Stumbling across the Same Phage: Comparative Genomics of Widespread Temperate Phages Infecting the Fish Pathogen Vibrio anguillarum

 ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Isolation, Purification and Propagation of Bacteriophages
2.3. Morphology, Life Cycle and Host Range of Bacteriophages
2.4. DNA Extraction and Sequencing
2.5. Annotation, Comparative Genomics, and Phylogeny
2.6. CRISPR Arrays and Prophage Detection in Vibrio
2.7. Integration of Phages in V. anguillarum Strain BA35
2.8. GenBank Accession Numbers
3. Results
3.1. Isolation and Characterization of Bacteriophages
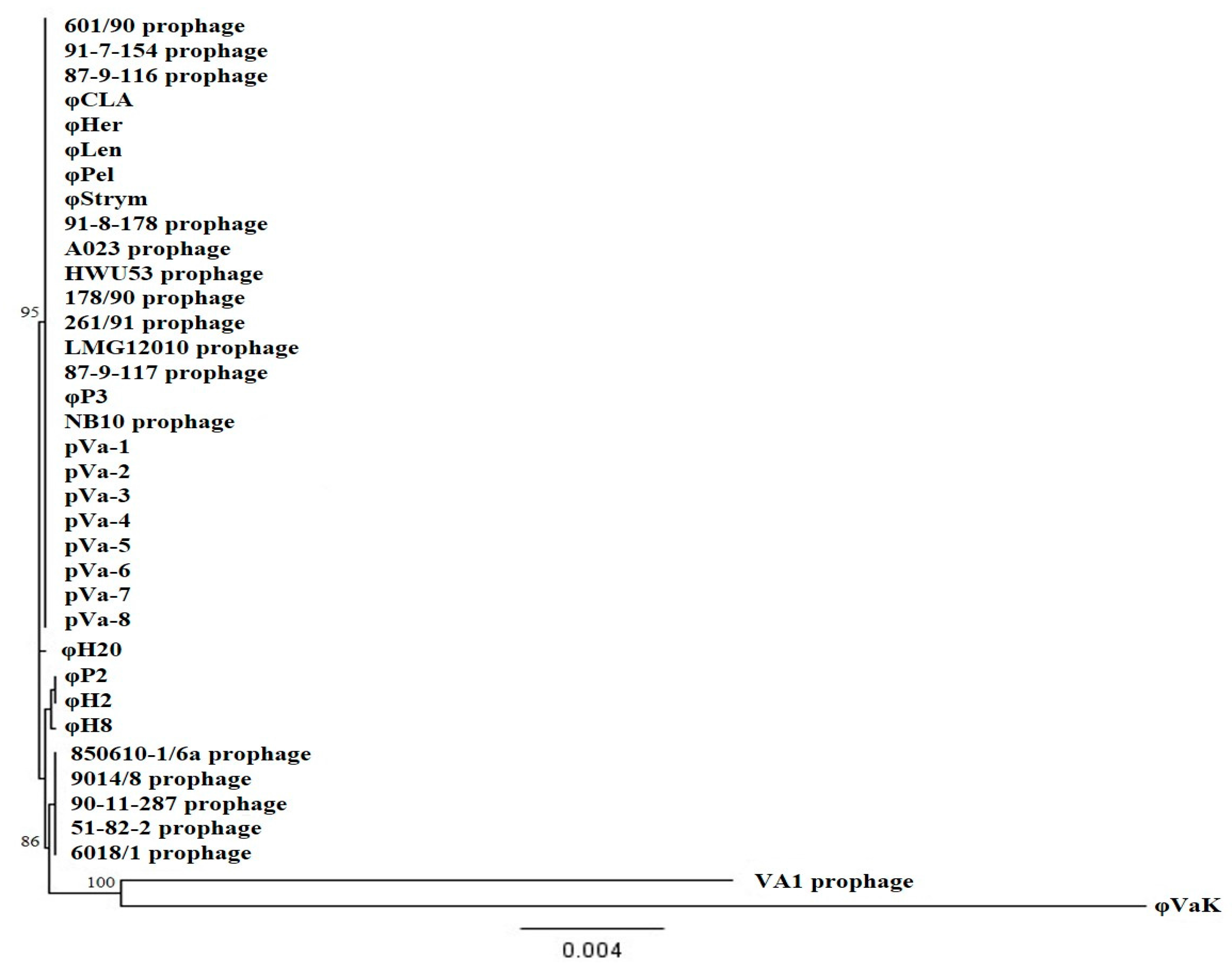
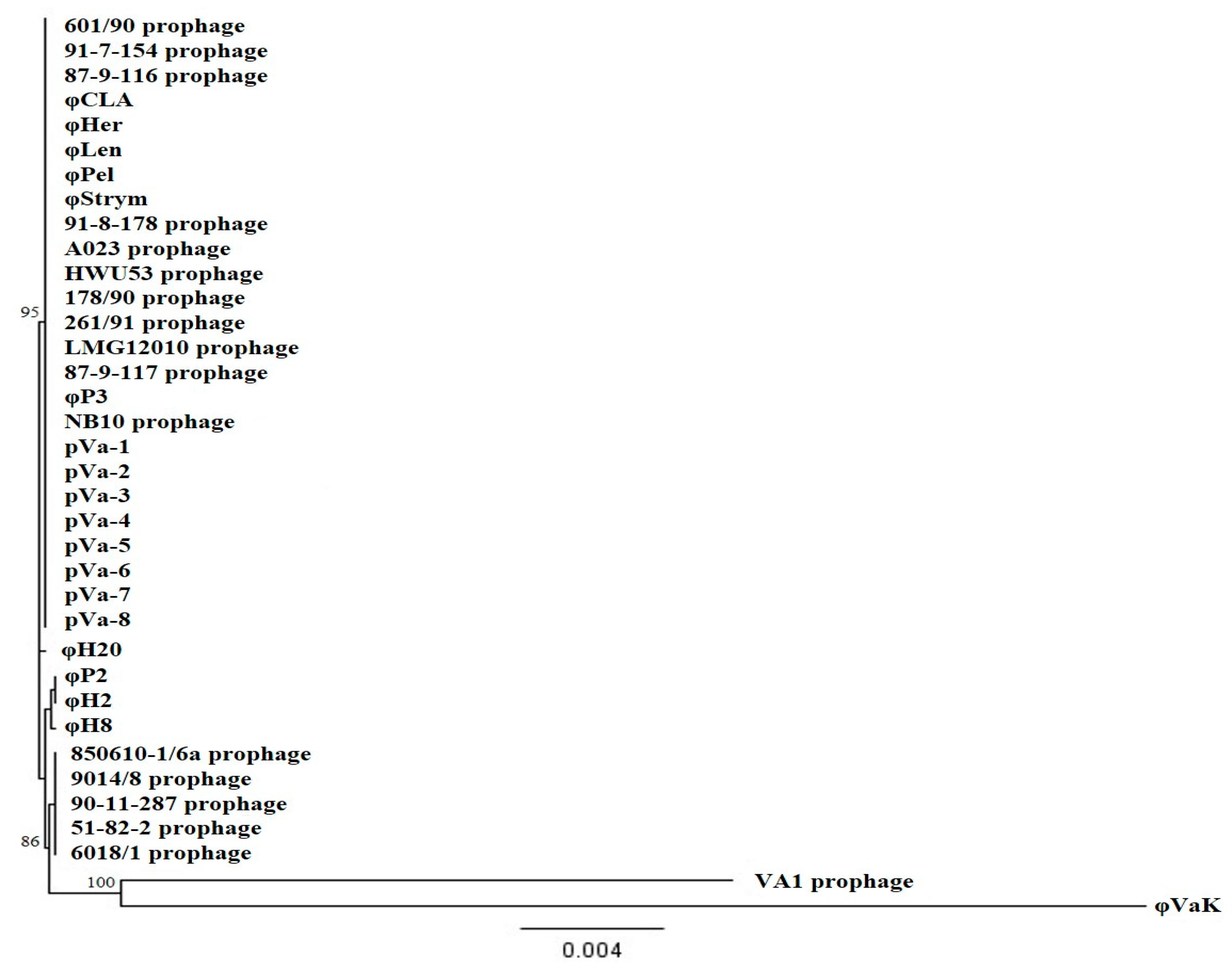
3.2. Host Range Analysis and Phylogeny
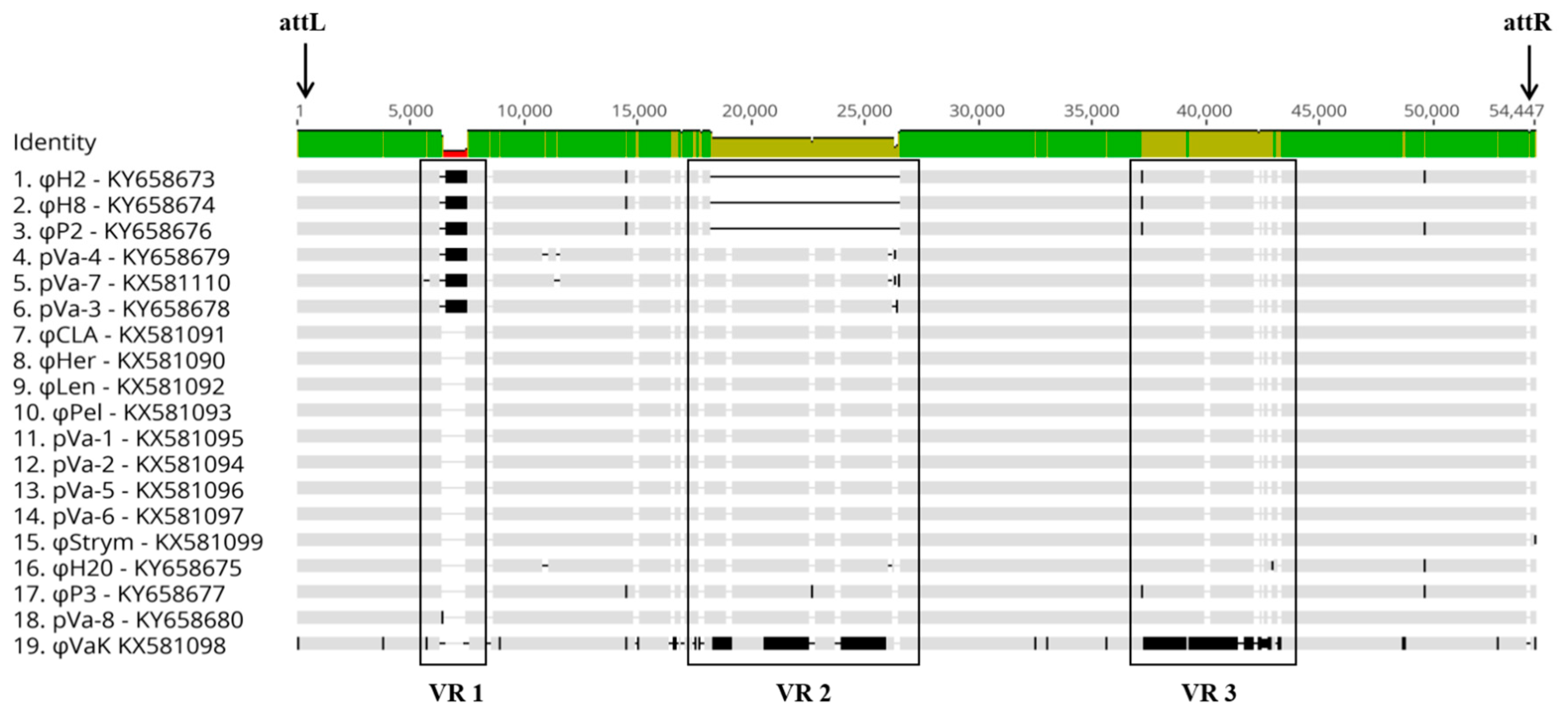
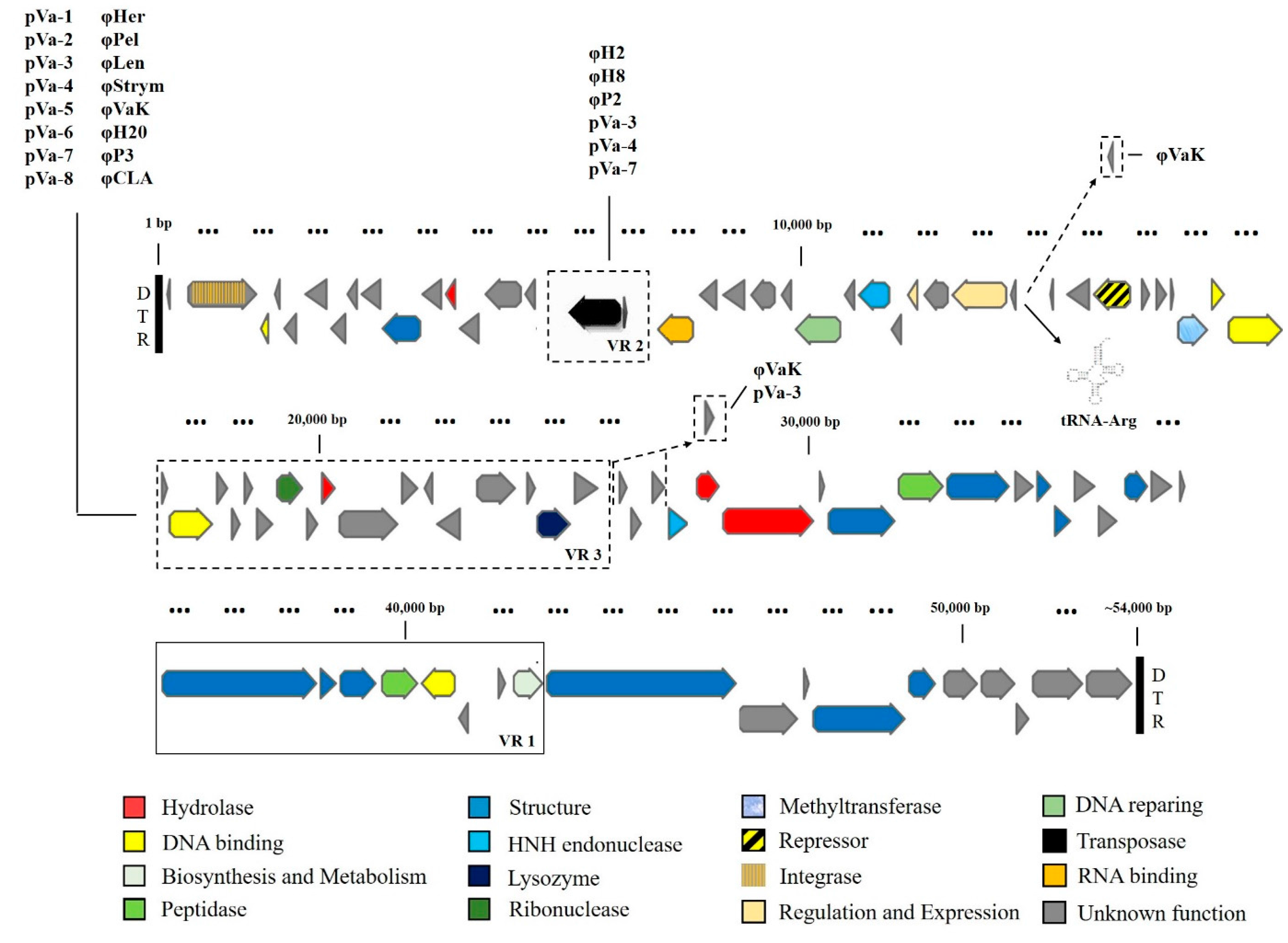
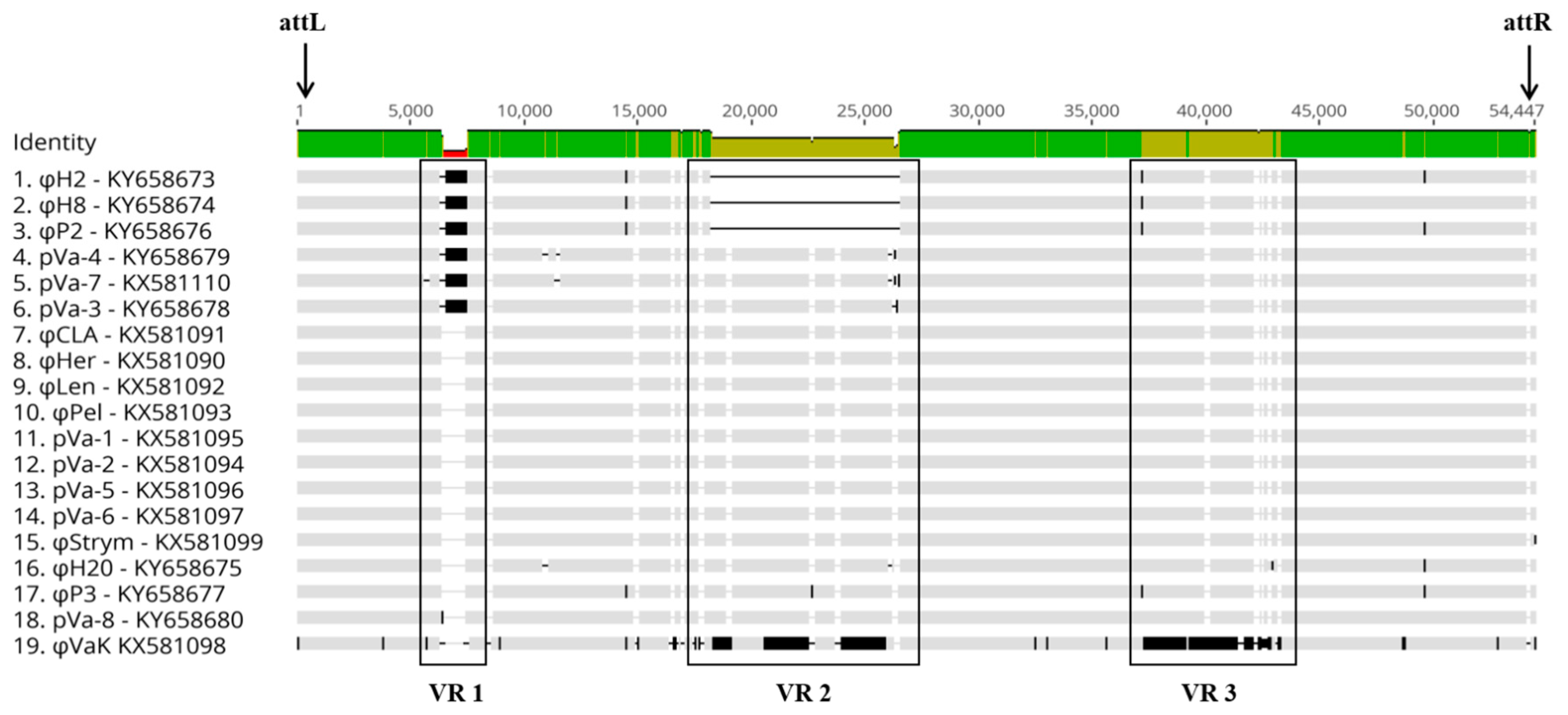
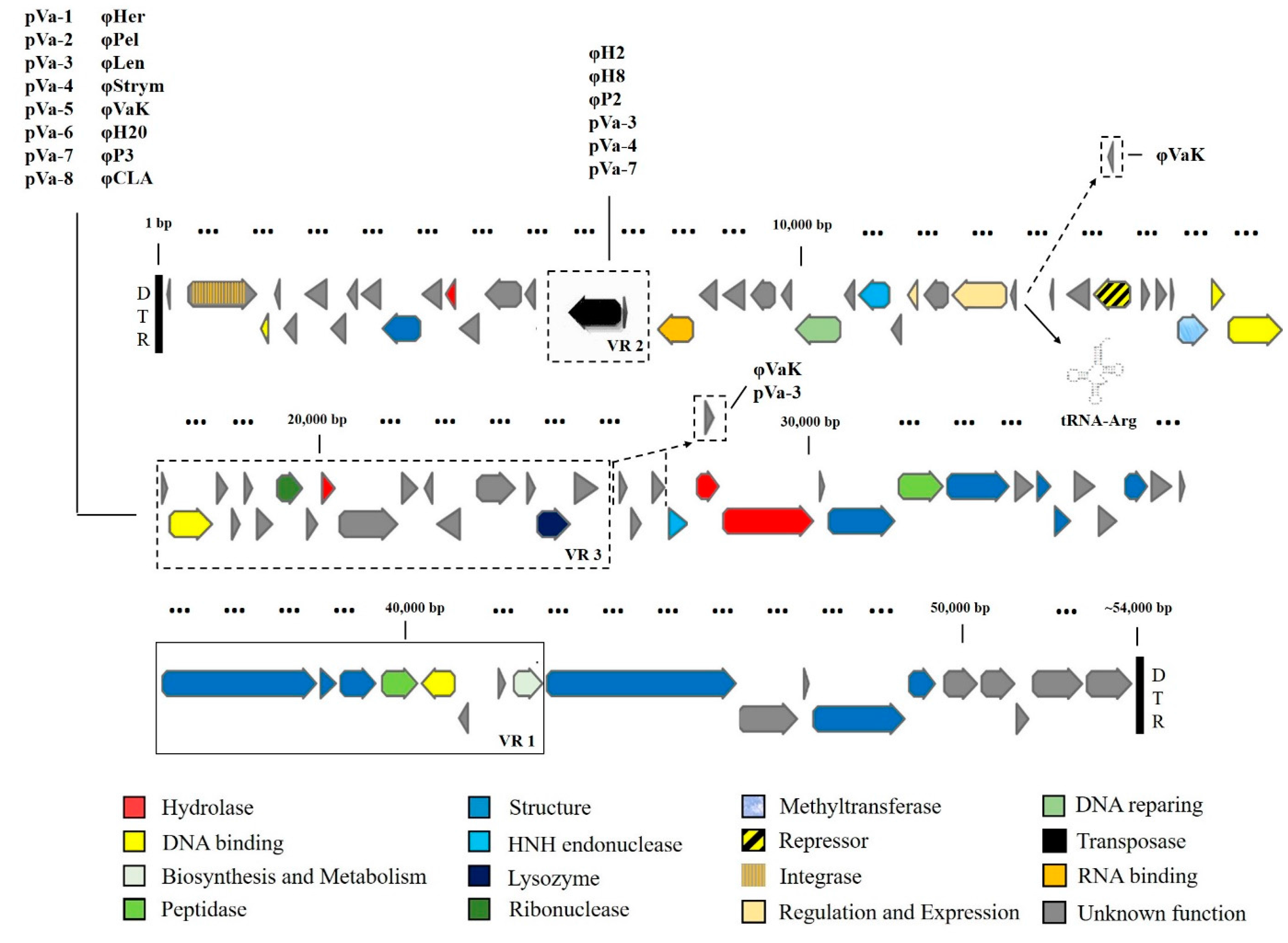
3.3. Comparative and Functional Phage Genomics
3.4. Vibrio CRISPR Spacers in H20-Like Phages
3.5. H20-Like Prophages Presence in V. anguillarum
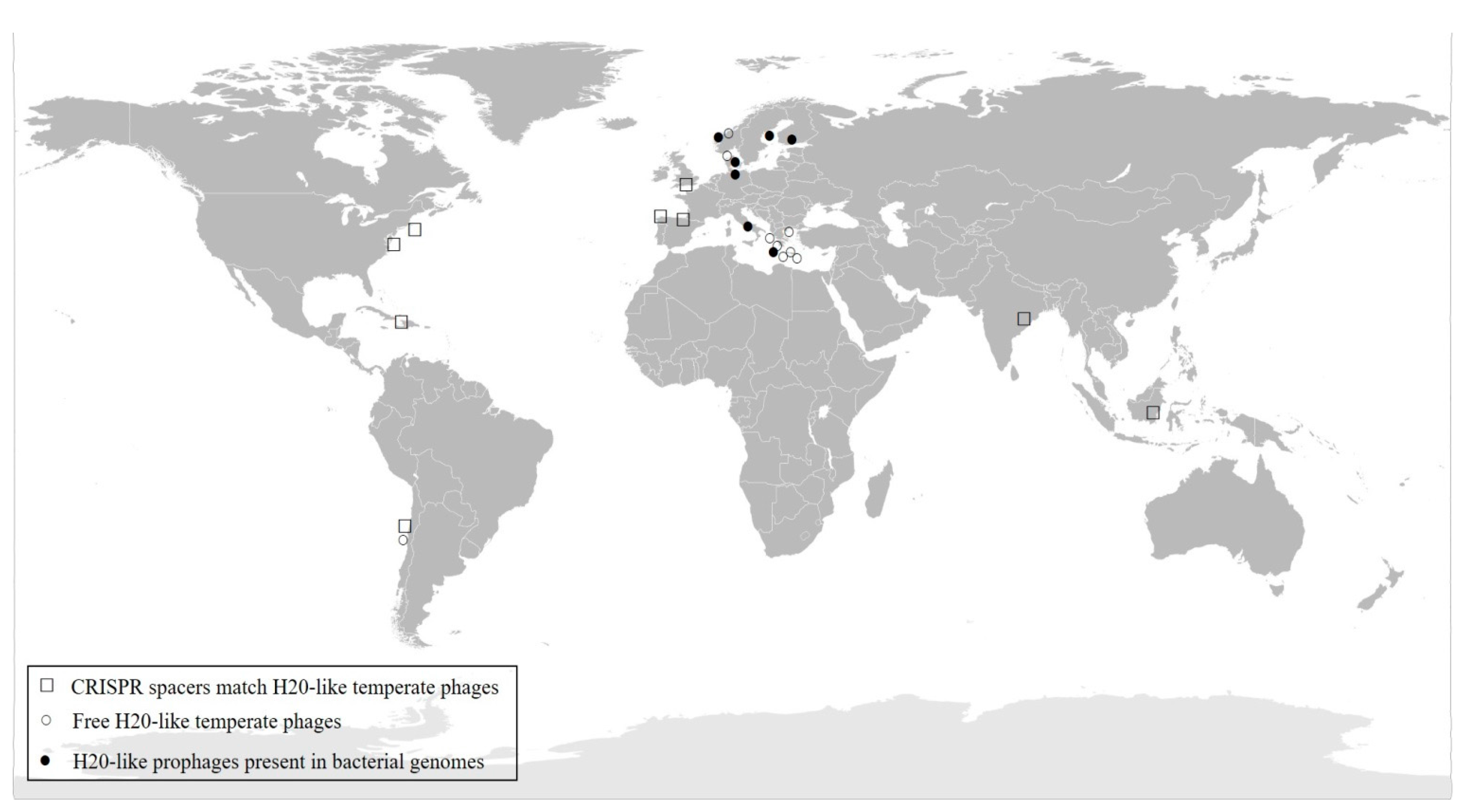
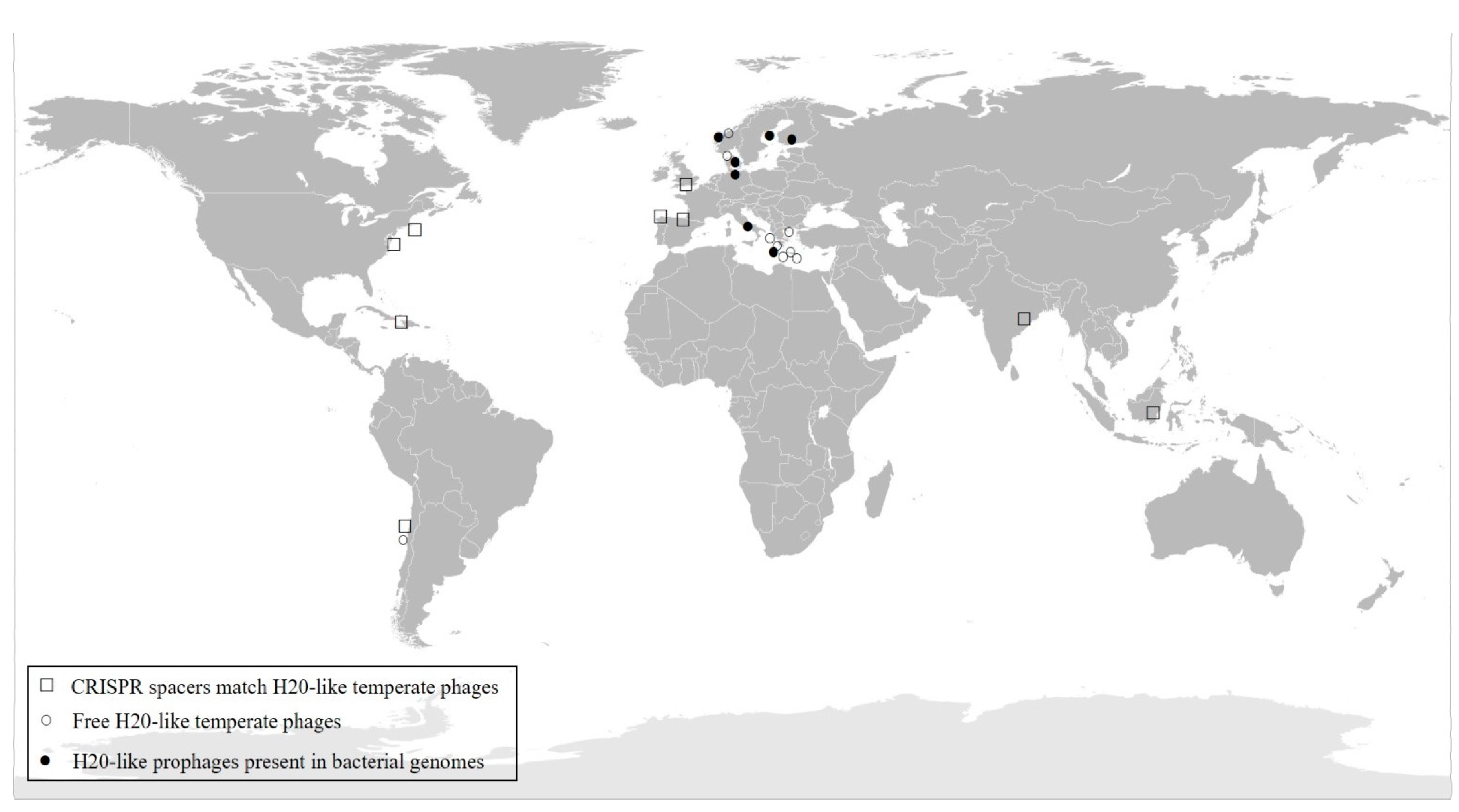
3.6. Distribution of H20-Like Phage on a Global Scale
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Takemura, A.F.; Chien, D.M.; Polz, M.F. Associations and dynamics of vibrionaceae in the environment, from the genus to the population level. Front. Microbiol. 2014, 5, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of Vibrios. Microbiol. Mol. Biol. Rev. 2004, 403–431. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Polz, M. Dynamics of Vibrio Populations and Their Role in Environmental Nutrient Cycling. In The Biology of Vibrios; Thompson, F., Austin, B., Swings, J., Eds.; ASM Press: Washington, DC, USA, 2006; pp. 190–203. [Google Scholar]
- Frans, I.; Michiels, C.W.; Bossier, P.; Willems, K.A.; Lievens, B.; Rediers, H. Vibrio anguillarum as a fish pathogen: Virulence factors, diagnosis and prevention. J. Fish Dis. 2011, 34, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Frans, I.; Dierckens, K.; Crauwels, S.; Van Assche, A.; Leisner, J.; Larsen, M.H.; Michiels, C.W.; Willems, K.A.; Lievens, B.; Bossier, P.; et al. Does Virulence Assessment of Vibrio anguillarum Using Sea Bass (Dicentrarchus labrax) Larvae Correspond with Genotypic and Phenotypic Characterization? PLoS One 2013, 8, 2–10. [Google Scholar] [CrossRef]
- Rønneseth, A.; Castillo, D.; D’Alvise, P.; Tønnesen, Ø.; Haugland, G.; Grotkjaer, T.; Engell-Sørensen, K.; Nørremark, L.; Bergh, Ø.; Wergeland, H.I.; et al. Comparative assessment of Vibrio virulence in marine fish larvae. J. Fish Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rodkhum, C.; Hirono, I.; Stork, M.; Di Lorenzo, M.; Crosa, J.H.; Aoki, T. Putative virulence-related genes in Vibrio anguillarum identified by random genome sequencing. J. Fish Dis. 2006, 29, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Naka, H.; Crosa, J.H. Genetic Determinants of Virulence in the Marine Fish Pathogen Vibrio anguillarum. Fish Pathol. 2011, 46, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Waldor, M.K. Bacteriophage Control of Bacterial Virulence. Infect. Immnunity 2002, 70, 3985–3993. [Google Scholar] [CrossRef]
- Davies, E.V.; Winstanley, C.; Fothergill, J.L.; James, C.E. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol. Lett. 2016, 363, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic Conversion by a Filamentous Phage Encoding Cholera Toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A; Baudry, B.; Pumplin, D.W.; Wasserman, S.S.; Tall, B.D.; Ketley, J.M.; Kaper, J.B. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 5242–5246. [Google Scholar] [CrossRef] [PubMed]
- Weynberg, K.D.; Voolstra, C.R.; Neave, M.J.; Buerger, P.; van Oppen, M.J.H. From cholera to corals: Viruses as drivers of virulence in a major coral bacterial pathogen. Sci. Rep. 2015, 5, 17889. [Google Scholar] [CrossRef] [PubMed]
- Oakey, H.J.; Owens, L. A new bacteriophage, VHML, isolated from a toxin-producing strain of Vibrio harveyi in tropical Australia. J. Appl. Microbiol. 2000, 89, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.; Oakey, J.; Bromage, E.; Owens, L. Experimental bacteriophage-mediated virulence in strains of Vibrio harveyi. Dis. Aquat. Organ. 2003, 54, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.A.; Grim, C.J.; Lipp, E.K.; Rivera, I.N.G.; Chun, J.; Haley, B.J.; Taviani, E.; Choi, S.Y.; Hoq, M.; Munk, A.C.; et al. Deep-sea hydrothermal vent bacteria related to human pathogenic Vibrio species. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, E2813–E2819. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.; Bru, H. Phages and the Evolution of Bacterial Pathogens: from Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Feiner, R.; Argov, T.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 2015, 13, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Obeng, N.; Pratama, A.A.; van Elsas, J.D. The Significance of Mutualistic Phages for Bacterial Ecology and Evolution. Trends Microbiol. 2016, 24, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Klumpp, J.; Mahony, J.; O’Connell-Motherway, M.; Nauta, A.; van Sinderen, D. Methyltransferases acquired by lactococcal 936-type phage provide protection against restriction endonuclease activity. BMC Genomics 2014, 15, 831. [Google Scholar] [CrossRef] [PubMed]
- Bochow, S.; Elliman, J.; Owens, L. Bacteriophage adenine methyltransferase: A life cycle regulator? Modelled using Vibrio harveyi myovirus like. J. Appl. Microbiol. 2012, 113, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.H. Prophages in marine bacteria: dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Spencer, R.G.M. Indigenous marine bacteriophages. J. Bacteriol. 1960, 614. [Google Scholar]
- Sonnenschein, E.C.; Nielsen, K.F.; D’Alvise, P.; Porsby, C.H.; Melchiorsen, J.; Heilmann, J.; Kalatzis, P.G.; Lopez-Perez, M.; Bunk, B.; Sproer, C.; et al. Global occurrence and heterogeneity of the Roseobacter-clade species Ruegeria mobilis. ISME J. 2016, 11, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V. Current insights into phage biodiversity and biogeography. Curr. Opin. Microbiol. 2009, 12, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, C.A.; Rose, J.B.; Jaing, S.C.; Paul, J.H. Genetic Diversity of Related Vibriophages Isolated from Marine Environments Around Florida and Hawaii, USA. Marine Sci. Fac. Pub. 1995, 120, 89–98. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends in Microbiol. 2005, 13, 243–293. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Suttle, C.A. Distribution, genetic richness and phage sensitivity of Vibrio spp. from coastal British Columbia. Environ. Microbiol. 2007, 9, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.; Higuera, G.; Gajardo, F.; Castillo, D.; Middelboe, M.; Garcia, K.; Ramirez, C.; Espejo, R.T. Complete Genome Sequence of Vibrio anguillarum Phage CHOED Successfully Used for Phage Therapy in Aquaculture. Genome Announc. 2014, 2, 2013–2014. [Google Scholar] [CrossRef] [PubMed]
- Higuera, G.; Bastías, R.; Tsertsvadze, G.; Romero, J.; Espejo, R.T. Recently discovered Vibrio anguillarum phages can protect against experimentally induced vibriosis in Atlantic salmon, Salmo salar. Aquaculture 2013, 392–395, 128–133. [Google Scholar] [CrossRef]
- Mateus, L.; Costa, L.; Silva, Y.J.; Pereira, C.; Cunha, A.; Almeida, A. Efficiency of phage cocktails in the inactivation of Vibrio in aquaculture. 2014, 424–425, 167–173. [Google Scholar] [CrossRef]
- Castillo, D.; Alvise, P.D.; Xu, R.; Zhang, F.; Middelboe, M.; Gram, L. Comparative genome analyses of Vibrio anguillarum strains reveal a link with pathogenicity traits. mSystems 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Chan, A.M.; Suttle, C.A. Genetic richness of vibriophages isolated in a coastal environment. Environ. Microbiol. 2006, 8, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Stenholm, A.R.; Dalsgaard, I.; Middelboe, M. Isolation and characterization of bacteriophages infecting the fish pathogen Flavobacterium psychrophilum. Appl. Environ. Microbiol. 2008, 74, 4070–4078. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Gram, L.; Middelboe, M. Vibriophages and their interactions with the fish pathogen Vibrio anguillarum. Appl. Environ. Microbiol. 2014, 80, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Practical methods for determining phage growth paramenters. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization and Interactions; Clokie, M., Kropinski, A., Eds.; Humana Press: New York, NY, USA, 2009; pp. 175–202. [Google Scholar]
- Castillo, D.; Christiansen, R.H.; Dalsgaard, I.; Madsen, L.; Middelboe, M. Bacteriophage resistance mechanisms in the fish pathogen Flavobacterium psychrophilum: Linking genomic mutations to changes in bacterial virulence factos. Appl. Environ. Microbiol. 2015, 81, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiong DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; Vonstein, V.; Wattam, A.R.; Xia, F.; Stevens, R. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.A.; Mezulis, S.; Yates, C.; Wass, M.; Sternberg, M. The Phyre2 web portal for protein modelling, prediction, and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Phyre2. Available online: http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index (accessed on 1 March 2017).
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Canese, K.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.O.; Nilsson, K.; Hjerde, E.; Willassen, N.-P.; Milton, D.L. Complete genome sequence of Vibrio anguillarum strain NB10, a virulent isolate from the Gulf of Bothnia. Stand. Genomic Sci. 2015, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- MacDonell, M.T.; Colwell, R.R. Phylogeny of the Vibrionaceae, and Recommendation for Two New Genera, Listonella and Shewanella. Syst. Appl. Microbiol. 1985, 6, 171–182. [Google Scholar] [CrossRef]
- Mikkelsen, H.; Schrøder, M.B.; Lund, V. Vibriosis and atypical furunculosis vaccines; efficacy, specificity and side effects in Atlantic cod, Gadus morhua L. Aquaculture 2004, 242, 81–91. [Google Scholar] [CrossRef]
- Samuelsen, O.B.; Bergh, Ø. Efficacy of orally administered florfenicol and oxolinic acid for the treatment of vibriosis in cod (Gadus morhua). Aquaculture 2004, 235, 27–35. [Google Scholar] [CrossRef]
- Pedersen, K.; Gram, L.; Austin, D.A.; Austin, B. Pathogenicity of Vibrio anguillarum serogroup O1 strains compared to plasmids, outer membrane protein profiles and siderophore production. J. Appl. Microbiol. 1997, 82, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Skov, M.N.; Pedersen, K.; Larsen, J.L. Comparison of Pulsed-Field Gel Electrophoresis, Ribotyping, and Plasmid Profiling for Typing of Vibrio anguillarum Serovar O1. Appl Environ Microbiol. 1995, 61, 1540–1545. [Google Scholar] [PubMed]
- Silva-Rubio, A.; Avendaño-Herrera, R.; Jaureguiberry, B.; Toranzo, A.E.; Magariños, B. First description of serotype O3 in Vibrio anguillarum strains isolated from salmonids in Chile. J. Fish Dis. 2008, 31, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Austin, B.; Alsina, M.; Austin, D.A.; Blanch, A.R.; Grimont, F.; Grimont, P.A.D.; Jofre, J.; Koblavi, S.; Larsen, J.L.; Pedersen, K.; Tiainen, T.; Verdonck, L.; Swings, J. Identification and Typing of Vibrio anguillarum: A Comparison of Different Methods. Syst. Appl. Microbiol. 1995, 18, 285–302. [Google Scholar] [CrossRef]
- Actis, L.A.; Fish, W.; Crosa, J.H.; Kellerman, K.; Ellenberger, S.R.; Hauser, F.M.; Sanders-Loehr, J. Characterization of anguibactin, a novel siderophore from Vibrio anguillarum 775(pJM1). J. Bacteriol. 1986, 167, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kosters, W.L.; Actisp, L.A.; Waldbeserp, L.S.; Tolmaskyp, M.E.; Crosapfl, J.H. Molecular characterization of the iron transport system mediated by the pJM1 plasmid in Vibrio anguillarum 775. J. Biol. Chem. 1991, 23829–23833. [Google Scholar]
- Rehnstam, A.S.; Norqvist, A.; Wolf-Watz, H.; Hagstrom, A. Identification of Vibrio anguillarum in fish by using partial 16S rRNA sequences and a specific 16S rRNA oligonucleotide probe. Appl. Envir. Microbiol. 1989, 55, 1907–1910. [Google Scholar]
- Oliveira, J.; Castilho, F.; Cunha, A.; Pereira, M.J. Bacteriophage therapy as a bacterial control strategy in aquaculture. Aquac. Int. 2012, 20, 879–910. [Google Scholar] [CrossRef]
- Williamson, S.J.; McLaughlin, M.R.; Paul, J.H. Interaction of the ΦHSIC virus with its host: lysogeny or pseudolysogeny? Appl. Environ. Microbiol. 2001, 67, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Thompson, F.L.; Klose, K.E.; Group, A. Vibrio2005: The First International Conference on the Biology of Vibrios. J. Bacteriol. 2006, 188, 4592–4596. [Google Scholar] [CrossRef] [PubMed]
- Jacobs-Sera, D.; Marinelli, L.; Bowman, C.; Broussard, G.; Guerrero, C.; Boyle, M.; Petrova, Z.; Dedrick, R.; Pope, W.; SEA-PHAGES; et al. On the nature of mycobacteriophage diversity and host preference. Virology 2012, 434, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Cumby, N.; Reimer, K.; Mengin-Lecreulx, D.; Davidson, A.R.; Maxwell, K.L. The phage tail tape measure protein, an inner membrane protein and a periplasmic chaperone play connected roles in the genome injection process of E.coli phage HK97. Mol. Microbiol. 2015, 96, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Wiener, M.; Freymann, D.; Ghosh, P.; Stroud, R.M. Crystal structure of colicin Ia. Nature 1997, 385, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Tarr, A.W.; Penfold, C.N. Colicin import into E. coli cells: A model system for insights into the import mechanisms of bacteriocins. Biochim. Biophys. Acta - Mol. Cell Res. 2014, 1843, 1717–1731. [Google Scholar] [CrossRef] [PubMed]
- Alonso, G. How bacteria protect themselves against channel-forming colicins. Int. Microbiol. 2000, 3, 81–88. [Google Scholar] [PubMed]
- Penfold, C.N.; Walker, D.; Kleanthous, C. How bugs kill bugs: progress and challenges in bacteriocin research. Biochem. Soc. Trans. 2012, 40, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rubio, L.; Gutiérrez, D.; Martínez, B.; Rodríguez, A.; Götz, F.; García, P. The tape measure protein of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA35 has an active muramidase domain. Appl. Environ. Microbiol. 2012, 78, 6369–6371. [Google Scholar] [CrossRef] [PubMed]
- Luria, S.E. Host-induced modifications of viruses. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Nathans, D.; Smith, H.O. Restriction endonucleases in the analysis and restructuring of dna molecules. Annu. Rev. Biochem. 1975, 44, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, G. Do; Choi, T.J. Molecular cloning and characterization of the DNA adenine methyltransferase gene in Feldmannia sp. virus. Virus Genes 2007, 34, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Wion, D.; Casadesús, J. N6-methyl-adenine: an epigenetic signal for DNA – protein interactions. Nat. Rev. Microbiol. 2006, 4, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Low, D.A.; Weyand, N.J.; Mahan, M.J. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect. Immun. 2001, 69, 7197–7204. [Google Scholar] [CrossRef] [PubMed]
- Portillo, F.G.-D.; Pucciarelli, M.G.; Casadesus, J. DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1999, 96, 11578–11583. [Google Scholar] [CrossRef]
- Oakey, H.J.; Cullen, B.R.; Owens, L. The complete nucleotide sequence of the Vibrio harveyi bacteriophage VHML. J. Appl. Microbiol. 2002, 93, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Mahony, J.; Ainsworth, S.; Nauta, A.; van Sinderen, D. Bacteriophage orphan DNA methyltransferases: Insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 2013, 79, 7547–7555. [Google Scholar] [CrossRef] [PubMed]
- van Houte, S.; Buckling, A.; Westra, E.R. Evolutionary Ecology of Prokaryotic Immune Mechanisms. Microbiol. Mol. Biol. Rev. 2016, 80, 745–763. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.H.; Jacobs-Sera, D.; Russel, D.A.; Peebles, C.L.; Al-Atrache, Z.; Alcoser, T.A.; Alexander, L.M.; Alfano, M.B.; Alford, S.T.; Amy, N.E.; et al. Expanding the diversity of mycobacteriophages: Insights into genome architecture and evolution. PLoS One 2011, 6, e16329. [Google Scholar] [CrossRef] [PubMed]
- Berngruber, T.W.; Weissing, F.J.; Gandon, S. Inhibition of superinfection and the evolution of viral latency. J. Virol. 2010, 84, 10200–10208. [Google Scholar] [CrossRef] [PubMed]
- Vidgen, M.; Carson, J.; Higgins, M.; Owens, L. Changes to the phenotypic profile of Vibrio harveyi when infected with the Vibrio harveyi myovirus-like (VHML) bacteriophage. J. Appl. Microbiol. 2006, 100, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Paul, J.H. Environmental factors that influence the transition from lysogenic to lytic existence in the HSIC/Listonella pelagia marine phage-host system. Microb. Ecol. 2006, 52, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, Y.; Vincent, M.; Sun, Y.; Yu, H.; Wang, J.; Bao, Q.; Kong, H.; Hu, S. Complete genome sequence of bacteriophage t5. Virology 2005, 332, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Zahn, K.; Landy, A. Modulation of lambda integrase synthesis by rare arginine tRNA. Mol. Microbiol. 1996, 21, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.G.; Landon, M.; Gregg, C.J.; Lajoie, M.J.; Govindarajan, L.; Mosberg, J.A.; Kuznetsov, G.; Goodman, D.B.; Vargas-Rodriguez, O.; Isaacs, F.J.; et al. Emergent rules for codon choice elucidated by editing rare arginine codons in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E5588–E5597. [Google Scholar] [CrossRef] [PubMed]
- Sano, E.; Carlson, S.; Wegley, L.; Rohwer, F. Movement of viruses between biomes. Appl. Environ. Microbiol. 2004, 70, 5842–5846. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Rusch, D.B.; Yooseph, S.; Halpern, A.L.; Heidelberg, K.B.; Glass, J.I.; Pfannkoch, C.A.; Fadrosh, D.; Miller, C.S.; Sutton, G.; Frazler, M.; Venter, J.C. The sorcerer II global ocean sampling expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS One 2008, 3, e1456. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteriophages | Origin | Environment | Host Strain | Genome Size (bp) | GC Content (%) | ORFs | GenBank Accession Number |
|---|---|---|---|---|---|---|---|
| φH2 | DK | Aq | A023 | 46,149 | 43.1 | 76 | KY658673 |
| φH8 | DK | Aq | T265 | 46,157 | 43.1 | 76 | KY658674 |
| φH20 | DK | Aq | BA35 | 53,224 | 43 | 91 | KY658675 |
| φP2 | N | Aq | BA35 | 46,149 | 43.1 | 76 | KY658676 |
| φP3 | N | Aq | BA35 | 53,242 | 43 | 91 | KY658677 |
| pVa-1 | GR | M | BA35 | 53,227 | 43.1 | 91 | KX581095 |
| pVa-2 | GR | M | BA35 | 53,286 | 43 | 91 | KX581094 |
| pVa-3 | GR | M | BA35 | 54,344 | 43.1 | 94 | KY658678 |
| pVa-4 | GR | M | BA35 | 54,295 | 43.1 | 93 | KY658679 |
| pVa-5 | GR | M | BA35 | 53,227 | 43 | 91 | KX581096 |
| pVa-6 | GR | M | BA35 | 53,2274 | 43 | 91 | KX581097 |
| pVa-7 | GR | M | BA35 | 54,268 | 43.1 | 93 | KX581110 |
| pVa-8 | GR | M | BA35 | 53,227 | 43 | 91 | KY658680 |
| φCLA | CL | M | BA35 | 53,226 | 43 | 91 | KX581091 |
| φHer | GR | M | BA35 | 53,226 | 43 | 91 | KX581090 |
| φLen | GR | M | BA35 | 53,226 | 43 | 91 | KX581092 |
| φPel | GR | M | BA35 | 53,227 | 43 | 91 | KX581093 |
| φStrym | GR | M | BA35 | 53,226 | 43 | 91 | KX581099 |
| φVaK | GR | Aq | VaKef | 53,216 | 43 | 92 | KX581098 |
| Bacteriophage | Latency Time (min) | Burst Size (Virions/Cell) | Adsorption Constant K30 |
|---|---|---|---|
| φH20 | 60 | 112 ± 9 | 1.18 × 10−7 ± 1.28 × 10−8 |
| pVa-3 | 60 | 100 ± 13 | 8.63 × 10−8 ± 2.46 × 10−9 |
| φP3 | 70 | 101 ± 16 | 6.40 × 10−8 ± 4.39 × 10−9 |
| φCla | 50 | 145 ± 24 | 2.11 × 10−7 ± 2.68 × 10−8 |
| Stain Code | Origin | Phylogenetic Group | H20-Like Prophage | Reference |
|---|---|---|---|---|
| DSM21597 | Norway | I | - | [51] |
| 4299 | Norway | II | - | [52] |
| HI610 | Norway | II | - | [53] |
| S2 2/9 | Denmark | III | - | [54] |
| 90-11-286 | Denmark | III | - | [54,55] |
| PF430-3 | Chile | IV | - | [36] |
| PF4 | Chile | IV | - | [56] |
| PF7 | Chile | IV | - | [56] |
| A023 | Spain | V | X | [57] |
| 90-11-287 | Denmark | V | X | [55] |
| 51/82/2 | Germany | V | X | [57] |
| T265 | UK | V | - | [57] |
| HWU53 | Denmark | V | X | [57] |
| 6018/1 | Denmark | V | X | [57] |
| 91-8-178 | Norway | V | X | [57] |
| 87-9-116 | Finland | V | X | [54,55] |
| 91-7-154 | Denmark | V | X | [54,55] |
| 601/90 | Italy | V | X | [54] |
| 178/90 | Italy | V | X | [54] |
| 9014/8 | Denmark | V | X | [54] |
| 261/91 | Italy | V | X | [57] |
| LMG12010 | n/a | V | X | [57] |
| 87-9-117 | Finland | V | X | [54,55] |
| BA35 | USA | V | - | [57] |
| 775 | USA | V | - | [58,59] |
| NB10 | North Baltic | V | X | [49,60] |
| VA1 | Denmark | V | X | [36] |
| 850610-1/6a | Denmark | n/a | X | [57] |
| VA2 | Denmark | n/a | - | [36] |
| VA3 | Denmark | n/a | - | [36] |
| VaKef | Greece | n/a | X | Clinical strain—Unpublished |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalatzis, P.G.; Rørbo, N.I.; Castillo, D.; Mauritzen, J.J.; Jørgensen, J.; Kokkari, C.; Zhang, F.; Katharios, P.; Middelboe, M. Stumbling across the Same Phage: Comparative Genomics of Widespread Temperate Phages Infecting the Fish Pathogen Vibrio anguillarum. Viruses 2017, 9, 122. https://doi.org/10.3390/v9050122
Kalatzis PG, Rørbo NI, Castillo D, Mauritzen JJ, Jørgensen J, Kokkari C, Zhang F, Katharios P, Middelboe M. Stumbling across the Same Phage: Comparative Genomics of Widespread Temperate Phages Infecting the Fish Pathogen Vibrio anguillarum. Viruses. 2017; 9(5):122. https://doi.org/10.3390/v9050122
Chicago/Turabian StyleKalatzis, Panos G., Nanna Iben Rørbo, Daniel Castillo, Jesper Juel Mauritzen, Jóhanna Jørgensen, Constantina Kokkari, Faxing Zhang, Pantelis Katharios, and Mathias Middelboe. 2017. "Stumbling across the Same Phage: Comparative Genomics of Widespread Temperate Phages Infecting the Fish Pathogen Vibrio anguillarum" Viruses 9, no. 5: 122. https://doi.org/10.3390/v9050122
APA StyleKalatzis, P. G., Rørbo, N. I., Castillo, D., Mauritzen, J. J., Jørgensen, J., Kokkari, C., Zhang, F., Katharios, P., & Middelboe, M. (2017). Stumbling across the Same Phage: Comparative Genomics of Widespread Temperate Phages Infecting the Fish Pathogen Vibrio anguillarum. Viruses, 9(5), 122. https://doi.org/10.3390/v9050122