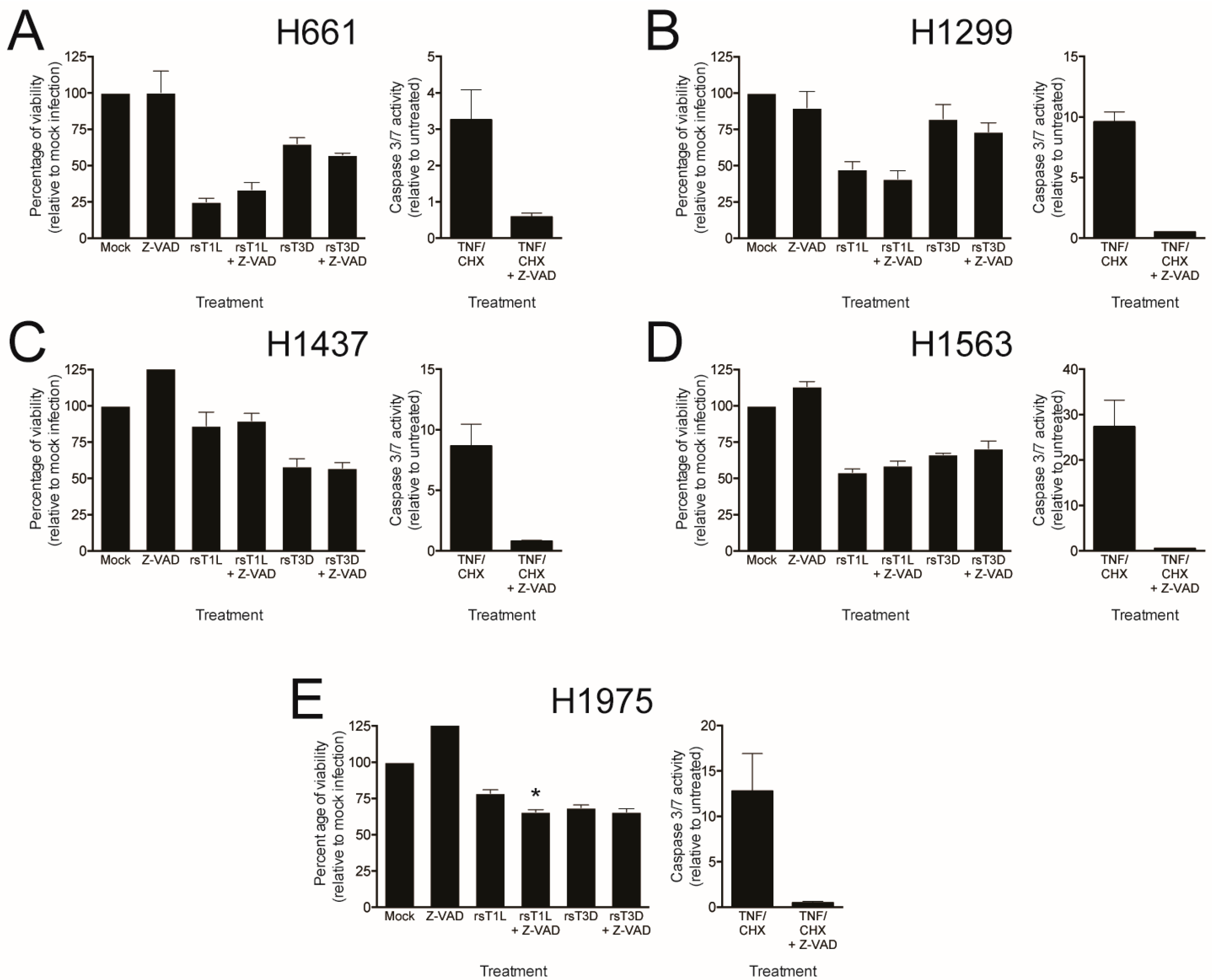
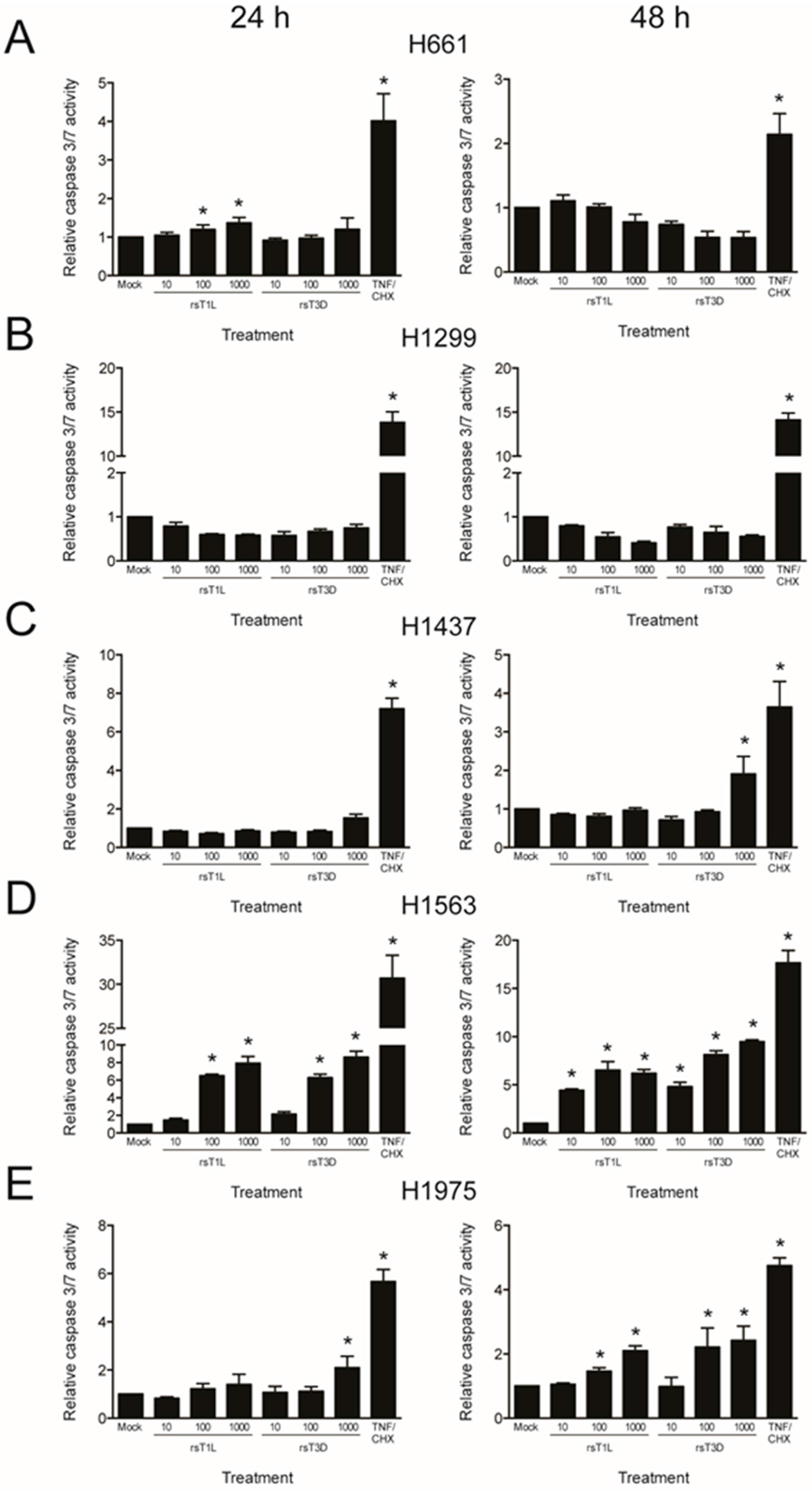
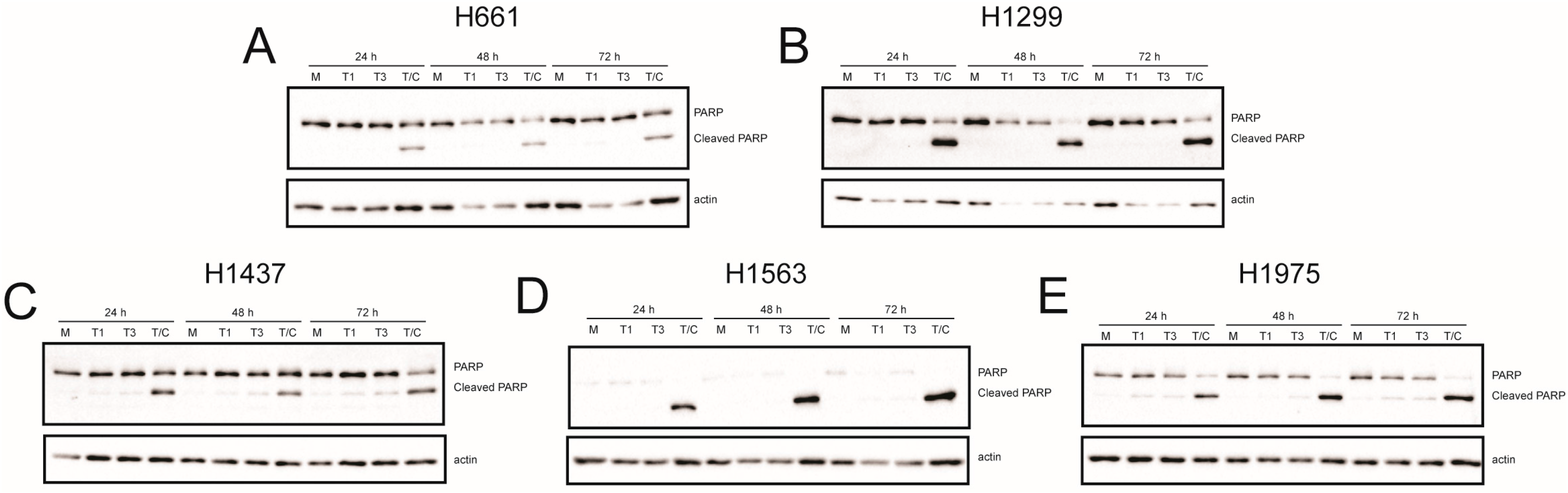
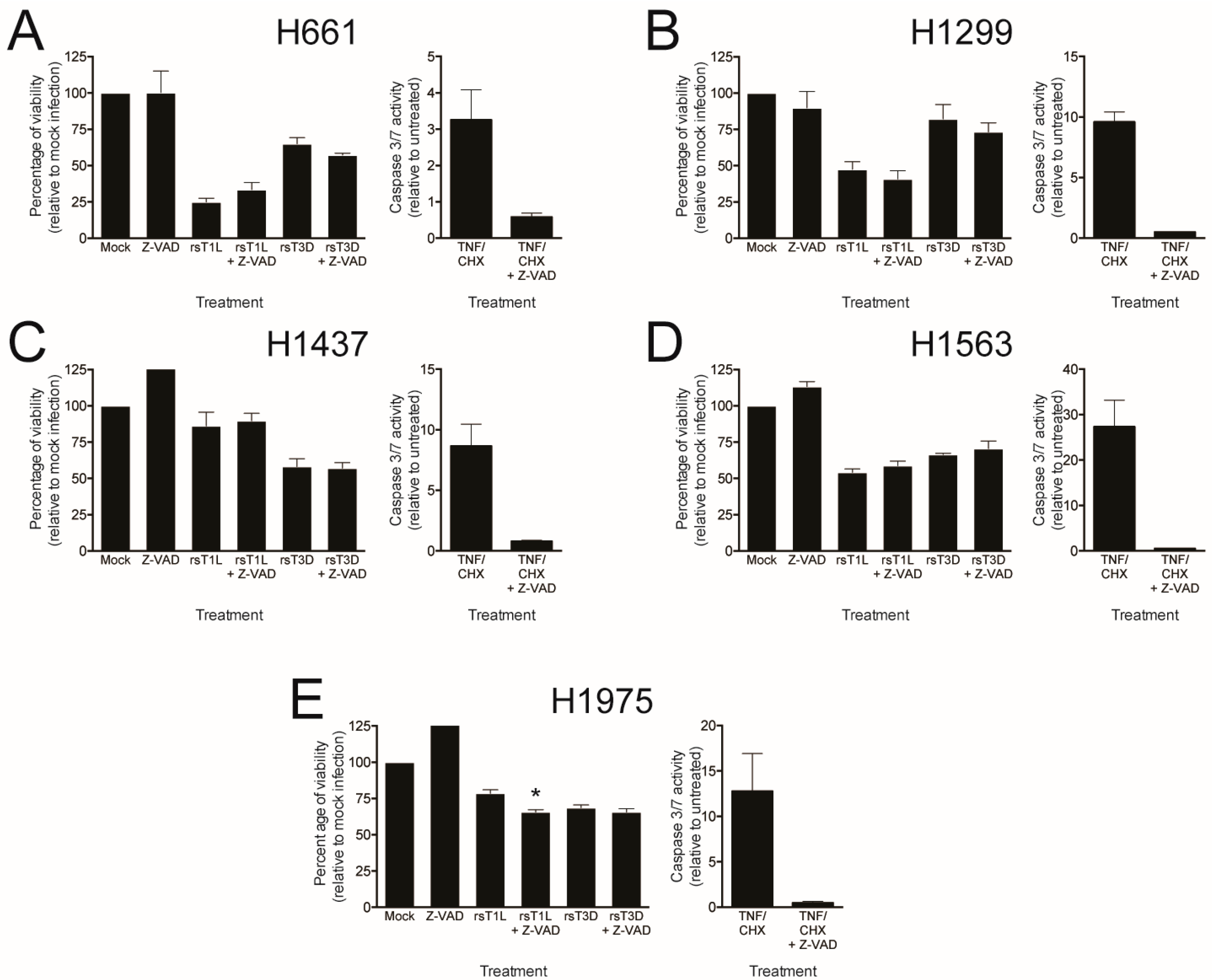
2.4. Recombinant Reoviruses Kill NSCLC Cell Lines by a Caspase-Independent Mechanism
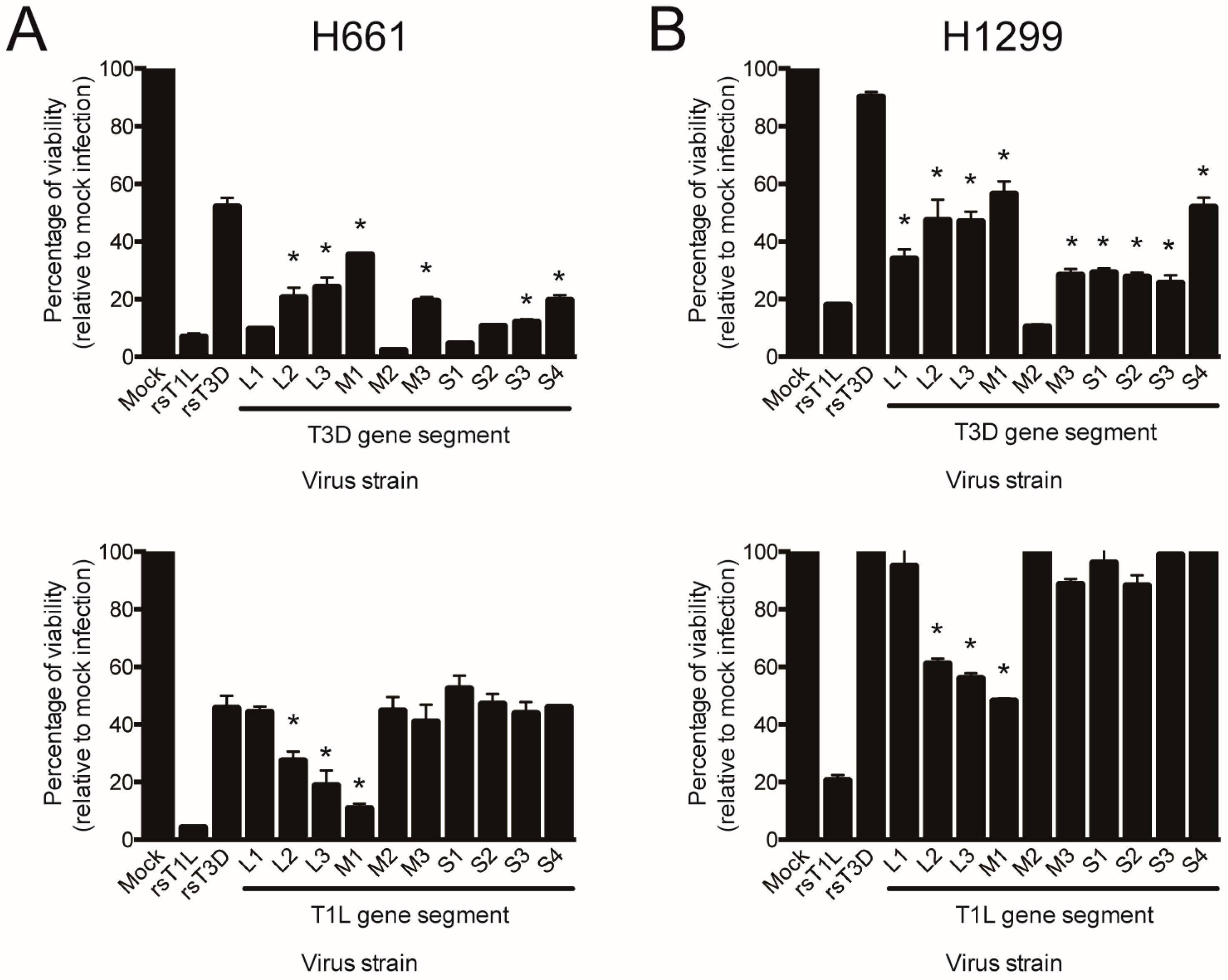
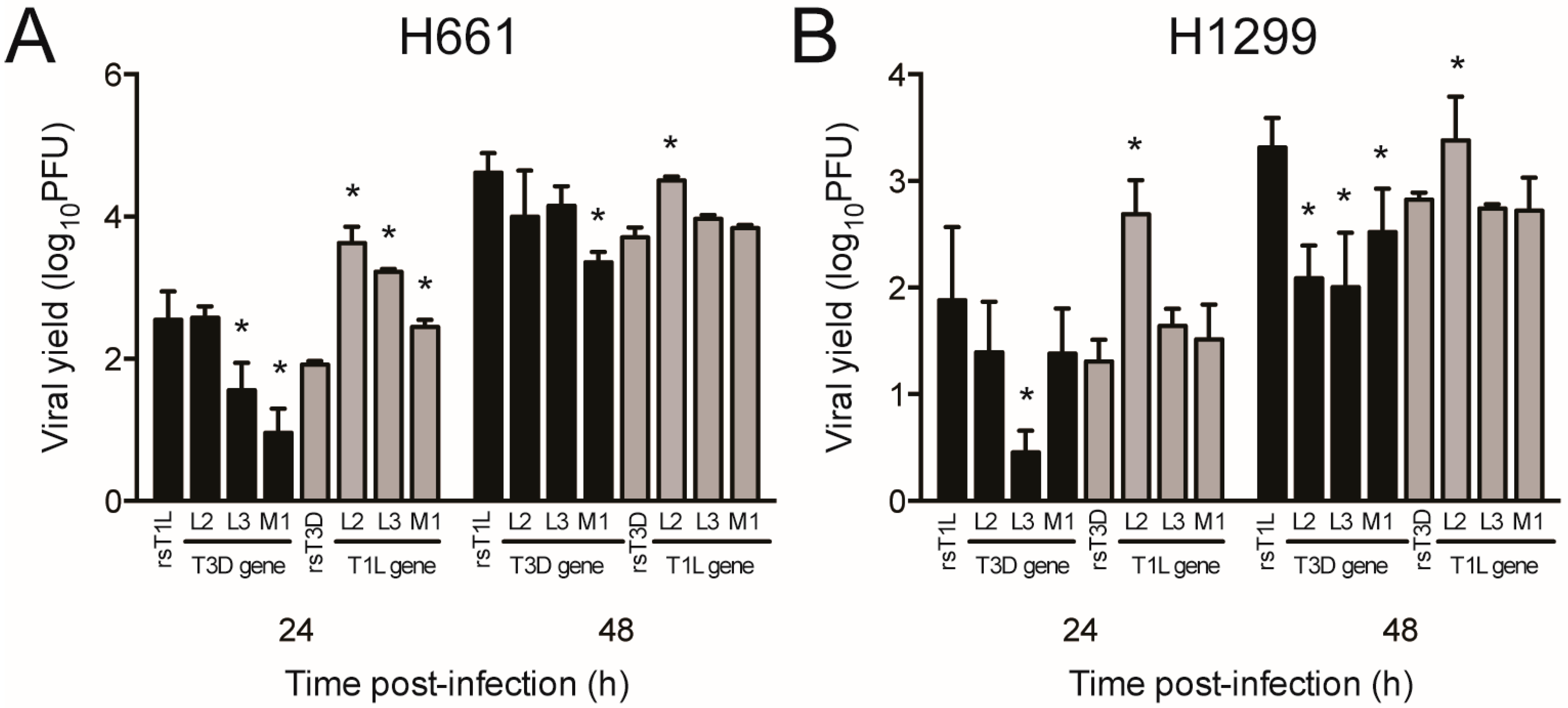
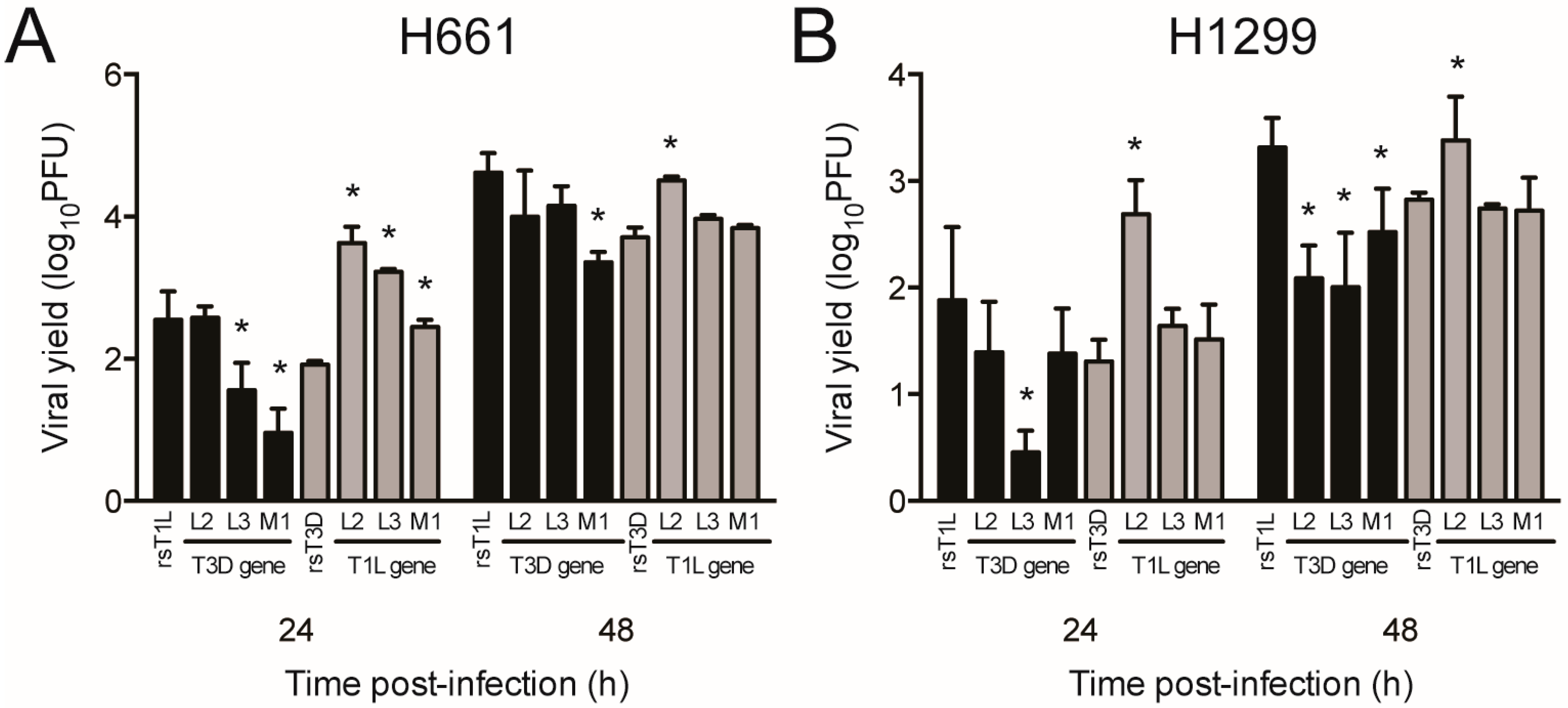
2.5. Reovirus Gene Segments L2, L3, and M1 Correlate with Strain-Specific Differences in Cell Killing
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Viruses
4.3. Cell Viability Assay
4.4. Virus Replication
4.5. Active Caspase-3/7 Assay
4.6. Immunoblot Assay
4.7. Indirect Immunofluorescence
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.R.; Stanish, S.M.; Cox, D.C. Differential sensitivity of normal and transformed human cells to reovirus infection. J. Virol. 1978, 28, 444–449. [Google Scholar] [PubMed]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, K.; Nishikawa, S.G.; Norman, K.L.; Alain, T.; Kossakowska, A.; Lee, P.W. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002, 62, 1696–1701. [Google Scholar] [PubMed]
- Norman, K.L.; Coffey, M.C.; Hirasawa, K.; Demetrick, D.J.; Nishikawa, S.G.; DiFrancesco, L.M.; Strong, J.E.; Lee, P.W. Reovirus oncolysis of human breast cancer. Hum. Gene. Ther. 2002, 13, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.E.; Yang, W.; Senger, D.; Rewcastle, N.B.; Morris, D.G.; Brasher, P.M.; Shi, Z.Q.; Johnston, R.N.; Nishikawa, S.; Lee, P.W.; et al. Reovirus as an oncolytic agent against experimental human malignant gliomas. J. Natl. Cancer Inst. 2001, 93, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Comins, C.; Heinemann, L.; Harrington, K.; Melcher, A.; De Bono, J.; Pandha, H. Reovirus: Viral therapy for cancer “as nature intended”. Clin. Oncol. 2008, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Comins, C.; Spicer, J.; Protheroe, A.; Roulstone, V.; Twigger, K.; White, C.M.; Vile, R.; Melcher, A.; Coffey, M.C.; Mettinger, K.L.; et al. REO-10: A phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin. Cancer Res. 2010, 16, 5564–5572. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, S.; West, E.J.; Scott, K.J.; Appleton, E.; Melcher, A.; Ralph, C. Evidence for Oncolytic Virotherapy: Where Have We Got to and Where Are We Going? Viruses 2015, 7, 6291–6312. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Immunostimulatory cytokines. Oncoimmunology 2013, 2, e24850. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.M.; Barrett, J.W.; Nazarian, S.H.; Werden, S.; McFadden, G. Oncolytic virotherapy synergism with signaling inhibitors: Rapamycin increases myxoma virus tropism for human tumor cells. J. Virol. 2007, 81, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Au, G.G.; Lindberg, A.M.; Barry, R.D.; Shafren, D.R. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int. J. Oncol. 2005, 26, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Grote, D.; Russell, S.J.; Cornu, T.I.; Cattaneo, R.; Vile, R.; Poland, G.A.; Fielding, A.K. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood 2001, 97, 3746–3754. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [PubMed]
- Mohamed, A.; Johnston, R.N.; Shmulevitz, M. Potential for Improving Potency and Specificity of Reovirus Oncolysis with Next-Generation Reovirus Variants. Viruses 2015, 7, 6251–6278. [Google Scholar] [CrossRef] [PubMed]
- Sabin, A.B. Reoviruses: A new group of respiratory and enteric viruses formerly classified as ECHO type 10 is described. Science 1959, 130, 1387–1389. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Markovic, S.N.; Suman, V.J.; Nuovo, G.J.; Vile, R.G.; Kottke, T.J.; Nevala, W.K.; Thompson, M.A.; Lewis, J.E.; Rumilla, K.M.; et al. Phase II trial of intravenous administration of Reolysin (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol. Ther. 2012, 20, 1998–2003. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.G.; Feng, X.; DiFrancesco, L.M.; Fonseca, K.; Forsyth, P.A.; Paterson, A.H.; Coffey, M.C.; Thompson, B. REO-001: A phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin) in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Gujar, S.A.; Ahn, D.G.; Mohamed, A.; Lee, P.W. Reovirus variants with mutations in genome segments S1 and L2 exhibit enhanced virion infectivity and superior oncolysis. J. Virol. 2012, 86, 7403–7413. [Google Scholar] [CrossRef] [PubMed]
- Dermody, T.S.; Parker, J.S.L.; Sherry, B. Orthoreovirus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1304–1346. [Google Scholar]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Antar, A.A.; Boehme, K.W.; Danthi, P.; Eby, E.A.; Guglielmi, K.M.; Holm, G.H.; Johnson, E.M.; Maginnis, M.S.; Naik, S.; et al. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 2007, 1, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ooms, L.S.; Ikizler, M.; Chappell, J.D.; Dermody, T.S. An improved reverse genetics system for mammalian orthoreoviruses. Virology 2010, 398, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Kawagishi, T.; Kobayashi, T.; Ikizler, M.; Iskarpatyoti, J.; Dermody, T.S.; Taniguchi, K. A plasmid-based reverse genetics system for mammalian orthoreoviruses driven by a plasmid-encoded T7 RNA polymerase. J. Virol. Methods 2014, 196, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Komoto, S.; Kawagishi, T.; Nouda, R.; Nagasawa, N.; Onishi, M.; Matsuura, Y.; Taniguchi, K.; Kobayashi, T. Entirely plasmid-based reverse genetics system for rotaviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- American Lung Association. Available online: http://www.lung.org (accessed on 29 May 2017).
- U.S. National Institutes of Health; National Cancer Institute. SEER Cancer Statistics Review, 1975–2013. Available online: https://seer.cancer.gov/archive/csr/1975_2013/ (accessed on 29 May 2017).
- American Cancer Society. Available online: http://www.cancer.org (accessed on 29 May 2017).
- Danthi, P.; Kobayashi, T.; Holm, G.H.; Hansberger, M.W.; Abel, T.W.; Dermody, T.S. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J. Virol. 2008, 82, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Roner, M.R.; Mutsoli, C. The use of monoreassortants and reverse genetics to map reovirus lysis of a ras-transformed cell line. J. Virol. Methods 2007, 139, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.Q.; Senger, D.L.; Lun, X.Q.; Muzik, H.; Shi, Z.Q.; Dyck, R.H.; Norman, K.; Brasher, P.M.; Rewcastle, N.B.; George, D.; et al. Reovirus as an experimental therapeutic for brain and leptomeningeal metastases from breast cancer. Gene Ther. 2004, 11, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Etoh, T.; Suzuki, K.; Mitui, M.T.; Nishizono, A.; Shiraishi, N.; Kitano, S. Efficacy of oncolytic reovirus against human gastric cancer with peritoneal metastasis in experimental animal model. Int. J. Oncol. 2010, 37, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.; Yatabe, Y.; Ishikawa, Y.; Wistuba, I.; Flieder, D.B.; Franklin, W.; et al. Diagnosis of lung cancer in small biopsies and cytology: Implications of the 2011 International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification. Arch. Pathol. Lab. Med. 2013, 137, 668–684. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.; Yatabe, Y.; Ishikawa, Y.; Wistuba, I.; Flieder, D.B.; Franklin, W.; et al. Diagnosis of lung adenocarcinoma in resected specimens: Implications of the 2011 International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification. Arch. Pathol. Lab. Med. 2013, 137, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Sei, S.; Mussio, J.K.; Yang, Q.E.; Nagashima, K.; Parchment, R.E.; Coffey, M.C.; Shoemaker, R.H.; Tomaszewski, J.E. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents in non-small cell lung cancer cells. Mol. Cancer 2009, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Himeno, Y.; Matsumoto, T.; Aramaki, M.; Kawano, K.; Nishizono, A.; Kitano, S. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin. Cancer Res. 2003, 9, 1218–1223. [Google Scholar] [PubMed]
- Twigger, K.; Roulstone, V.; Kyula, J.; Karapanagiotou, E.M.; Syrigos, K.N.; Morgan, R.; White, C.; Bhide, S.; Nuovo, G.; Coffey, M.; et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the EGFR pathway. BMC Cancer 2012, 12, 368. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L.; Squier, M.K.; Brown, A.L.; Pike, B.; Willis, D.; Oberhaus, S.M.; Dermody, T.S.; Cohen, J.J. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: Role of the S1 and M2 genes. J. Virol. 1996, 70, 7984–7991. [Google Scholar] [PubMed]
- Tyler, K.L.; Squier, M.K.; Rodgers, S.E.; Schneider, B.E.; Oberhaus, S.M.; Grdina, T.A.; Cohen, J.J.; Dermody, T.S. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J. Virol. 1995, 69, 6972–6979. [Google Scholar] [PubMed]
- Fleeton, M.N.; Contractor, N.; Leon, F.; Wetzel, J.D.; Dermody, T.S.; Kelsall, B.L. Peyer’s patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J. Exp. Med. 2004, 200, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P.; Coffey, C.M.; Parker, J.S.; Abel, T.W.; Dermody, T.S. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog. 2008, 4, e1000248. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.M.; Nodwell, M.J.; Hirasawa, K.; Shi, Z.Q.; Diaz, R.; Luider, J.; Johnston, R.N.; Forsyth, P.A.; Magliocco, A.M.; Lee, P.; et al. Oncolytic viral therapy for prostate cancer: Efficacy of reovirus as a biological therapeutic. Cancer Res. 2010, 70, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Espitia, C.M.; Mahalingam, D.; Oyajobi, B.O.; Coffey, M.; Giles, F.J.; Carew, J.S.; Nawrocki, S.T. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene 2012, 31, 3023–3038. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Espitia, C.M.; Zhao, W.; Kelly, K.R.; Coffey, M.; Freeman, J.W.; Nawrocki, S.T. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013, 4, e728. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.K.; Danthi, P. Reovirus activates a caspase-independent cell death pathway. mBio 2013, 4, e00178-13. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus modulates autophagy during oncolysis of multiple myeloma. Autophagy 2013, 9, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Harvey, J.D.; Bellamy, A.R. Subunit structure of the reovirus spike. J. Virol. 1980, 36, 894–896. [Google Scholar] [PubMed]
- White, C.K.; Zweerink, H.J. Studies on the structure of reovirus cores: Selective removal of polypeptide lambda 2. Virology 1976, 70, 171–180. [Google Scholar] [CrossRef]
- Schiff, L.A.; Nibert, M.L.; Tyler, K.L. Orthoreoviruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 1853–1915. [Google Scholar]
- Dryden, K.A.; Wang, G.; Yeager, M.; Nibert, M.L.; Coombs, K.M.; Furlong, D.B.; Fields, B.N.; Baker, T.S. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: Analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J. Cell Biol. 1993, 122, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.R.; Zarbl, H.; Millward, S. Reovirus guanylyltransferase is L2 gene product lambda 2. J. Virol. 1986, 60, 307–311. [Google Scholar] [PubMed]
- Coombs, K.M. Stoichiometry of reovirus structural proteins in virus, ISVP, and core particles. Virology 1998, 243, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Nibert, M.L. Core protein mu2 is a second determinant of nucleoside triphosphatase activities by reovirus cores. J. Virol. 1997, 71, 7728–7735. [Google Scholar] [PubMed]
- Noble, S.; Nibert, M.L. Characterization of an ATPase activity in reovirus cores and its genetic association with core-shell protein lambda1. J. Virol. 1997, 71, 2182–2191. [Google Scholar] [PubMed]
- Bisaillon, M.; Bergeron, J.; Lemay, G. Characterization of the nucleoside triphosphate phosphohydrolase and helicase activities of the reovirus lambda1 protein. J. Biol. Chem. 1997, 272, 18298–18303. [Google Scholar] [CrossRef] [PubMed]
- Bisaillon, M.; Lemay, G. Characterization of the reovirus lambda1 protein RNA 5′-triphosphatase activity. J. Biol. Chem. 1997, 272, 29954–29957. [Google Scholar] [CrossRef] [PubMed]
- Sherry, B.; Baty, C.J.; Blum, M.A. Reovirus-induced acute myocarditis in mice correlates with viral RNA synthesis rather than generation of infectious virus in cardiac myocytes. J. Virol. 1996, 70, 6709–6715. [Google Scholar] [PubMed]
- Sherry, B.; Torres, J.; Blum, M.A. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J. Virol. 1998, 72, 1314–1323. [Google Scholar] [PubMed]
- Zurney, J.; Kobayashi, T.; Holm, G.H.; Dermody, T.S.; Sherry, B. Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J. Virol. 2009, 83, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.; Tran, H.; Fortin, Y.; Yu, Z.; Shen, S.H.; Kolman, J.; Onions, D.; Voyer, R.; Hagerman, A.; Serl, S.; et al. Evaluation of homogeneity and genetic stability of REOLYSIN (pelareorep) by complete genome sequencing of reovirus after large scale production. Appl. Microbiol. Biotechnol. 2014, 98, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Sandekian, V.; Lim, D.; Prud’homme, P.; Lemay, G. Transient high level mammalian reovirus replication in a bat epithelial cell line occurs without cytopathic effect. Virus Res. 2013, 173, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Furlong, D.B.; Nibert, M.L.; Fields, B.N. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J. Virol. 1988, 62, 246–256. [Google Scholar] [PubMed]
- Smith, R.E.; Zweerink, H.J.; Joklik, W.K. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 1969, 39, 791–810. [Google Scholar] [CrossRef]
- Virgin, H.W.T.; Bassel-Duby, R.; Fields, B.N.; Tyler, K.L. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J. Virol. 1988, 62, 4594–4604. [Google Scholar] [PubMed]
- Mainou, B.A.; Zamora, P.F.; Ashbrook, A.W.; Dorset, D.C.; Kim, K.S.; Dermody, T.S. Reovirus cell entry requires functional microtubules. mBio 2013, 4, e00405-13. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.K.; Hiller, B.E.; Thete, D.; Snyder, A.J.; Perez, E., Jr.; Upton, J.W.; Danthi, P. Viral RNA at Two Stages of Reovirus Infection Is Required for the Induction of Necroptosis. J. Virol. 2017, 91, e02404-16. [Google Scholar] [CrossRef] [PubMed]
- Van den Hengel, S.K.; Balvers, R.K.; Dautzenberg, I.J.; van den Wollenberg, D.J.; Kloezeman, J.J.; Lamfers, M.L.; Sillivis-Smit, P.A.; Hoeben, R.C. Heterogeneous reovirus susceptibility in human glioblastoma stem-like cell cultures. Cancer Gene Ther. 2013, 20, 507–513. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Histology | Tumor Source |
|---|---|---|
| NCI-H661 | Large cell carcinoma | Lymph node metastasis |
| NCI-H1299 | Large cell carcinoma | Lymph node metastasis |
| NCI-H1437 | Adenocarcinoma | Pleural effusion metastasis |
| NCI-H1563 | Adenocarcinoma | Primary |
| NCI-H1975 | Adenocarcinoma | Primary |
| Cell Line | Cell Killing | Infectivity | Replication | Apoptosis Induction | ||||
|---|---|---|---|---|---|---|---|---|
| rsT1L | rsT3D | rsT1L | rsT3D | rsT1L | rsT3D | rsT1L | rsT3D | |
| H661 | +++ | ++ | +++ | +++ | +++ | +++ | − | − |
| H1299 | +++ | + | +++ | ++ | ++ | ++ | − | − |
| H1437 | ++ | ++ | + | ++ | + | ++ | − | + |
| H1563 | +++ | +++ | ++ | ++ | +++ | ++ | +++ | +++ |
| H1975 | ++ | ++ | + | + | ++ | ++ | + | + |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, E.J.; Howells, M.A.; Stuart, J.D.; Boehme, K.W. Serotype-Specific Killing of Large Cell Carcinoma Cells by Reovirus. Viruses 2017, 9, 140. https://doi.org/10.3390/v9060140
Simon EJ, Howells MA, Stuart JD, Boehme KW. Serotype-Specific Killing of Large Cell Carcinoma Cells by Reovirus. Viruses. 2017; 9(6):140. https://doi.org/10.3390/v9060140
Chicago/Turabian StyleSimon, Emily J., Morgan A. Howells, Johnasha D. Stuart, and Karl W. Boehme. 2017. "Serotype-Specific Killing of Large Cell Carcinoma Cells by Reovirus" Viruses 9, no. 6: 140. https://doi.org/10.3390/v9060140
APA StyleSimon, E. J., Howells, M. A., Stuart, J. D., & Boehme, K. W. (2017). Serotype-Specific Killing of Large Cell Carcinoma Cells by Reovirus. Viruses, 9(6), 140. https://doi.org/10.3390/v9060140