
Variation in the Genetic Repertoire of Viruses Infecting Micromonas pusilla Reflects Horizontal Gene Transfer and Links to Their Environmental Distribution


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequencing and Annotation of Micromonas pusilla Viruses
2.2. Comparing Prasinovirus Genomes and Inferring Viral Phylogeny
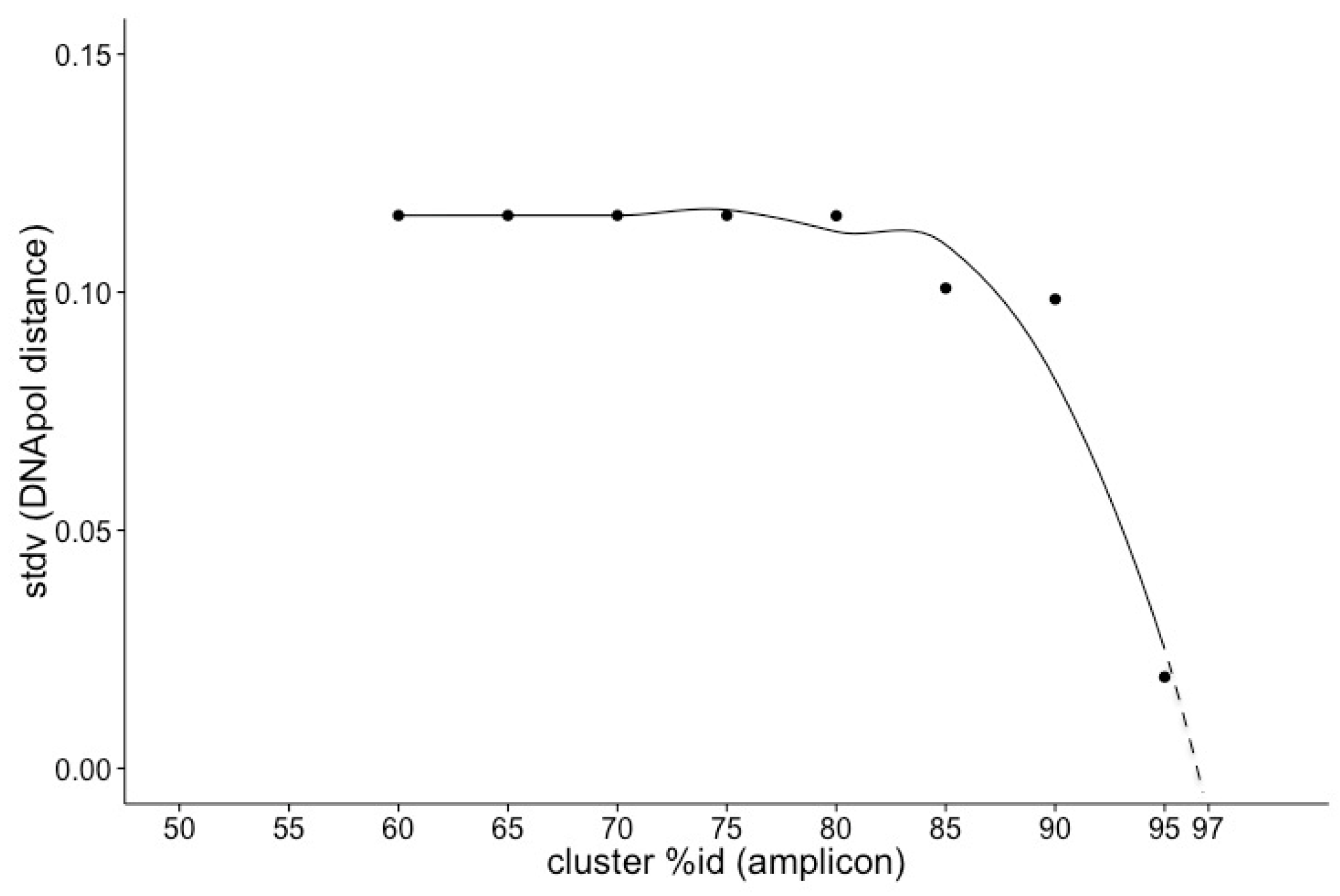
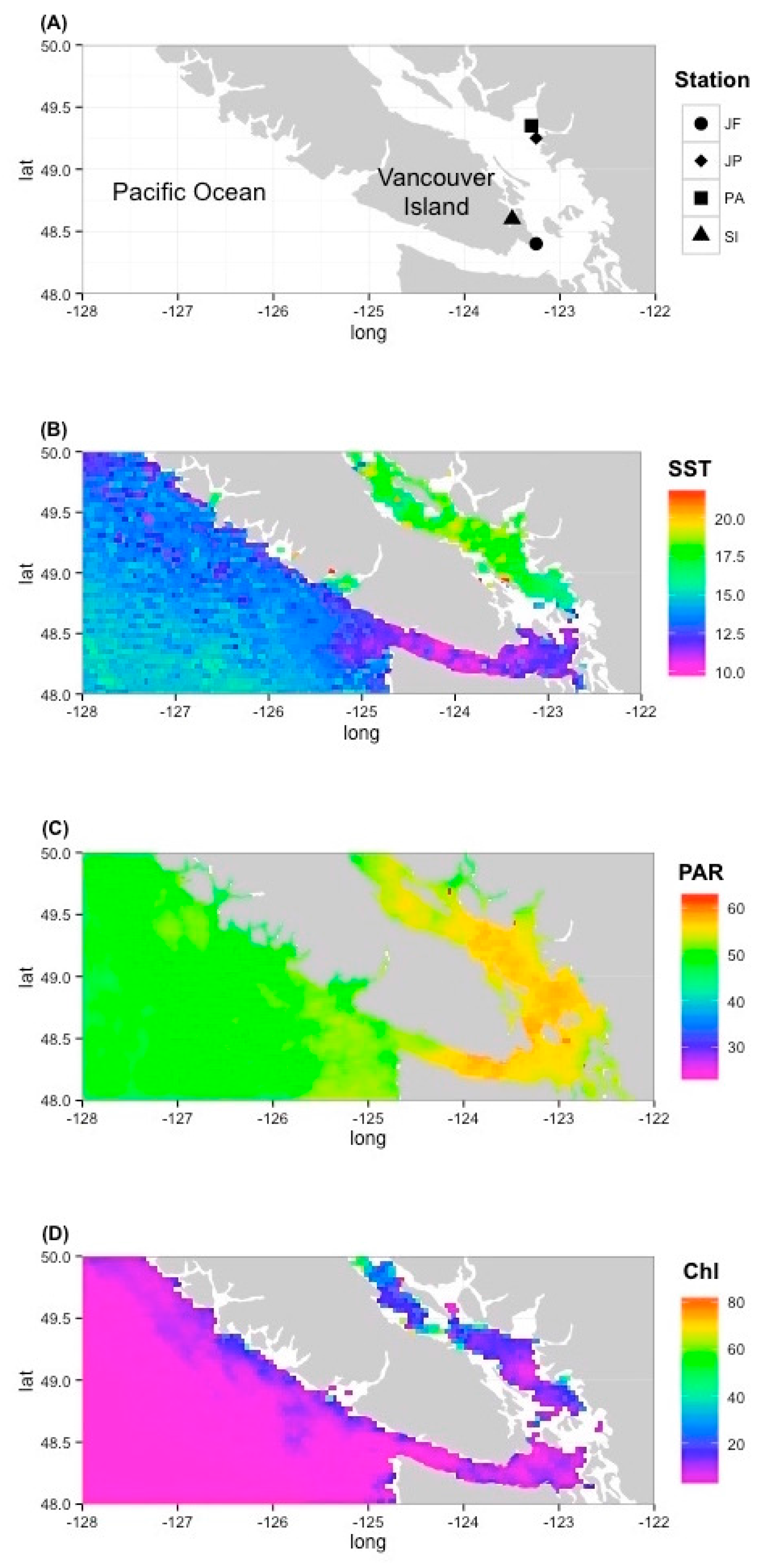
2.3. Assessing the Prevalence of Prasinoviruses in Environmental Samples
3. Results
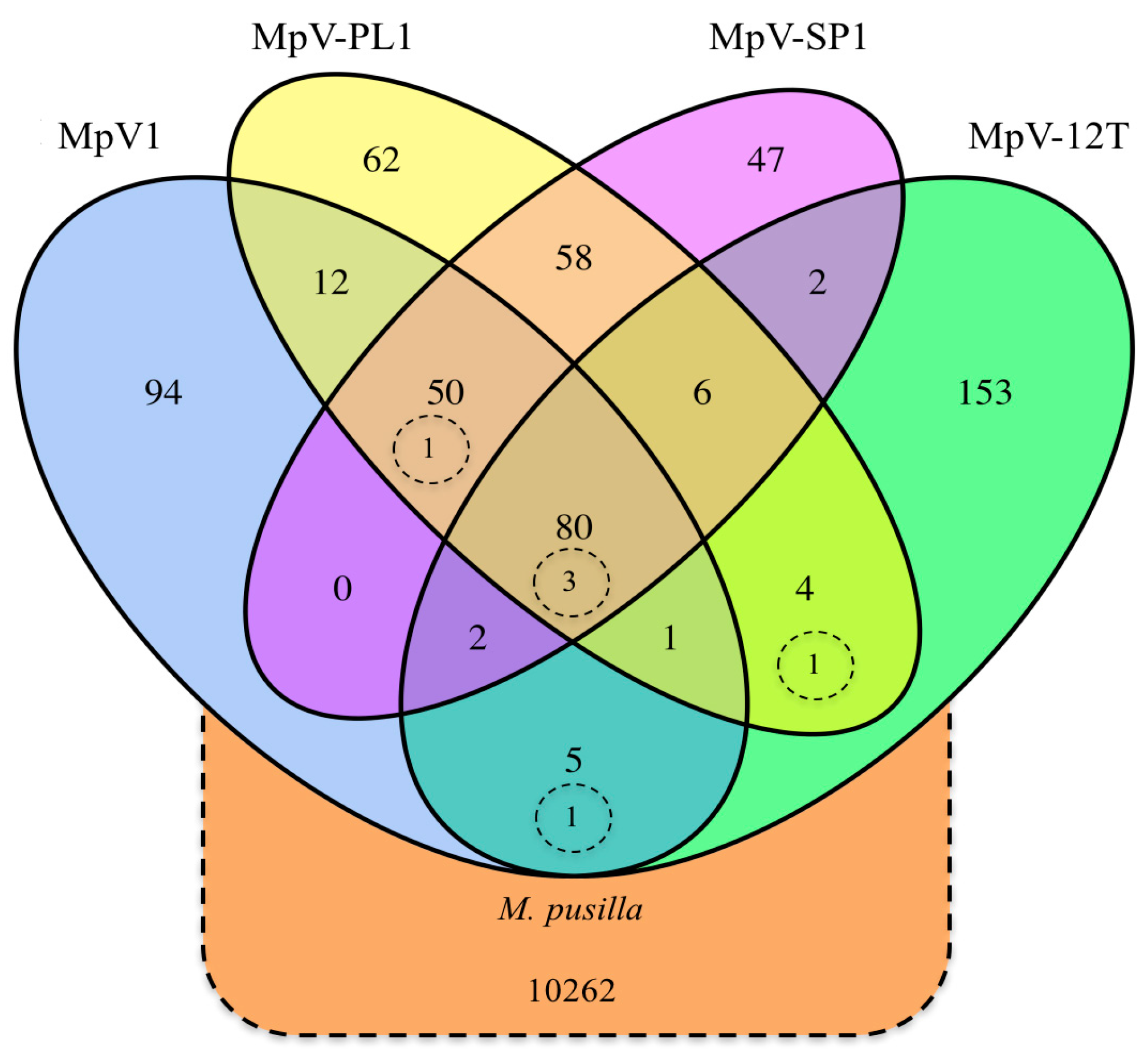
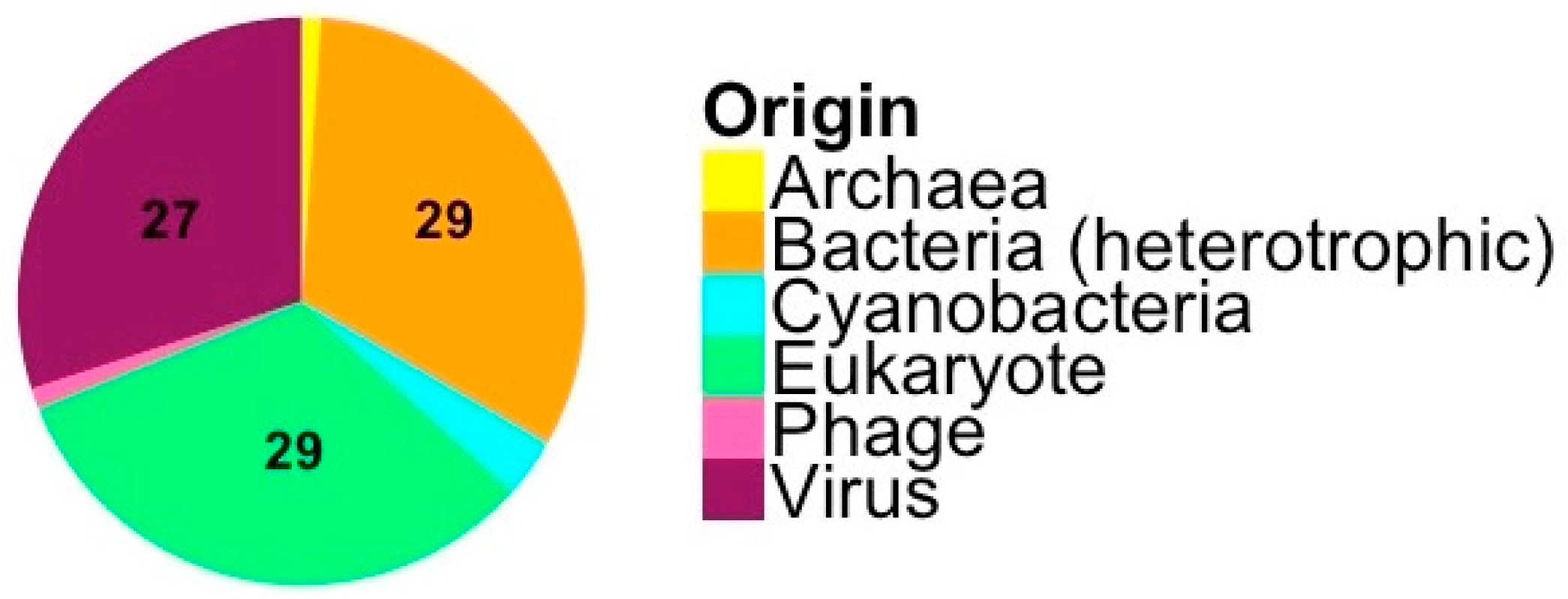
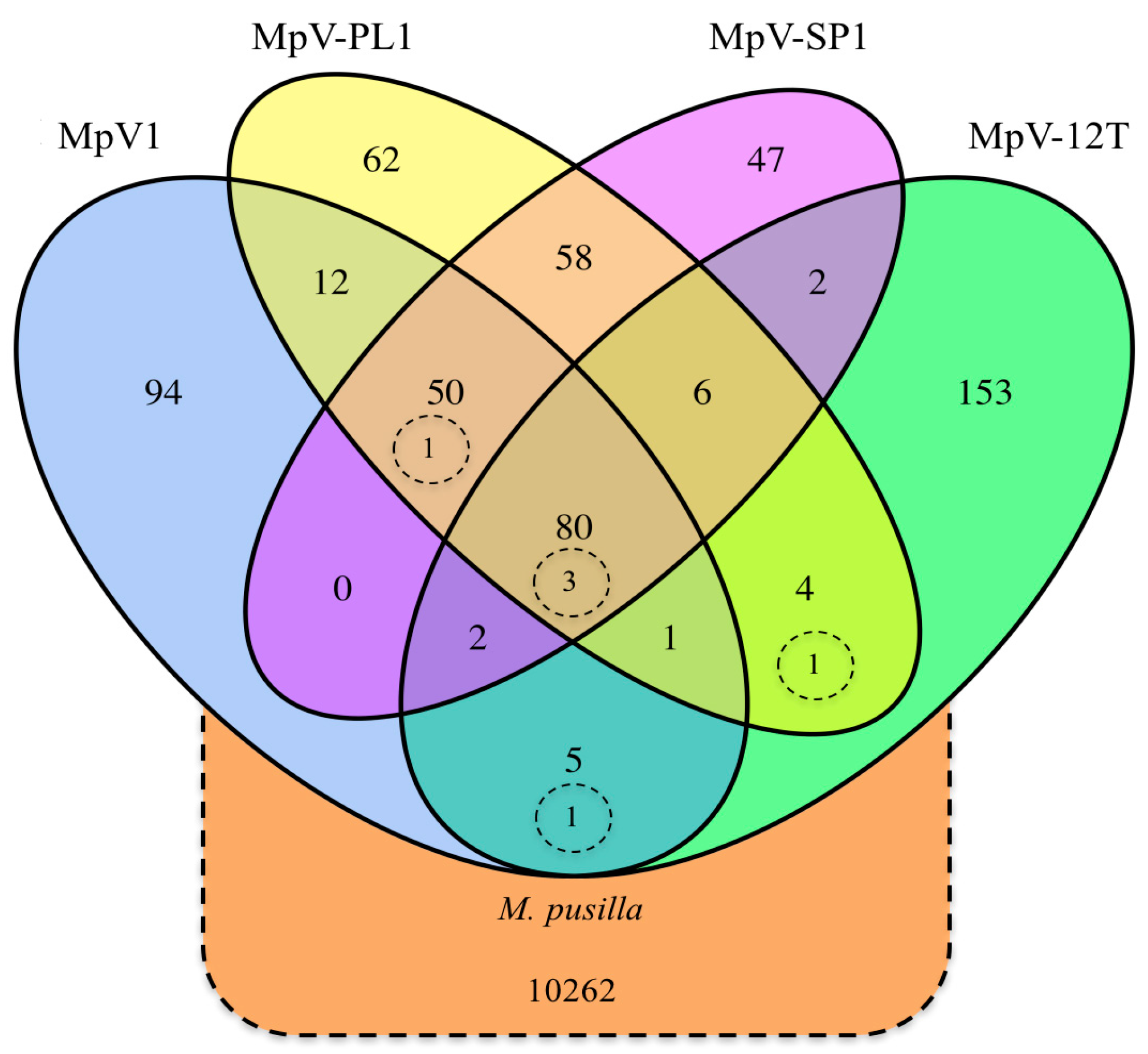
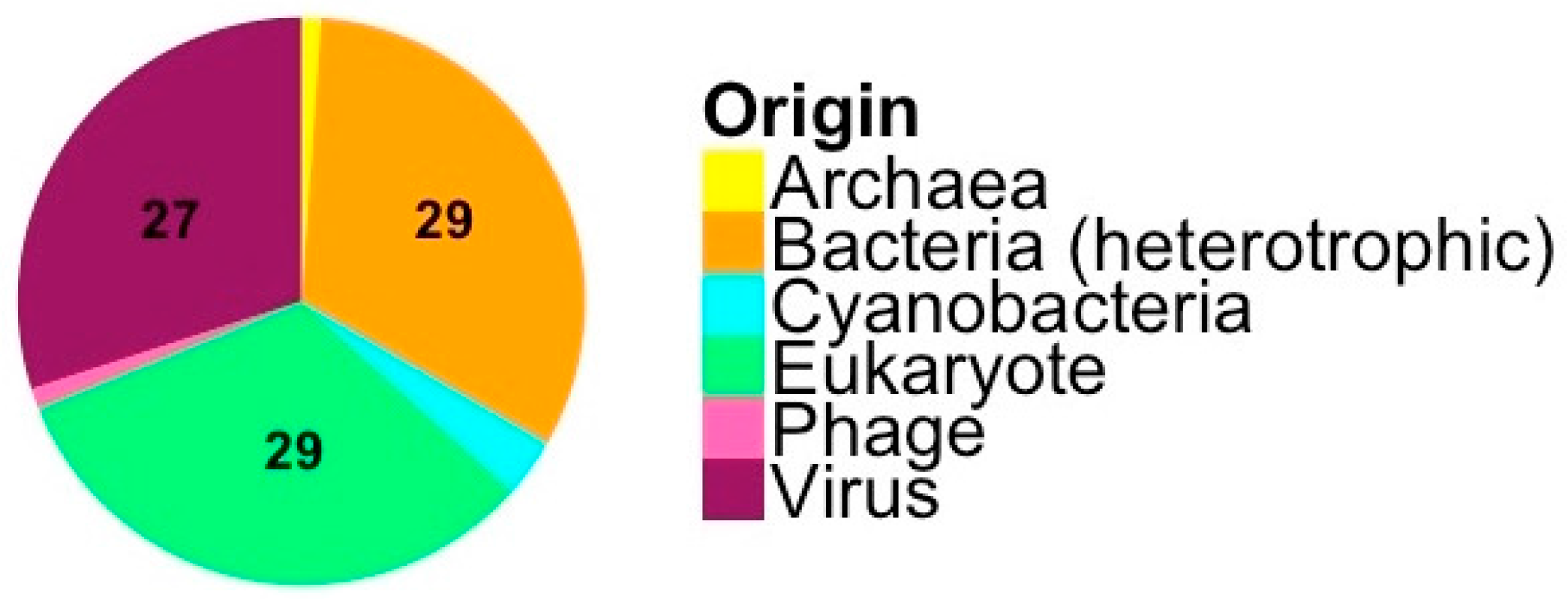
3.1. Origin and Distribution of Genes in Micromonas Viruses
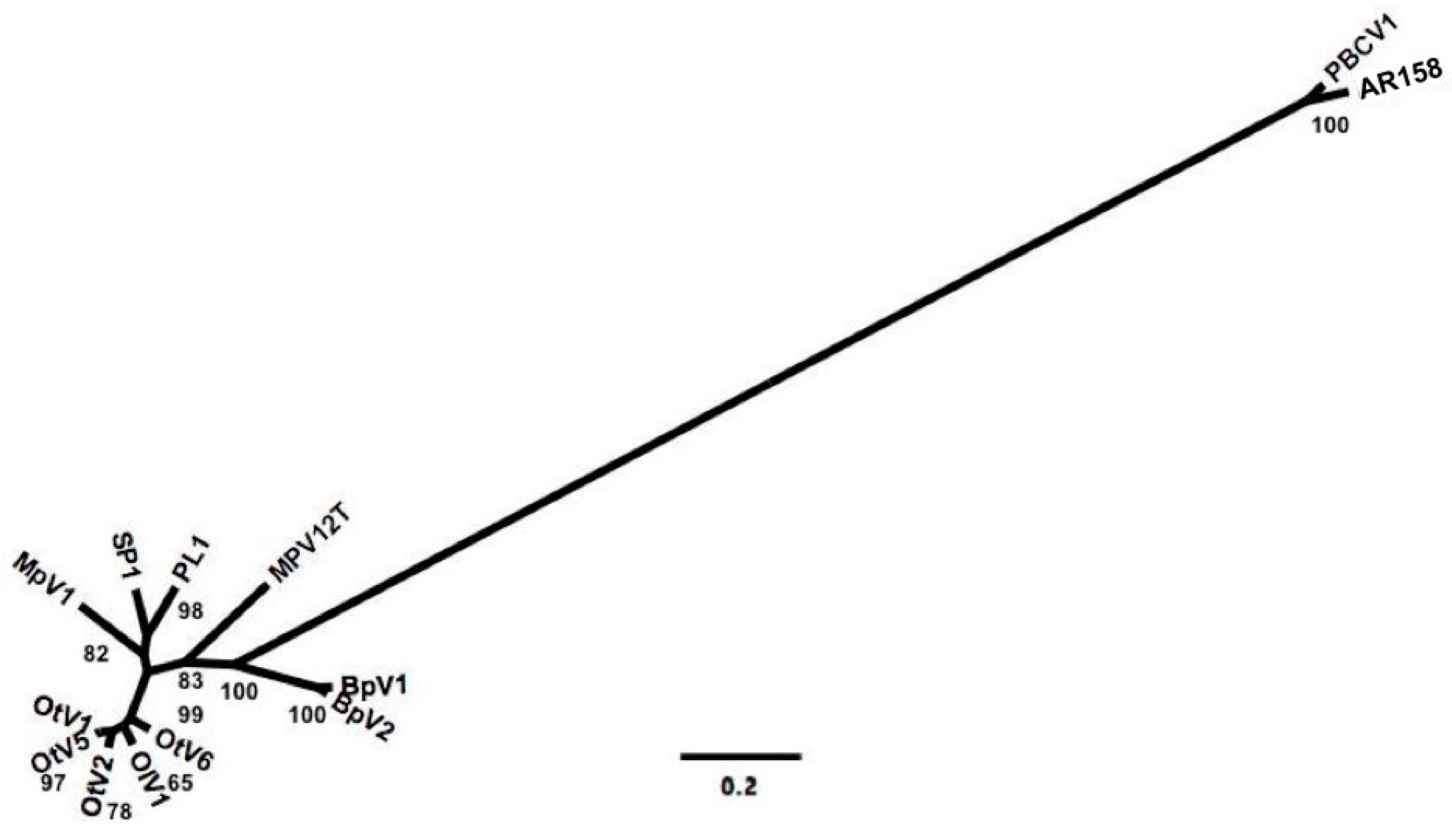
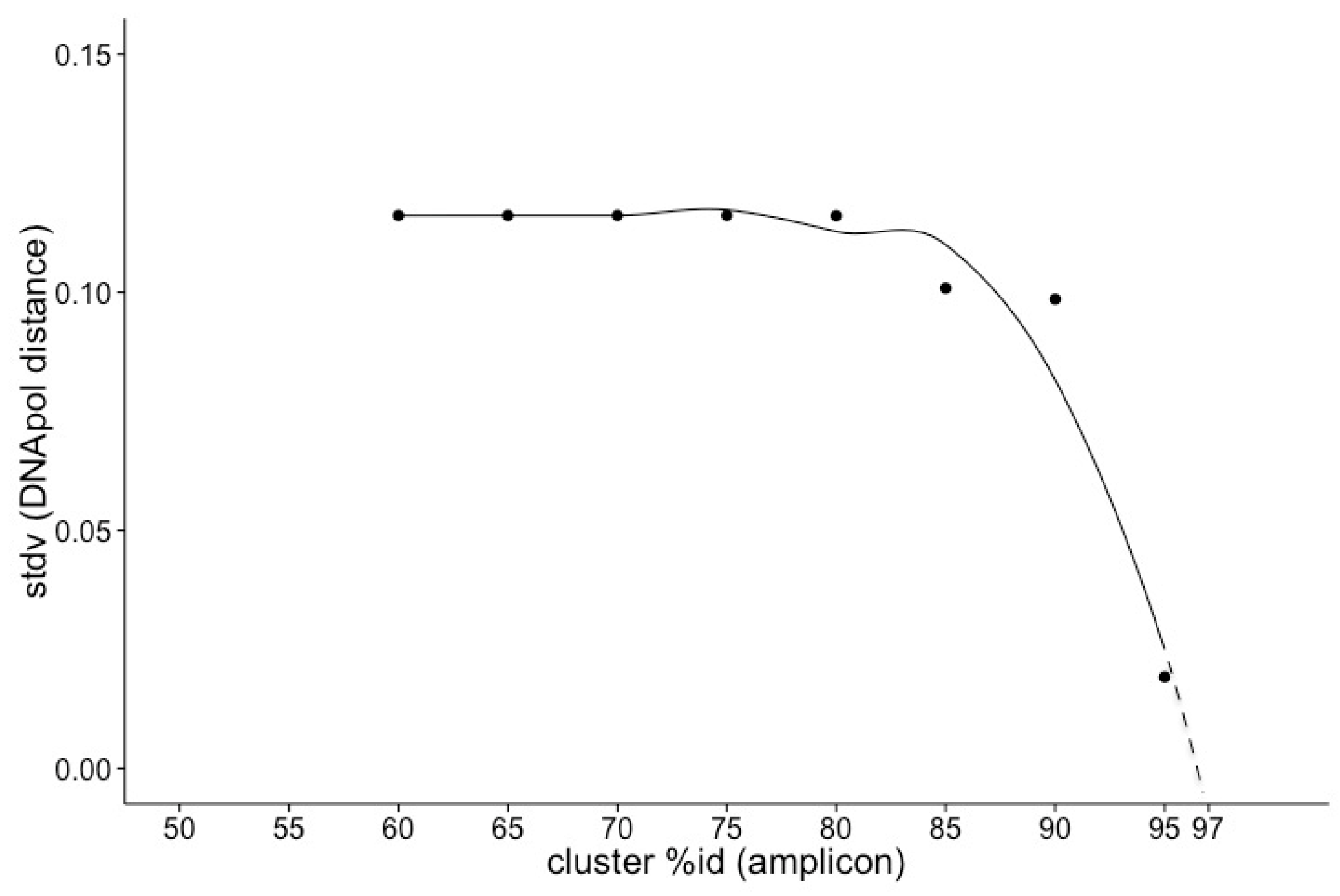
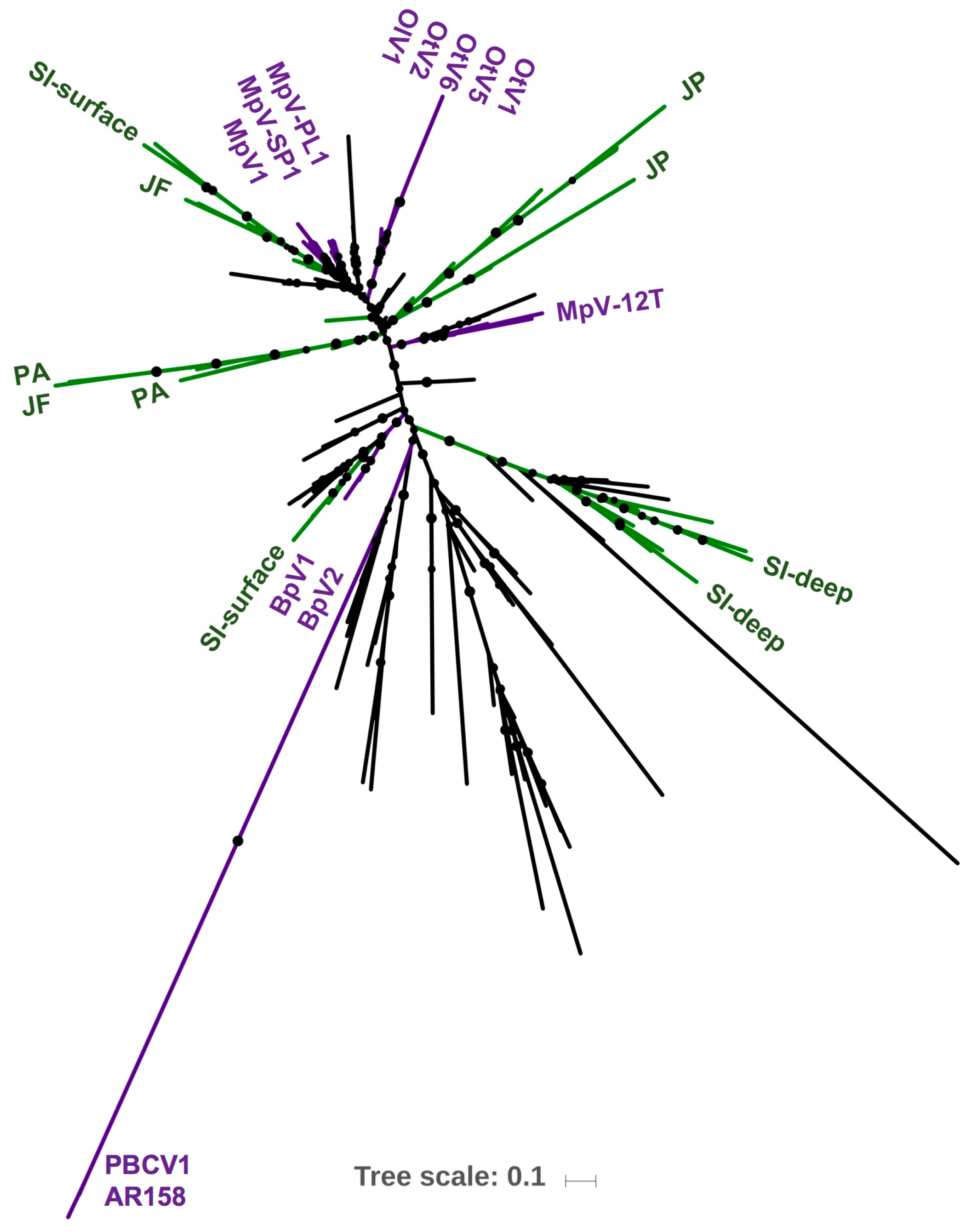
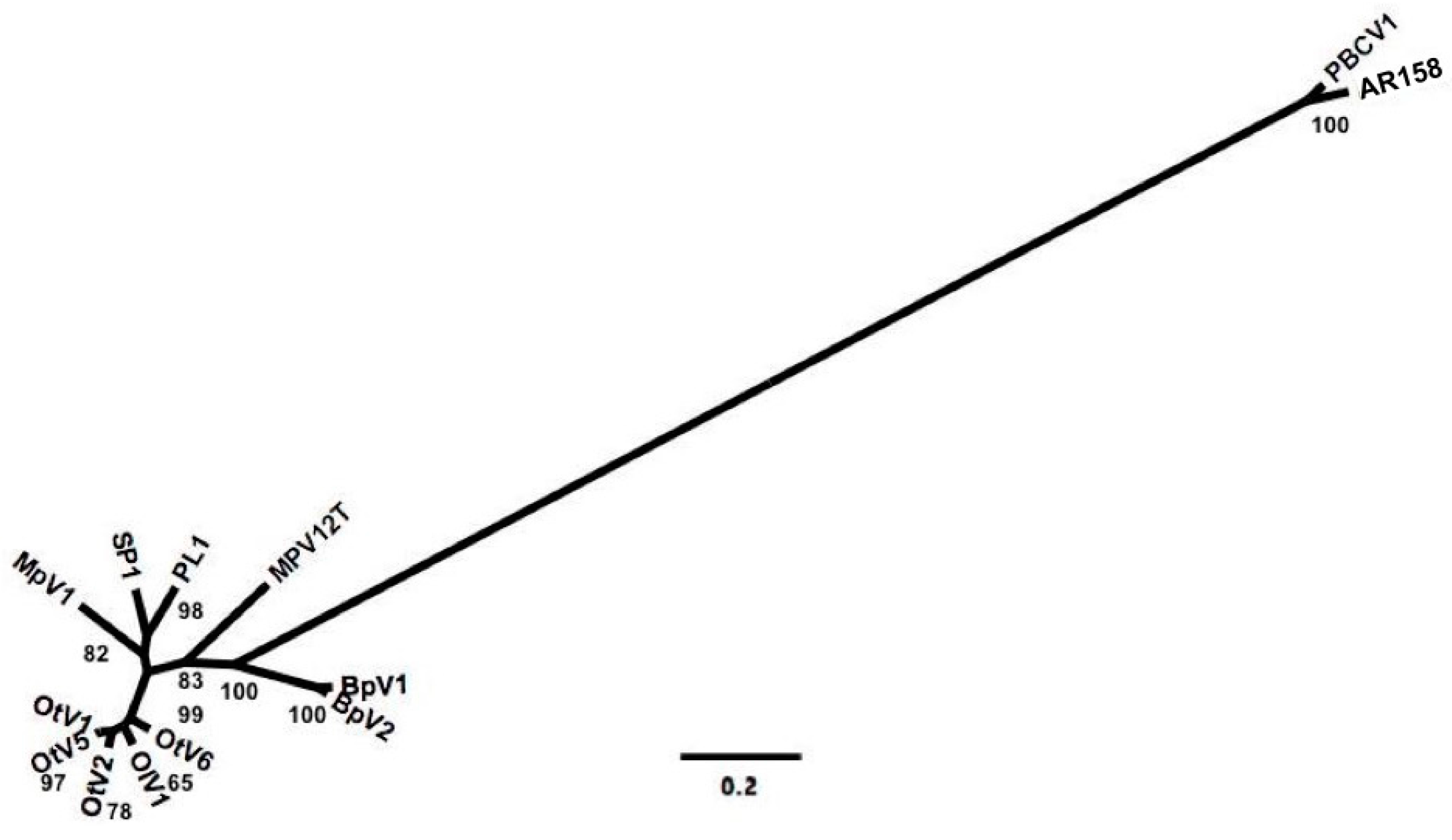
3.2. Deriving Similarity in Gene Content from DNApol
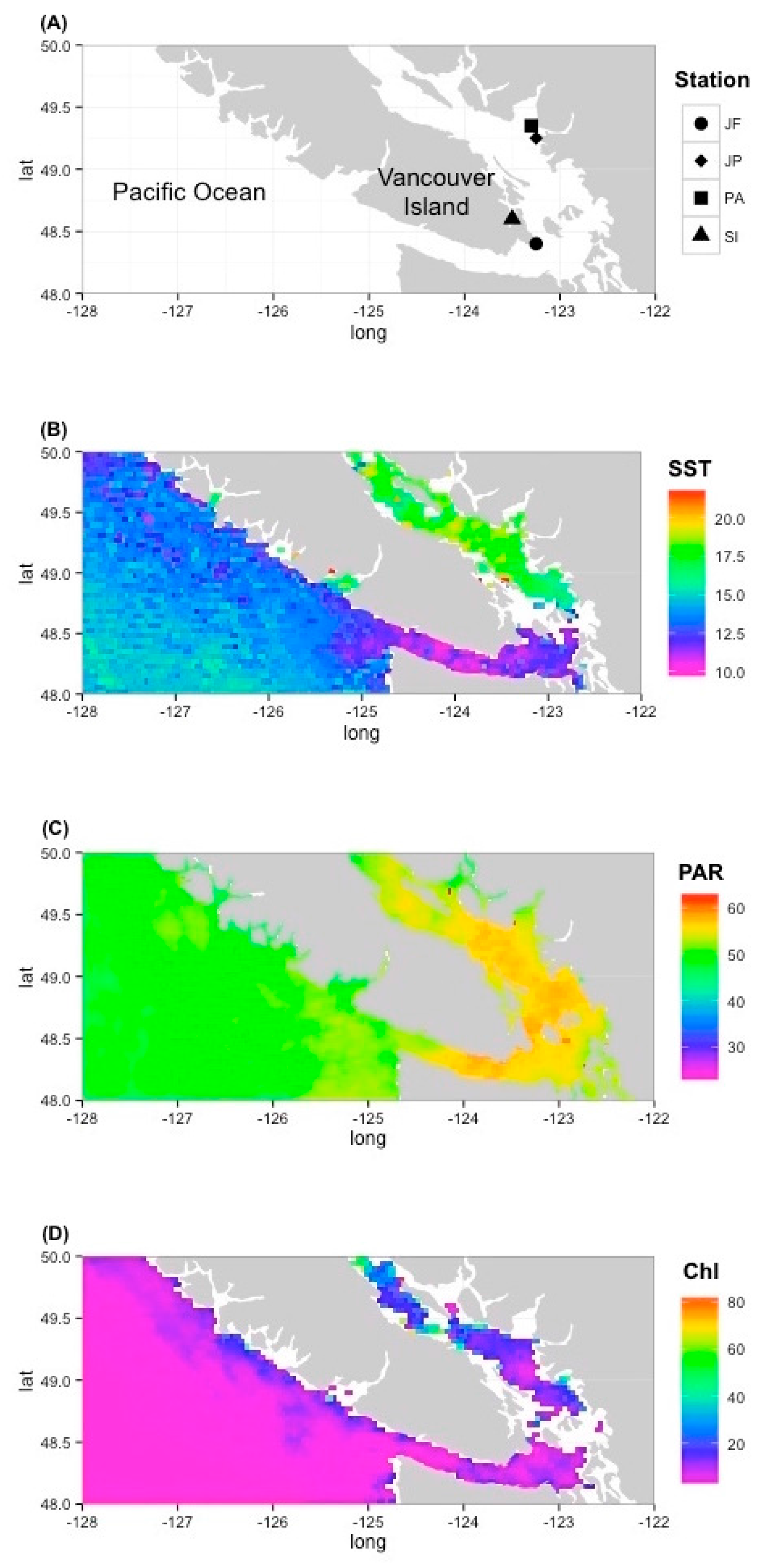
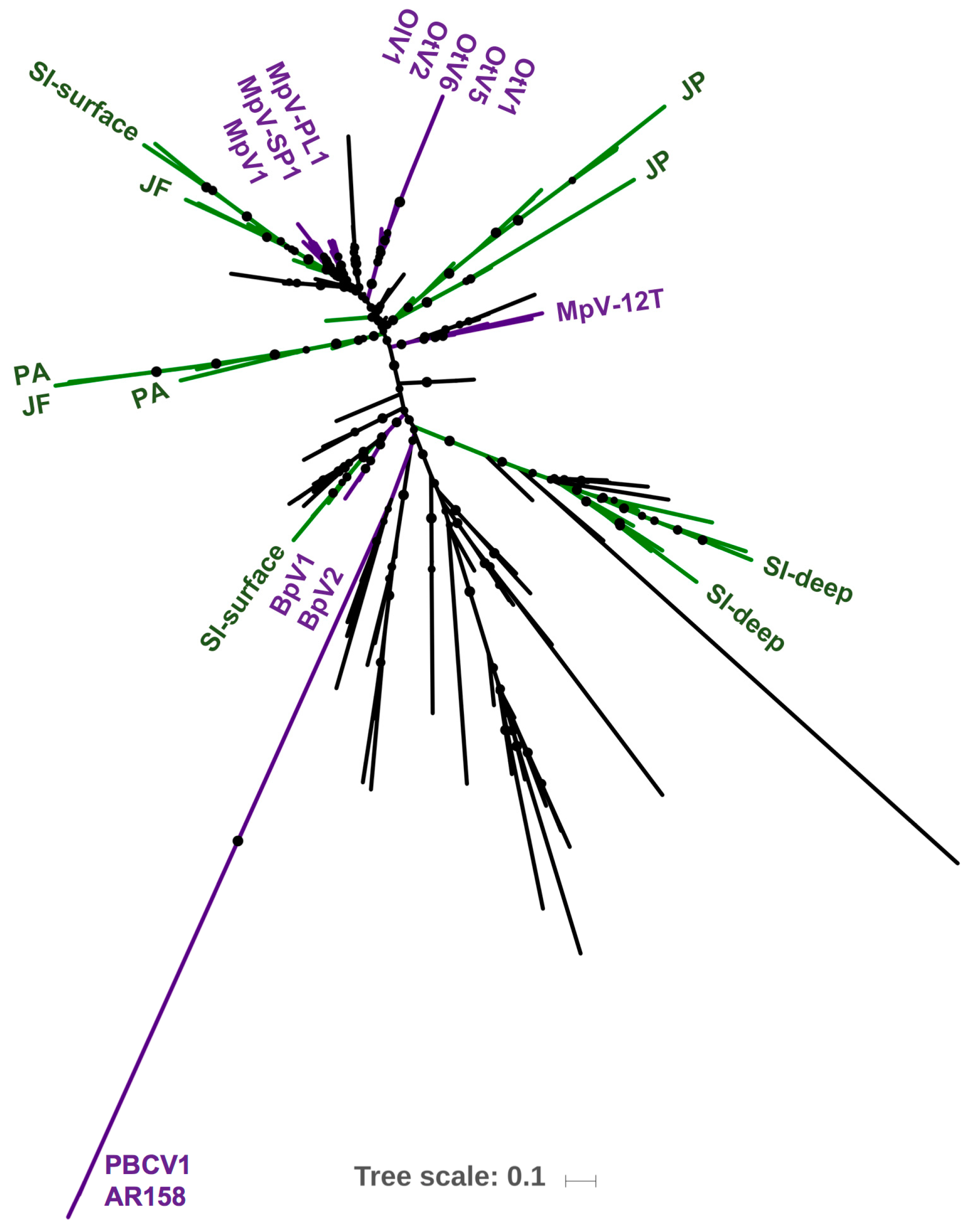
3.3. Environmental Prevalence of Prasinoviruses Show Adaptation to Environmental Conditions
4. Discussion
4.1. Origin and Distribution of Genes in Micromonas Viruses
4.2. Deriving Similarity in Gene Content from DNA Polymerase
4.3. Environmental Prevalence of Prasinoviruses Show Adaptation to Environmental Conditions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Leliaert, F.; Smith, D.R.; Herron, M.D.; Verbruggen, H.; Delwiche, C.F.; De Clerck, O. Phylogeny and molecular evolution of the green algae. CRC Crit. Rev. Plant Sci. 2012, 31, 1–46. [Google Scholar] [CrossRef]
- Marin, B.; Melkonian, M. Molecular phylogeny and classification of the Mamiellophyceae class. nov. (Chlorophyta) based on sequence comparisons of the nuclear- and plastid-encoded rRNA operons. Protist 2010, 161, 304–336. [Google Scholar] [CrossRef] [PubMed]
- Worden, A.Z.; Nolan, J.K.; Palenik, B. Assessing the dynamics and ecology of marine picophytoplankton: The importance of the eukaryotic component. Limnol. Oceanogr. 2004, 49, 168–179. [Google Scholar] [CrossRef]
- Van Etten, J.L.; Graves, M.V.; Boland, W.; Delaroque, N. Phycodnaviridae—Large DNA algal viruses. Arch. Virol. 2002, 147, 1479–1516. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Koonin, E.V.; Yutin, N. Origin and evolution of eukaryotic large nucleo-cytoplasmic DNA viruses. Intervirology 2010, 53, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Lane, L.C.; Dunigan, D.D. DNA viruses: The really big ones (Giruses). Annu. Rev. Microbiol. 2010, 64, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.D. Viral control of phytoplankton populations, a review. J. Eukaryot. Microbiol. 2004, 51, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Dunigan, D.D.; Fitzgerald, L.A.; Van Etten, J.L. Phycodnaviruses: A peek at genetic diversity. Virus Res. 2006, 117, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Derelle, E.; Ferraz, C.; Escande, M.-L.; Eychenie, S.; Cooke, R.; Piganeau, G.; Desdives, Y.; Bellec, L.; Moreau, H.; Grimsley, N. Life-cycle and genome of OtV5, a large DNA virus of the pelagic marine unicellular green alga Ostreococcus tauri. PLoS ONE 2008, 3, e2250. [Google Scholar] [CrossRef] [PubMed]
- Derelle, E.; Monier, A.; Cooke, R.; Worden, A.Z.; Nigel, H.; Moreau, H. Diversity of viruses infecting the green micro-alga Ostreococcus lucimarinus. J. Virol. 2015, 89, 5812–5821. [Google Scholar] [CrossRef] [PubMed]
- Moreau, H.; Piganeau, G.; Desdevises, Y.; Cooke, R.; Derelle, E.; Grimsley, N. Marine prasinovirus genomes show low evolutionary divergence and acquisition of protein metabolism genes by horizontal gene transfer. J. Virol. 2010, 84, 12555–12563. [Google Scholar] [CrossRef] [PubMed]
- Weynberg, K.D.; Allen, M.J.; Ashelford, K.; Scanlan, D.J.; Wilson, W.H. From small hosts come big viruses: The complete genome of a second Ostreococcus tauri virus, OtV-1. Environ. Microbiol. 2009, 11, 2821–2839. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.M.; Boere, A.; Gilg, I.; Van Lent, J.W.M. New lipid envelope-containing dsDNA virus isolates infecting Micromonas pusilla reveal a separate phylogenetic group. Aquat. Microb. Ecol. 2015, 74, 17–28. [Google Scholar] [CrossRef]
- Iyer, L.M.; Balaji, S.; Koonin, E.V.; Aravind, L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006, 117, 156–184. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Jeudy, S.; Bartoli, J.; Poirot, O.; Lescot, M.; Abergel, C.; Barbe, V.; Wommack, K.E.; Noordeloos, A.A.M.; Brussaard, C.P.D.; et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc. Natl. Acad. Sci. USA 2013, 110, 10800–10805. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Thompson, L.R.; Suttle, C.A.; Sullivan, M.B. Exploring the vast diversity of marine viruses. Oceanography 2007, 20, 135–139. [Google Scholar] [CrossRef]
- Siotto, F.; Martin, C.; Rauh, O.; Van Etten, J.L.; Schroeder, I.; Moroni, A.; Thiel, G. Viruses infecting marine picoplancton encode functional potassium ion channels. Virology 2014, 466–467, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Suttle, C.A. Evolutionary relationships among large double-stranded DNA viruses that infect microalgae and other organisms as inferred from DNA polymerase genes. Virology 1996, 219, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Suttle, C.A.; Short, S.M. Genetic diversity in marine algal virus communities as revealed by sequence analysis of DNA polymerase genes. Appl. Environ. Microbiol. 1996, 62, 2869–2874. [Google Scholar] [PubMed]
- Short, S.M.; Suttle, C.A. Sequence analysis of marine virus communities reveals that groups of related algal viruses are widely distributed in nature. Appl. Environ. Microbiol. 2002, 68, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Suttle, C.A. Temporal dynamics of natural communities of marine algal viruses and eukaryotes. Aquat. Microb. Ecol. 2003, 32, 107–119. [Google Scholar] [CrossRef]
- Short, S.M.; Short, C.M. Diversity of algal viruses in various North American freshwater environments. Aquat. Microb. Ecol. 2008, 51, 13–21. [Google Scholar] [CrossRef]
- Clasen, J.L.; Suttle, C.A. Identification of freshwater Phycodnaviridae and their potential phytoplankton hosts, using DNA pol sequence fragments and a genetic-distance analysis. Appl. Environ. Microbiol. 2009, 75, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Gimenes, M.V.; Zanotto, P.M.; Suttle, C.A.; da Cunha, H.B.; Mehnert, D.U. Phylodynamics and movement of phycodnaviruses among aquatic environments. ISME J. 2012, 6, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Larsen, A.; Bratbak, G.; Sandaa, R. Phylogenetic analysis of members of the Phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl. Environ. Microbiol. 2008, 74, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.M.; Fabre, M.F.; Gobena, D.; Wilson, W.H.; Wilhelm, S.W. Application of the major capsid protein as a marker of the phylogenetic diversity of Emiliania huxleyi viruses. FEMS Micriobiol. Ecol. 2011, 76, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Clerissi, C.; Grimsley, N.; Ogata, H.; Hingap, P.; Poulain, J.; Desdevises, Y. Unveiling of the diversity of prasinoviruses (Phycodnaviridae) in marine samples by using high-throughput sequencing analyses of PCR-amplified DNA polymerase and major capsid protein genes. Appl. Environ. Microbiol. 2014, 80, 3150–3160. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Jacquet, S. Contrasting diversity of phycodnavirus signature genes in two large and deep western European lakes. Environ. Microbiol. 2014, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alampic, J.M.; Ramus, C.; Bruley, C.; Labadie, K.; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, E5327–E5335. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Wolf, Y.I.; Koonin, E.V. Origin of giant viruses from smaller DNA viruses not from a fourth domain of cellular life. Virology 2014, 466–467, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.T.; Suttle, C.A. Biogeography of Viruses in the Sea. Annu. Rev. Virol. 2015, 2, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Marston, M.F.; Martiny, J.B.H. Genomic diversification of marine cyanophages into stable ecotypes. Environ. Microbiol. 2016, 18, 4240–4253. [Google Scholar] [CrossRef] [PubMed]
- Bellec, L.; Grimsley, N.; Derelle, E.; Moreau, H.; Desdevises, Y. Abundance, spatial distribution and genetic diversity of Ostreococcus tauri viruses in two different environments. Environ. Microbiol. Rep. 2010, 2, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Clerissi, C.; Grimsley, N.; Subirana, L.; Maria, E.; Oriol, L.; Ogata, H.; Moreau, H.; Desdives, Y. Prasinovirus distribution in the northwest Mediterranean Sea is affected by the environment and particularly by phosphate availability. Virology 2014, 466, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, M.T.; Suttle, C.A. Genetic diversity of algal viruses which lyse the photosynthetic picoflagellate Micromonas pusilla (Prasinophyceae). Appl. Environ. Microbiol. 1995, 61, 3088–3091. [Google Scholar] [PubMed]
- Fischer, M.G.; Kelly, I.; Foster, L.J.; Suttle, C.A. The virion of Cafeteria roenbergensis virus (CroV) contains a complex suite of proteins for transcription and DNA repair. Virology 2014, 466–467, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.; Barrell, B.G.; Parkhill, J. ACT: The Artemis comparison tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Tree Figure Drawing Tool Version 1.4.2; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2014. [Google Scholar]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Suttle, C.A. Amplification of DNA polymerase gene fragments from viruses infecting microalgae. Appl. Environ. Microbiol. 1995, 61, 1274–1278. [Google Scholar] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzales-Pena, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Rho, M.; Tang, H.; Ye, Y. FragGeneScan: Predicting genes in short and error-prone reads. Nucleic Acids Res. 2010, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, G.F.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community ecology package. R package version 2.3–3. 2016. [Google Scholar]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.B.; Crosti, G.; Dwivedi, B.; Mcdaniel, L.D.; Varsani, A.; Suttle, C.A.; Weinbauer, M.G.; Sandaa, R.A.; Breitbart, M. Development of phoH as a novel signature gene for assessing marine phage diversity. Appl. Environ. Microbiol. 2011, 77, 7730–7739. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.B.; Parsons, R.J.; Beyene, D. Deep sequencing of the viral phoH gene reveals temporal variation, depth-specific composition, and persistent dominance of the same viral phoH genes in the Sargasso Sea. PeerJ 2015, 3, e997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhou, J.; Liu, T.; Yu, Y.; Pan, Y.; Yan, S.; Wang, Y. Four novel algal virus genomes discovered from Yellowstone Lake metagenomes. Sci. Rep. 2015, 5, 15131. [Google Scholar] [CrossRef] [PubMed]
- Bachy, C.; Charlesworth, C.J.; Finke, J.F.; Chan, A.M.; Wong, C.H.; Ngan, C.Y.; Wei, C.-L.; Kunde-Ramamoorthy, G.; Coleman, M.L.; Suttle, C.A.; et al. Transcriptional responses of the marine alga Micromonas pusilla and an infecting prasino virus to different phosphate conditions. (Manuscript in preparation). 2017. [Google Scholar]
- Thompson, L.R.; Zeng, Q.; Kelly, L.; Huang, K.H.; Singer, A.U.; Stubbe, J.; Chisholm, S.W. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, E757–E764. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.; Li, Y.; Que, Q.; Bhattacharya, M.; Lane, L.C.; Chaney, W.G.; VanEtten, J. Evidence for virus-encoded glycosylation specificity. Proc. Natl. Acad. Sci. USA 1993, 90, 3840–3844. [Google Scholar] [CrossRef] [PubMed]
- Filée, J. Genomic comparison of closely related giant viruses supports an accordion-like model of evolution. Front. Microbiol. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Pagarete, A.; de Vargas, C.; Allen, M.J.; Read, B.; Claverie, J.-M.; Ogata, H. Horizontal gene transfer of an entire metabolic pathway between a eukaryotic alga and its DNA virus. Genome Res. 2009, 19, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Jeanniard, A.; Dunigan, D.D.; Gurnon, J.R.; Agarkova, I.V.; Kang, M.; Vitek, J.; Duncan, G.; Mcclung, O.W.; Larsen, M.; Claverie, J.; et al. Towards defining the chloroviruses: A genomic journey through a genus of large DNA viruses. BMC Genom. 2013, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, M.T.; Suttle, C.A. Wide-spread occurrence and clonal variation in viruses which cause lysis of a cosmopolitan, eukaryotic marine phytoplankter, Micromonas pusilla. Mar. Ecol. Prog. Ser. 1991, 78, 1–9. [Google Scholar] [CrossRef]
- Mayer, J.A.; Taylor, F.J.R. A virus which lyses the marine nanoflagellate Micromonas pusilla. Nature 1979, 281, 299–301. [Google Scholar] [CrossRef]
- Zaikova, E.; Walsh, D.A.; Stilwell, C.P.; Mohn, W.W.; Tortell, P.D.; Hallam, S.J. Microbial community dynamics in a seasonally anoxic fjord: Saanich Inlet, British Columbia. Environ. Microbiol. 2010, 12, 172–191. [Google Scholar] [CrossRef] [PubMed]
- Masson, D.; Pena, A. Chlorophyll distribution in a temperate estuary: The Strait of Georgia and Juan de Fuca Strait. Estuarine Coast. Shelf Sci. 2009, 82, 19–28. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MpV-PL1 | MpV-SP1 | MpV1 | MpV-12T | |
|---|---|---|---|---|
| Genome size (bp) | 196,960 | 173,350 | 184,095 | 205,622 |
| Host | MpUTEX991 | MpUTEX991 | MpRCC1109 | MpLAC38 |
| # ORF | 275 | 248 | 244 | 253 |
| ORF length | 684 | 659 | 715 | 749 |
| % GC | 43.3 | 40.6 | 41.0 | 39.8 |
| Asn-tRNA | 1 | 1 | 1 | 2 |
| Gle-tRNA | 1 | 1 | 1 | 1 |
| Ile-tRNA | 1 | 1 | 1 | 1 |
| Leu-tRNA | 0 | 1 | 1 | 1 |
| Thr-tRNA | 1 | 1 | 1 | 1 |
| Tyr-tRNA | 1 | 1 | 1 | 1 |
| Core-Genes | Pan-Genes | ||
|---|---|---|---|
| Class | Putative Function | Class | Putative Function |
| DNA replication | DNA polymerase | AA synthesis | Acetolacetate synthase |
| DNA topoisomerase | Acetolactate synthase | ||
| DNA ligase | Aminotransferase | ||
| DNA primase | DNA repair | Heat shock protein 70 | |
| Nucleotide metabolism | RNase/ | Site specific DNA methylases/ | |
| Ribonuclease | methyltransferases | ||
| Ribonucleotide reductase | |||
| Transcription | mRNA capping enzyme | Sugar manipulation | dTDP-d-glucose 4,6-dehydratase |
| Transcription initiation factor | UDP-glucose 6-dehydrogenase | ||
| Transcription elongation factor | 6-phosphofructokinase | ||
| Structural genes | Capsid protein | Transketolase N-terminal | |
| Major capsid protein | Transketolase B subunit | ||
| Metabolism | PhoH | Total Shared | 108 |
| Total Core | 119 | Total Unique | 327 |
| Presence-Absence (aa id 50%) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CDS Clusters | MpV1 | MPV-12T | MpV-PL1 | MpV-SP1 | BpV1 | BpV2 | OtV1 | OtV2 | OtV5 | OtV6 | OlV1 | PBCV1 | AR158 | |
| DNApol | MpV1 | 244 | 0.98 | 0.64 | 0.66 | 1.11 | 1.08 | 0.63 | 0.72 | 0.69 | 0.57 | 0.68 | 4.99 | 5.40 |
| MPV-12T | 0.36 | 252 | 1.02 | 0.99 | 1.18 | 1.16 | 1.06 | 1.05 | 1.11 | 1.00 | 1.06 | 5.41 | 5.01 | |
| MpV-PL1 | 0.26 | 0.36 | 271 | 0.30 | 1.12 | 1.10 | 0.71 | 0.75 | 0.75 | 0.67 | 0.73 | 5.04 | 5.45 | |
| MpV-SP1 | 0.23 | 0.36 | 0.18 | 244 | 1.09 | 1.10 | 0.70 | 0.75 | 0.77 | 0.69 | 0.71 | 4.99 | 5.40 | |
| BpV1 | 0.38 | 0.42 | 0.38 | 0.39 | 202 | 0.24 | 1.07 | 1.16 | 1.17 | 1.15 | 1.13 | 4.89 | 4.90 | |
| BpV2 | 0.39 | 0.42 | 0.38 | 0.39 | 0.05 | 209 | 1.05 | 1.17 | 1.13 | 1.11 | 1.13 | 4.91 | 4.92 | |
| OtV1 | 0.27 | 0.33 | 0.24 | 0.25 | 0.37 | 0.38 | 230 | 0.29 | 0.25 | 0.42 | 0.24 | 5.36 | 5.37 | |
| OtV2 | 0.26 | 0.33 | 0.23 | 0.24 | 0.41 | 0.41 | 0.05 | 235 | 0.33 | 0.48 | 0.22 | 6.07 | 6.08 | |
| OtV5 | 0.27 | 0.33 | 0.25 | 0.25 | 0.37 | 0.38 | 0.01 | 0.06 | 260 | 0.46 | 0.28 | 5.42 | 5.43 | |
| OtV6 | 0.26 | 0.35 | 0.25 | 0.24 | 0.38 | 0.38 | 0.09 | 0.10 | 0.09 | 249 | 0.45 | 5.40 | 5.41 | |
| OlV1 | 0.28 | 0.35 | 0.25 | 0.24 | 0.39 | 0.40 | 0.08 | 0.08 | 0.08 | 0.09 | 246 | 6.09 | 6.10 | |
| PBCV1 | 2.09 | 2.15 | 2.19 | 2.05 | 2.03 | 2.05 | 2.21 | 2.21 | 2.22 | 2.13 | 2.16 | 789 | 0.81 | |
| AR158 | 2.15 | 2.18 | 2.23 | 2.10 | 2.07 | 2.08 | 2.27 | 2.27 | 2.30 | 2.22 | 2.25 | 0.12 | 806 | |
| Mantel Test | 0.96, p = 0.01 | 0.99, p = 0.01 | ||||||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finke, J.F.; Winget, D.M.; Chan, A.M.; Suttle, C.A. Variation in the Genetic Repertoire of Viruses Infecting Micromonas pusilla Reflects Horizontal Gene Transfer and Links to Their Environmental Distribution. Viruses 2017, 9, 116. https://doi.org/10.3390/v9050116
Finke JF, Winget DM, Chan AM, Suttle CA. Variation in the Genetic Repertoire of Viruses Infecting Micromonas pusilla Reflects Horizontal Gene Transfer and Links to Their Environmental Distribution. Viruses. 2017; 9(5):116. https://doi.org/10.3390/v9050116
Chicago/Turabian StyleFinke, Jan F., Danielle M. Winget, Amy M. Chan, and Curtis A. Suttle. 2017. "Variation in the Genetic Repertoire of Viruses Infecting Micromonas pusilla Reflects Horizontal Gene Transfer and Links to Their Environmental Distribution" Viruses 9, no. 5: 116. https://doi.org/10.3390/v9050116
APA StyleFinke, J. F., Winget, D. M., Chan, A. M., & Suttle, C. A. (2017). Variation in the Genetic Repertoire of Viruses Infecting Micromonas pusilla Reflects Horizontal Gene Transfer and Links to Their Environmental Distribution. Viruses, 9(5), 116. https://doi.org/10.3390/v9050116