
Regulated Entry of Hepatitis C Virus into Hepatocytes


{kind=link}
{kind=link}
Abstract
:1. Introduction
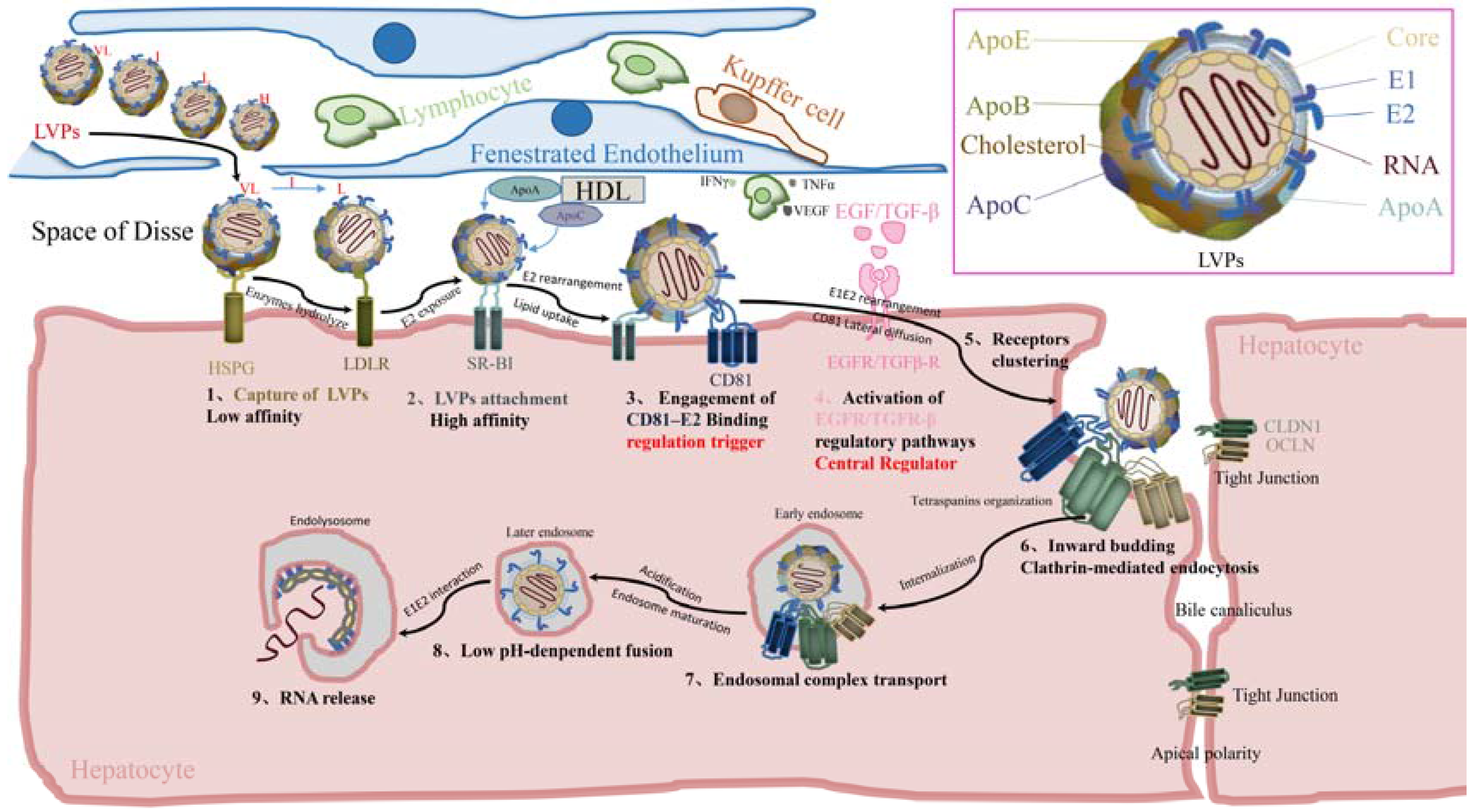
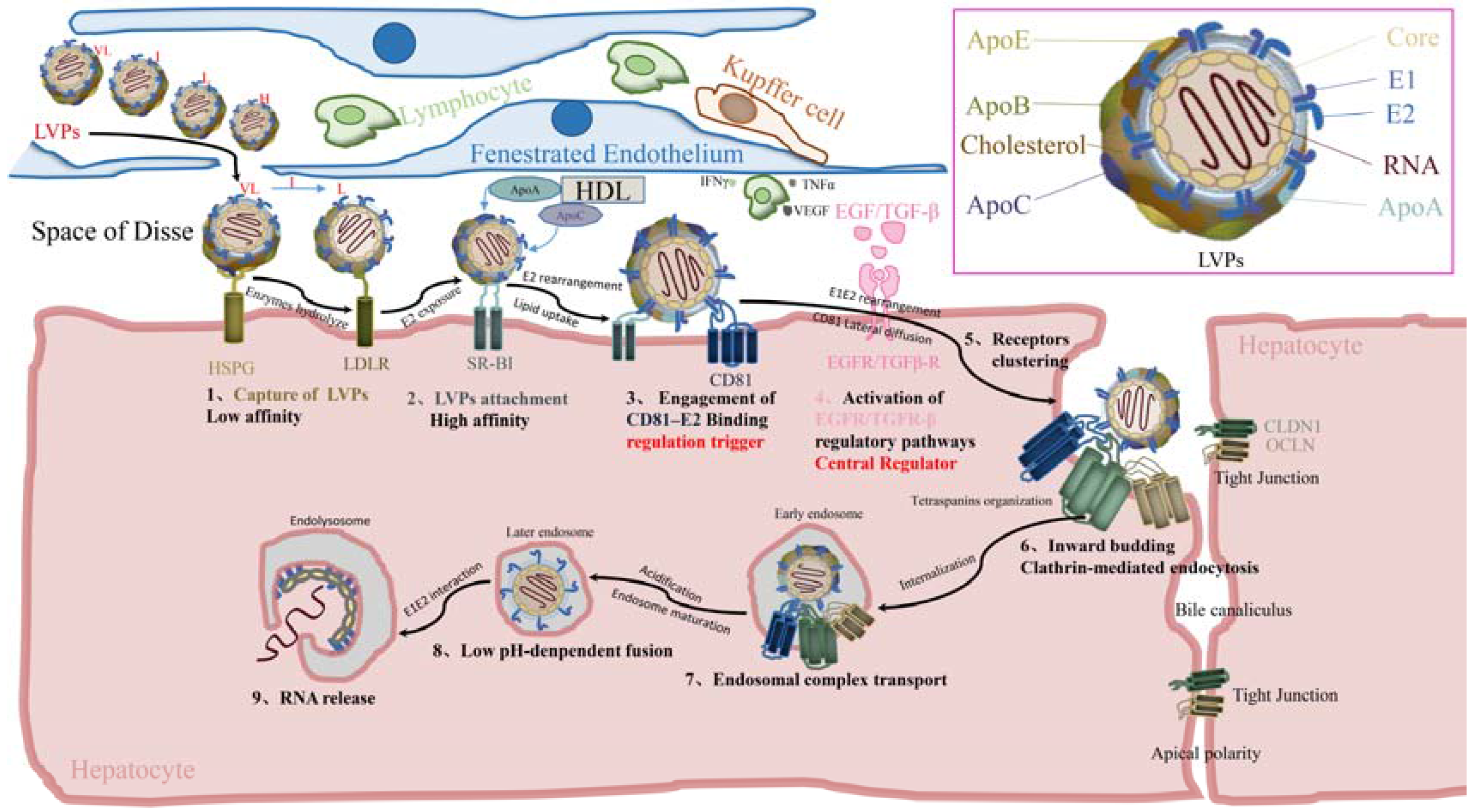
2. Cell-Free Entry
2.1. Before Binding
2.1.1. Virus Landing
2.1.2. Capture of Viral Particles
2.1.3. Viral Particles Attachment
2.2. E2–CD81 Binding and Activation of Regulatory Pathways
2.2.1. Engagement of E2–CD81 Binding
2.2.2. Activation of Regulatory Pathways
2.3. Postbinding
Lateral Diffusion of CD81 and Receptor Clustering
2.4. Internalization
2.4.1. Formation of Primary Endocytic Vesicles
2.4.2. Signal Degradation and Endosome Maturation
2.5. Fusion
2.5.1. Endosome Acidification
2.5.2. Fusion of Viral Envelope with Endosome Membrane
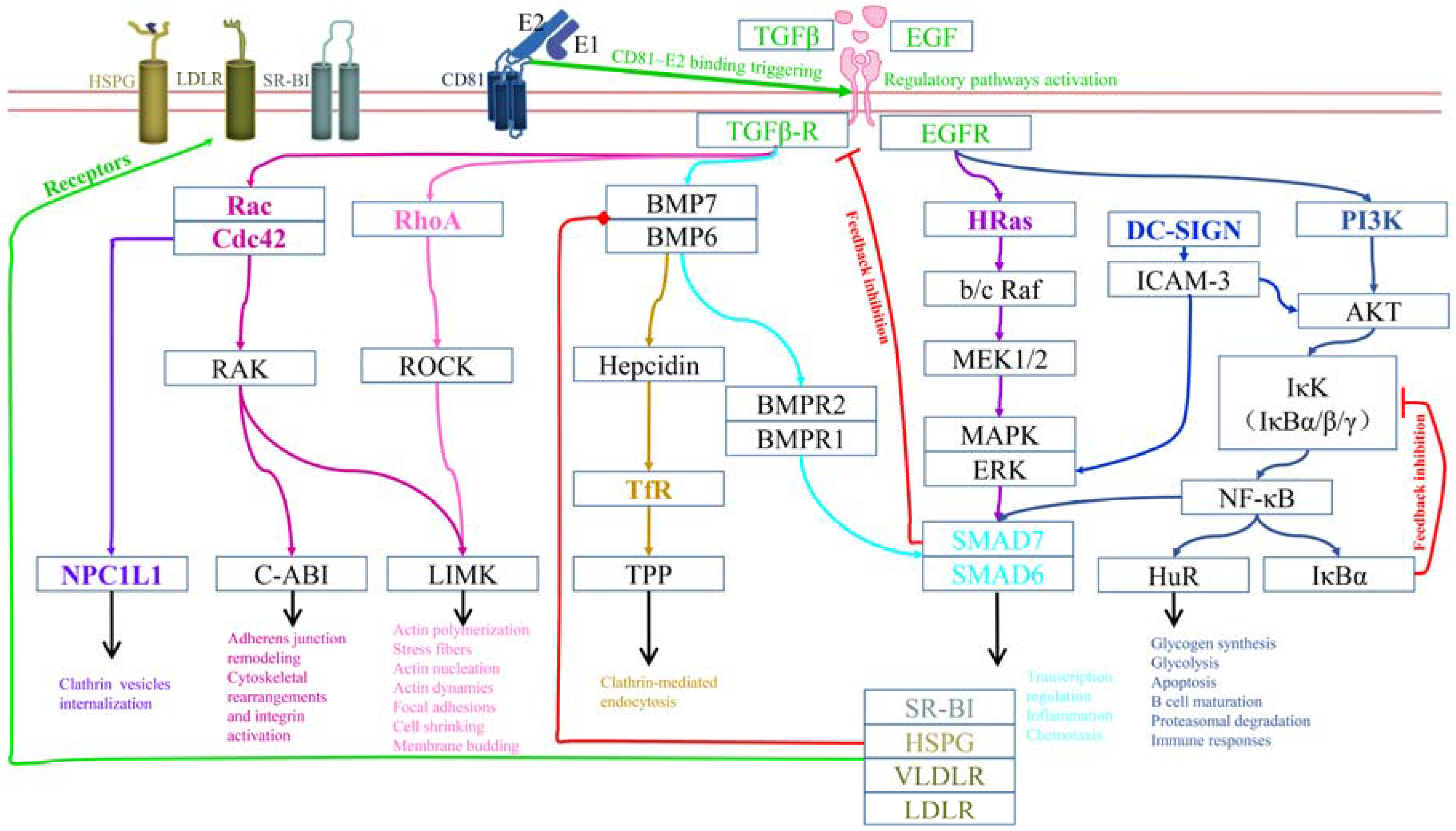
3. Regulatory Mechanisms for HCV Entry
3.1. Regulation for CD81 Lateral Diffusion and Receptors Clustering
3.2. Regulation of Particle Internalization and Endosome Maturation
3.3. Other Regulatory Mechanisms
4. Cell-to-Cell Spread
4.1. Cell-to-Cell Spread Shares the Same Receptors with Cell-Free Entry
4.2. Cell-to-Cell Spread Leads to Immune Evasion and Resistance to Direct-Acting Antiviral (DAA) Agents
5. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liang, T.J.; Rehermann, B.; Seeff, L.B.; Hoofnagle, J.H. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Annu. Int. Med. 2000, 132, 296–305. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; André, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.U.; Bassendine, M.F.; Burt, A.D.; Martin, C.; Pumeechockchai, W.; Toms, G.L. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 2006, 80, 2418–2428. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, D.J.; Sheridan, D.A.; Bridge, S.H.; Nielsen, S.U.; Milne, R.W.; Packard, C.J.; Caslake, M.J.; McLauchlan, J.; Toms, G.L.; Neely, R.D.; et al. Intravascular transfer contributes to postprandial increase in numbers of very-low-density hepatitis C virus particles. Gastroenterology 2010, 139, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, X.; Chi, X.; Zhao, F.; Guo, J.; Ma, P.; Zhong, J.; Niu, J.; Pan, X.; Long, G. Neglected but important role of apolipoprotein E exchange in hepatitis C virus infection. J. Virol. 2016, 90, 9632–9643. [Google Scholar] [CrossRef] [PubMed]
- Fauvelle, C.; Felmlee, D.J.; Crouchet, E.; Lee, J.; Heydmann, L.; Lefevre, M.; Magri, A.; Hiet, M.S.; Fofana, I.; Habersetzer, F.; et al. Apolipoprotein E mediates evasion from hepatitis C virus neutralizing antibodies. Gastroenterology 2016, 150, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Acosta, E.G.; Stoeck, I.K.; Long, G.; Hiet, M.S.; Mueller, B.; Fackler, O.T.; Kallis, S.; Bartenschlager, R. Apolipoprotein E likely contributes to a maturation step of infectious hepatitis C virus particles and interacts with viral envelope glycoproteins. J. Virol. 2014, 88, 12422–12437. [Google Scholar] [CrossRef] [PubMed]
- Scull, M.A.; Ploss, A. Exiting from uncharted territory: Hepatitis C virus assembles in mouse cell lines. Hepatology 2012, 55, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Merz, A.; Long, G.; Hiet, M.S.; Brugger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Long, G.; Hiet, M.S.; Windisch, M.P.; Lee, J.Y.; Lohmann, V.; Bartenschlager, R. Mouse hepatic cells support assembly of infectious hepatitis C virus particles. Gastroenterology 2011, 141, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.C.; Russell, R.S.; Engle, R.E.; Faulk, K.N.; Purcell, R.H.; Emerson, S.U. Apolipoprotein C1 association with hepatitis C virus. J. Virol. 2008, 82, 9647–9656. [Google Scholar] [CrossRef] [PubMed]
- Vieyres, G.; Thomas, X.; Descamps, V.; Duverlie, G.; Patel, A.H.; Dubuisson, J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J. Virol. 2010, 84, 10159–10168. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Dorner, M. Advances in experimental systems to study hepatitis C virus in vitro and in vivo. Virology 2015, 479, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology 2012, 142, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Li, H.C.; Lo, S.Y. Hepatitis C virus: Virology, diagnosis and treatment. World J. Hepatol. 2015, 7, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, Y.Y.; Chiu, S.; Hu, Z.; Lan, K.H.; Cha, H.; Sodroski, C.; Zhang, F.; Hsu, C.S.; Thomas, E.; et al. Integrative functional genomics of hepatitis C virus infection identifies host dependencies in complete viral replication cycle. PLoS Pathog. 2014, 10, e1004163. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Bakal, C.; Perrimon, N. Genomic screening with RNAi: Results and challenges. Annu. Rev. Biochem. 2010, 79, 37–64. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Schafer, C.; Adah, M.I.; Zhang, F.; Linhardt, R.J.; Toyoda, H.; Kinoshita-Toyoda, A.; Toida, T.; Van Kuppevelt, T.H.; Depla, E.; et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003, 278, 41003–41012. [Google Scholar] [CrossRef] [PubMed]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other Flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell entry of hepatitis c virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 2003, 278, 41624–41630. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Lozach, P.Y.; Lortat-Jacob, H.; de Lacroix de Lavalette, A.; Staropoli, I.; Foung, S.; Amara, A.; Houles, C.; Fieschi, F.; Schwartz, O.; Virelizier, J.L.; et al. DC-SIGN and l-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 2003, 278, 20358–20366. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, S.; Zhang, J.; Baribaud, F.; Chen, Z.; Leslie, G.J.; Lin, G.; Granelli-Piperno, A.; Doms, R.W.; Rice, C.M.; McKeating, J.A. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J. Virol. 2003, 77, 4070–4080. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.P.; Durso, R.J.; Arrigale, R.R.; Donovan, G.P.; Maddon, P.J.; Dragic, T.; Olson, W.C. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 2003, 100, 4498–4503. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B., Jr.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.N.; Uprichard, S.L. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. USA 2013, 110, 10777–10782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sodroski, C.; Cha, H.; Li, Q.; Liang, T.J. Infection of hepatocytes with HCV increases cell surface levels of heparan sulfate proteoglycans, uptake of cholesterol and lipoprotein, and virus entry by upregulating SMAD6 and SMAD7. Gastroenterology 2017, 152, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Forton, D.M. Hepatitis C—The brain strain. Liver Int. 2016, 36, 1415–1417. [Google Scholar] [CrossRef] [PubMed]
- Meredith, L.W.; Wilson, G.K.; Fletcher, N.F.; McKeating, J.A. Hepatitis Cvirus entry: Beyond receptors. Rev. Med. Virol. 2012, 22, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Mee, C.J.; Harris, H.J.; Farquhar, M.J.; Wilson, G.; Reynolds, G.; Davis, C.; van, I.S.C.; Balfe, P.; McKeating, J.A. Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J. Virol. 2009, 83, 6211–6221. [Google Scholar] [CrossRef] [PubMed]
- Brazzoli, M.; Bianchi, A.; Filippini, S.; Weiner, A.; Zhu, Q.; Pizza, M.; Crotta, S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 2008, 82, 8316–8329. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.H.; Kwon, D.S.; Torensma, R.; Vliet, S.J.V.; van Duijnhoven, G.C.F.; Middel, J.; Cornelissen, I.L.M.H.A.; Nottet, H.S.L.M.; Kewalramani, V.N.; Dan, R.L. DC-SIGN, a dendritic cell–specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587. [Google Scholar] [CrossRef]
- Mahley, R.W.; Ji, Z.S. Remnant lipoprotein metabolism: Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 1999, 40, 1–16. [Google Scholar] [PubMed]
- Albecka, A.; Belouzard, S.; Op de Beeck, A.; Descamps, V.; Goueslain, L.; Bertrand-Michel, J.; Terce, F.; Duverlie, G.; Rouille, Y.; Dubuisson, J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 2012, 55, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Jong, M.C.; Hofker, M.H.; Havekes, L.M. Role of ApoCs in lipoprotein metabolism: Functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, M.; Felmlee, D.J.; Parnot, M.; Baumert, T.F.; Schuster, C. Syndecan 4 is involved in mediating HCV entry through interaction with lipoviral particle-associated apolipoprotein E. PLoS ONE 2014, 9, e95550. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Jiang, J.; Luo, G. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J. Virol. 2013, 87, 6866–6875. [Google Scholar] [CrossRef] [PubMed]
- Murao, K.; Terpstra, V.; Green, S.R.; Kondratenko, N.; Steinberg, D.; Quehenberger, O. Characterization of CLA-1, a human homologue of rodent scavenger receptor bi, as a receptor for high density lipoprotein and apoptotic thymocytes. J. Biol. Chem. 1997, 272, 17551–17557. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Lavillette, D.; Cosset, F.L. The mechanism of HCV entry into host cells. Prog. Mol. Biol. Transl. Sci. 2015, 129, 63–107. [Google Scholar] [PubMed]
- Eck, M.V. Scavenger receptor BI facilitates the metabolism of VLDL lipoproteins in vivo. J. Lipid Res. 2008, 49, 136–146. [Google Scholar] [PubMed]
- Krieger, M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J. Clin. Investig. 2001, 108, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Boson, B.; Ricard-Blum, S.; Molle, J.; Lavillette, D.; Bartosch, B.; Pecheur, E.I.; Cosset, F.L. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J. Biol. Chem. 2007, 282, 32357–32369. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.C.; Engle, R.E.; Faulk, K.; Zhao, M.; Bartosch, B.; Alter, H.; Emerson, S.U.; Cosset, F.L.; Purcell, R.H.; Bukh, J. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. USA 2005, 102, 4560–4565. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.L.; Wang, N.; Xiao, X.; Tall, A.R. High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type 1 results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion. J. Biol. Chem. 2001, 276, 25287–25293. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Graziani, R.; von Hahn, T.; Moreau, M.; Huby, T.; Paonessa, G.; Santini, C.; Luzzago, A.; Rice, C.M.; Cortese, R.; et al. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J. Virol. 2007, 81, 8063–8071. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Granier, C.; Zeisel, M.B.; Guerin, M.; Mancip, J.; Granio, O.; Penin, F.; Lavillette, D.; Bartenschlager, R.; Baumert, T.F.; et al. Characterization of hepatitis C virus particle subpopulations reveals multiple usage of the scavenger receptor BI for entry steps. J. Biol. Chem. 2012, 287, 31242–31257. [Google Scholar] [CrossRef] [PubMed]
- Bankwitz, D.; Steinmann, E.; Bitzegeio, J.; Ciesek, S.; Friesland, M.; Herrmann, E.; Zeisel, M.B.; Baumert, T.F.; Keck, Z.Y.; Foung, S.K.; et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol. 2010, 84, 5751–5763. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Cormier, E.G.; Tsamis, F.; Kajumo, F.; Durso, R.J.; Gardner, J.P.; Dragic, T. CD81 is an entry coreceptor for hepatitis C virus. Proc. Natl. Acad. Sci. USA 2004, 101, 7270–7274. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Ansuini, H.; Graziani, R.; Huby, T.; Moreau, M.; Ball, J.K.; Paonessa, G.; Rice, C.M.; Cortese, R.; Vitelli, A. Role of scavenger receptor class B type I in hepatitis C virus entry: Kinetics and molecular determinants. J. Virol. 2010, 84, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F.L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Samreen, B.; Khaliq, S.; Ashfaq, U.A.; Khan, M.; Afzal, N.; Shahzad, M.A.; Riaz, S.; Jahan, S. Hepatitis C virus entry: Role of host and viral factors. Infect. Genet. Evol. 2012, 12, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Drummer, H.E.; Wilson, K.A.; Poumbourios, P. Identification of the hepatitis C virus E2 glycoprotein binding site on the large extracellular loop of CD81. J. Virol. 2002, 76, 11143–11147. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S.; Witteveldt, J.; Gatherer, D.; Owsianka, A.M.; Zeisel, M.B.; Zahid, M.N.; Rychłowska, M.; Foung, S.K.H.; Baumert, T.F.; Angus, A.G.N. Mutations within a conserved region of the hepatitis C virus E2 glycoprotein that influence virus-receptor interactions and sensitivity to neutralizing antibodies. J. Virol. 2010, 84, 5494–5507. [Google Scholar] [CrossRef] [PubMed]
- Owsianka, A.M.; Tarr, A.W.; Keck, Z.Y.; Li, T.K.; Witteveldt, J.; Adair, R.; Foung, S.K.H.; Ball, J.K.; Patel, A.H. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J. Gen. Virol. 2008, 89, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Owsianka, A.M.; Timms, J.M.; Tarr, A.W.; Brown, R.J.; Hickling, T.P.; Szwejk, A.; Bienkowska-Szewczyk, K.; Thomson, B.J.; Patel, A.H.; Ball, J.K. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 2006, 80, 8695–8704. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Dao Thi, V.L.; Maurin, G.; Fresquet, J.; Mompelat, D.; Zeisel, M.B.; Baumert, T.F.; Cosset, F.L.; Lavillette, D. Critical interaction between E1 and E2 glycoproteins determines binding and fusion properties of hepatitis C virus during cell entry. Hepatology 2014, 59, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.J.; Digman, M.A.; Lander, A.D. Heparan sulfate acts as a bone morphogenetic protein coreceptor by facilitating ligand-induced receptor hetero-oligomerization. Mol. Biol. Cell 2010, 21, 4028–4041. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell 1997, 89, 1165–1173. [Google Scholar] [CrossRef]
- Benedicto, I.; Molinajiménez, F.; Bartosch, B.; Cosset, F.L.; Lavillette, D.; Prieto, J.; Morenootero, R.; Valenzuelafernández, A.; Aldabe, R.; Lópezcabrera, M. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J. Virol. 2009, 83, 8012–8020. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, W.; Shen, L.; Turner, J.R.; Coyne, C.B.; Wang, T. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J. Virol. 2009, 83, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Davis, C.; Mullins, J.G.; Hu, K.; Goodall, M.; Farquhar, M.J.; Mee, C.J.; McCaffrey, K.; Young, S.; Drummer, H.; et al. Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem. 2010, 285, 21092–21102. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.J.; Hu, K.; Harris, H.J.; Davis, C.; Brimacombe, C.L.; Fletcher, S.J.; Baumert, T.F.; Rappoport, J.Z.; Balfe, P.; McKeating, J.A. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 2012, 86, 4305–4316. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Harris, H.J.; Hu, K.; Drummer, H.E.; Mckeating, J.A.; Mullins, J.G.L.; Balfe, P. In silico directed mutagenesis identifies the CD81/claudin-1 hepatitis C virus receptor interface. Cell. Microbiol. 2012, 14, 1892–1903. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Randall, G. Hepatitis C virus-host interactions, replication, and viral assembly. Curr. Opin. Virol. 2012, 2, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kuo, W.; Yang, W.; Liu, W.; Gibson, G.A.; Dorko, K.; Watkins, S.C.; Strom, S.C.; Wang, T. The second extracellular loop dictates occludin-mediated HCV entry. Virology 2010, 407, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Benedicto, I.; Molina-Jimenez, F.; Barreiro, O.; Maldonado-Rodriguez, A.; Prieto, J.; Moreno-Otero, R.; Aldabe, R.; Lopez-Cabrera, M.; Majano, P.L. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology 2008, 48, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Michta, M.L.; Hopcraft, S.E.; Narbus, C.M.; Kratovac, Z.; Israelow, B.; Sourisseau, M.; Evans, M.J. Species-specific regions of occludin required by hepatitis C virus for cell entry. J. Virol. 2010, 84, 11696–11708. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Shen, L.; Turner, J.R.; Bergelson, J.M. Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2007, 2, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.M.; Harris, H.J.; Jennings, A.; Hu, K.; Grove, J.; Lalor, P.F.; Adams, D.H.; Balfe, P.; Hubscher, S.G.; McKeating, J.A. Hepatitis C virus receptor expression in normal and diseased liver tissue. Hepatology 2008, 47, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Zegers, M.M.; Hoekstra, D. Mechanisms and functional features of polarized membrane traffic in epithelial and hepatic cells. Biochem. J. 1998, 336, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Mee, C.J.; Grove, J.; Harris, H.J.; Hu, K.; Balfe, P.; McKeating, J.A. Effect of cell polarization on hepatitis C virus entry. J. Virol. 2008, 82, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Musch, M.W.; Arvans, D.L.; Walshreitz, M.M.; Uchiyama, K.; Fukuda, M.; Chang, E.B. Synaptotagmin I binds intestinal epithelial NHE3 and mediates cAMP- and Ca2+-induced endocytosis by recruitment of AP2 and clathrin. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Coller, K.E.; Berger, K.L.; Heaton, N.S.; Cooper, J.D.; Yoon, R.; Randall, G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009, 5, e1000702. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Lu, T.Y.; Bair, C.H.; Chang, Y.S.; Jwo, J.K.; Chang, W. A novel cellular protein, VPEF, facilitates vaccinia virus penetration into Hela cells through fluid phase endocytosis. J. Virol. 2008, 82, 7988–7999. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Ewers, H.; Rajamäki, M.L.; Day, P.M.; Schiller, J.T.; Helenius, A. Human papillomavirus type 16 entry: Retrograde cell surface transport along actin-rich protrusions. PLoS Pathog. 2008, 4, e1000148. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Oh, M.J.; Kovacs, M.; Shukla, S.Y.; Valyinagy, T.; Shukla, D. Role for nectin-1 in herpes simplex virus 1 entry and spread in human retinal pigment epithelial cells. FEBS J. 2008, 275, 5272–5285. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Püntener, D.; Helenius, A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 2002, 296, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Van, M.G.; Van de Walle, G.R.; Favoreel, H.W.; Nauwynck, H.J.; Pensaert, M.B. Temporary disturbance of actin stress fibers in swine kidney cells during pseudorabies virus infection. Vet. Microbiol. 2002, 86, 89–94. [Google Scholar]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Urbé, S.; Mills, I.G.; Stenmark, H.; Kitamura, N.; Clague, M.J. Endosomal localization and receptor dynamics determine tyrosine phosphorylation of hepatocyte growth factor-regulated tyrosine kinase substrate. Mol. Cell. Biol. 2000, 20, 7685–7692. [Google Scholar] [CrossRef] [PubMed]
- Bean, A.J.; Davanger, S.; Chou, M.F.; Gerhardt, B.; Tsujimoto, S.; Chang, Y. Hrs-2 regulates receptor-mediated endocytosis via interactions with Eps15. J. Biol. Chem. 2000, 275, 15271–15278. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.S.; Raynor, M.C.; Wei, X.; Chen, H.Q.; Li, L. Hrs interacts with sorting nexin 1 and regulates degradation of epidermal growth factor receptor. J. Biol. Chem. 2001, 276, 7069–7078. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Bache, K.G.; Mehlum, A.; Stang, E.; Stenmark, H. Hrs recruits clathrin to early endosomes. EMBO J. 2001, 20, 5008–5021. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, T.E.; Atkinson, R.; Wu, M.N.; Zhou, Y.; Pennetta, G.; Bellen, H.J. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in drosophila. Cell 2002, 108, 261–269. [Google Scholar] [CrossRef]
- Meertens, L.; Bertaux, C.; Dragic, T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 2006, 80, 11571–11578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, A.; Zhang, X.; Xiao, H. A novel TIP30 protein complex regulates EGF receptor signaling and endocytic degradation. J. Biol. Chem. 2011, 286, 9373–9381. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar] [CrossRef] [PubMed]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 2006, 4, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Haid, S.; Pietschmann, T.; Pécheur, E.I. Low pH-dependent hepatitis C virus membrane fusion depends on E2 integrity, target lipid composition, and density of virus particles. J. Biol. Chem. 2009, 284, 17657–17667. [Google Scholar] [CrossRef] [PubMed]
- Krey, T.; Thiel, H.J.; Rümenapf, T. Acid-resistant bovine pestivirus requires activation for pH-triggered fusion during entry. J. Virol. 2005, 79, 4191–4200. [Google Scholar] [CrossRef] [PubMed]
- Moro, L.; Venturino, M.; Bozzo, C.; Silengo, L.; Altruda, F.; Beguinot, L.; Tarone, G.; Defilippi, P. Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998, 17, 6622–6632. [Google Scholar] [CrossRef] [PubMed]
- Murakami, G.; Watabe, T.; Takaoka, K.; Miyazono, K.; Imamura, T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell 2003, 14, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Pantua, H.; Ngu, H.; Komuves, L.; Diehl, L.; Schaefer, G.; Kapadia, S.B. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J. Virol. 2012, 86, 10935–10949. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tian, Y.; Machida, K.; Lai, M.M.; Luo, G.; Foung, S.K.; Ou, J.H. Transient activation of the PI3K-AKT pathway by hepatitis C virus to enhance viral entry. J. Biol. Chem. 2012, 287, 41922–41930. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. Hras signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Li, N.; Chen, Z.J.; Li, B.L.; Song, B.L. The small GTPase Cdc42 interacts with Niemann-Pick C1-like 1 (NPC1L1) and controls its movement from endocytic recycling compartment to plasma membrane in a cholesterol-dependent manner. J. Biol. Chem. 2011, 286, 35933–35942. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Qi, W.; Wang, L.J.; Miao, H.H.; Qu, Y.X.; Li, B.L.; Song, B.L. Flotillins play an essential role in Niemann-Pick C1-like 1-mediated cholesterol uptake. Proc. Natl. Acad. Sci. USA 2011, 108, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.Y.; Yeh, M.L.; Huang, C.F.; Hou, C.H.; Hsieh, M.Y.; Huang, J.F.; Lin, I.L.; Lin, Z.Y.; Chen, S.C.; Wang, L.Y.; et al. Chronic hepatitis c infection is associated with insulin resistance and lipid profiles. J. Gastroenterol. Hepatol. 2015, 30, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.M.; Zhang, C.L.; Song, Y.; Zhao, P.; Feng, Y.; Wang, B.; Li, Z.; Liu, L.; Xia, X. Genetic variations of the NPC1L1 gene associated with hepatitis C virus (HCV) infection and biochemical characteristics of HCV patients in china. Int. J. Infect. Dis. 2016, 53, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Yoshimoto, T.; Enjoji, M.; Fukushima, N.; Fukuizumi, K.; Nakamura, T.; Kurokawa, M.; Fujimori, N.; Sasaki, Y.; Shimonaka, Y.; et al. Hepcidin/ferroportin expression levels involve efficacy of pegylated-interferon plus ribavirin in hepatitis C virus-infected liver. World J. Gastroenterol. 2015, 21, 3291–3299. [Google Scholar] [PubMed]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [PubMed]
- Moruno Manchon, J.F.; Uzor, N.E.; Dabaghian, Y.; Furr-Stimming, E.E.; Finkbeiner, S.; Tsvetkov, A.S. Cytoplasmic sphingosine-1-phosphate pathway modulates neuronal autophagy. Sci. Rep. 2015, 5, 15213. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Dutta, D.; Iqbal, J.; Ansari, M.A.; Roy, A.; Chikoti, L.; Pisano, G.; Veettil, M.V.; Chandran, B. ESCRT-I protein Tsg101 plays a role in the post-macropinocytic trafficking and infection of endothelial cells by Kaposi’s sarcoma-associated herpesvirus. PLoS Pathog. 2016, 12, e1005960. [Google Scholar] [CrossRef] [PubMed]
- Barken, D.; Wang, C.J.; Kearns, J.; Cheong, R.; Hoffmann, A.; Levchenko, A. Comment on “oscillations in NF-κB signaling control the dynamics of gene expression”. Science 2005, 306, 704–708. [Google Scholar]
- Hiscott, J.; Kwon, H.; Génin, P. Hostile takeovers: Viral appropriation of the NF-κB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Schulz, S.; Borgmann, S.; Autenrieth, I.B.; Grobner, S. Differential roles of yersinia outer protein P-mediated inhibition of nuclear factor-κ Bin the induction of cell death in dendritic cells and macrophages. J. Med. Microbiol. 2008, 57, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N.F.; Sutaria, R.; Jo, J.; Barnes, A.; Blahova, M.; Meredith, L.W.; Cosset, F.L.; Curbishley, S.M.; Adams, D.H.; Bertoletti, A.; et al. Activated macrophages promote hepatitis C virus entry in a tumor necrosis factor-dependent manner. Hepatology 2014, 59, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N.F.; Clark, A.R.; Balfe, P.; McKeating, J.A. Tnf superfamily members promote hepatitis C virus entry via an NF-κB and myosin light chain kinase dependent pathway. J. Gen. Virol. 2017, 98, 405–412. [Google Scholar] [PubMed]
- Mossman, K.L.; Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.Y.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; et al. IL-1β production through the NRLP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar]
- Benchabane, H.; Wrana, J.L. GATA- and Smad1-dependent enhancers in the Smad7 gene differentially interpret bone morphogenetic protein concentrations. Mol. Cell. Biol. 2003, 23, 6646–6661. [Google Scholar] [CrossRef] [PubMed]
- Irie, A.; Habuchi, H.; Kimata, K.; Sanai, Y. Heparan sulfate is required for bone morphogenetic protein-7 signaling. Biochem. Biophys. Res. Commun. 2003, 308, 858–865. [Google Scholar] [CrossRef]
- Brkljacic, J.; Pauk, M.; Erjavec, I.; Cipcic, A.; Grgurevic, L.; Zadro, R.; Inman, G.J.; Vukicevic, S. Exogenous heparin binds and inhibits bone morphogenetic protein 6 biological activity. Int. Orthop. 2013, 37, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Sainz, B., Jr.; Hussain, S.; Uprichard, S.L. Determining the involvement and therapeutic implications of host cellular factors in hepatitis C virus cell-to-cell spread. J. Virol. 2014, 88, 5050–5061. [Google Scholar] [CrossRef] [PubMed]
- Timpe, J.M.; Stamataki, Z.; Jennings, A.; Hu, K.; Farquhar, M.J.; Harris, H.J.; Schwarz, A.; Desombere, I.; Roels, G.L.; Balfe, P.; et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 2008, 47, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Brimacombe, C.L.; Grove, J.; Meredith, L.W.; Hu, K.; Syder, A.J.; Flores, M.V.; Timpe, J.M.; Krieger, S.E.; Baumert, T.F.; Tellinghuisen, T.L.; et al. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J. Virol. 2011, 85, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Fofana, I.; Heydmann, L.; Barth, H.; Soulier, E.; Habersetzer, F.; Doffoel, M.; Bukh, J.; Patel, A.H.; Zeisel, M.B.; et al. Hepatitis C virus cell-cell transmission and resistance to direct-acting antiviral agents. PLoS Pathog. 2014, 10, e1004128. [Google Scholar] [CrossRef] [PubMed]
- Ujino, S.; Nishitsuji, H.; Hishiki, T.; Sugiyama, K.; Takaku, H.; Shimotohno, K. Hepatitis C virus utilizes VLDLR as a novel entry pathway. Proc. Natl. Acad. Sci. USA 2016, 113, 188. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Fukuhara, T.; Ono, C.; Uemura, K.; Kawachi, Y.; Shiokawa, M.; Mori, H.; Wada, M.; Shima, R.; Okamoto, T.; et al. Lipoprotein Receptors Redundantly Participate in Entry of Hepatitis C Virus. PLoS Pathog. 2016, 12, e1005610. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, Z.; Xie, Z.; Miao, J.; Ran, J.; Feng, Y.; Xia, X. Regulated Entry of Hepatitis C Virus into Hepatocytes. Viruses 2017, 9, 100. https://doi.org/10.3390/v9050100
Miao Z, Xie Z, Miao J, Ran J, Feng Y, Xia X. Regulated Entry of Hepatitis C Virus into Hepatocytes. Viruses. 2017; 9(5):100. https://doi.org/10.3390/v9050100
Chicago/Turabian StyleMiao, Zhijiang, Zhenrong Xie, Jing Miao, Jieyu Ran, Yue Feng, and Xueshan Xia. 2017. "Regulated Entry of Hepatitis C Virus into Hepatocytes" Viruses 9, no. 5: 100. https://doi.org/10.3390/v9050100
APA StyleMiao, Z., Xie, Z., Miao, J., Ran, J., Feng, Y., & Xia, X. (2017). Regulated Entry of Hepatitis C Virus into Hepatocytes. Viruses, 9(5), 100. https://doi.org/10.3390/v9050100