1. Introduction
Human cytomegalovirus (HCMV) is a widespread human β-herpesvirus with seroprevalence from 40 to 100%, which is closely related to socioeconomic and geographical conditions [
1]. Life-long latent or persistent HCMV infection can be established in immunocompetent populations after the primary infection, and productive replication can occur from spontaneous reactivation due to immune abnormalities. Thus, HCMV is a major viral cause of mental retardation and sensorineural deafness in newborns, and particularly causes morbidity and mortality in transplant recipients and AIDS patients with an immunocompromised state [
2]. Current studies also tie HCMV to multiple common chronic inflammatory diseases, such as cardiovascular disease, autoimmune diseases, inflammatory bowel diseases (IBDs), and certain cancers [
3].
HCMV has a large linear dsDNA genome, with a size of 240-kbp encoding more than 150 open reading frames (ORFs) and 26 mature miRNAs encoded from 14 pre-miRNAs [
4]. The large size of the viral genome has enabled co-evolution with its host for millions of years, to acquire various and even redundant strategies for immune evasion, to establish a life-long latent or persistent infection. As a result, a special balanced immune microenvironment for HCMV, from the interplay between the viral reactivation and immune response, was established, and intense research on its mechanisms could potentially contribute to a better understanding of the pathogenesis and biology of this virus.
Since most of the key immune cells, such as NK, T, and B cells, are not productively infected by HCMV, previous studies mainly focused on how the infection of permissive myeloid lineage or stromal cells alters the ability of these cells to prime and activate NK (natural killer cell) and T cells by down-modulating MHC (Major Histocompatibility Complex) and costimulatory molecules, suppression of activating signals, and provision of inhibitory molecules [
5]. In fact, HCMV also uses diverse tactics to modulate the early immune response. Type I interferons (IFNs) and pro-inflammatory cytokines are rapidly expressed and secreted by infected cells upon the detection of HCMV binding and entry [
6]. Gene expression profile assays have revealed that large numbers of antiviral genes, belonging to the interferon stimulated gene (ISG) family and the inflammatory cytokine family, are strongly induced by early events during HCMV entry [
7]. Binding to cellular receptors by HCMV envelopeglycoproteins can initiate signal transduction pathways, and lead to the activation of NF-κB and interferon regulatory factor 3 (IRF3), which participates in the subsequent transcription of antiviral Type I IFNs and pro-inflammatory cytokines [
8]. The early immune response is a pivotal time for the host to defend against HCMV infection. Recombinant IFN-α has been therapeutically used to control HCMV-induced retinitis in patients with AIDS [
9] and congenital viremia with HCMV infection [
10]. HCMV proteins have been shown to target early antiviral immune responses. For example, IE86, encoded by the HCMV immediate-early 2 (IE2) gene, can efficiently suppress HCMV or Sendai virus (Sev)-induced expression of IFN-β [
11]. The HCMV tegument protein UL82 has been reported to inhibit Stimulator of Interferon Genes (STING)-mediated signaling for evading antiviral innate immunity [
12].
Cellular receptor mediated signaling pathways are generally assumed to play an important role during the earliest immune response to virus infection. In previous studies, Toll-like receptor 2 (TLR2) has been reported to be critical in the early response to HCMV infection by recognizing the viral surface glycoproteins gB and gH [
13]. Here, CD147/EMMPRIN, a type-I transmembrane glycoprotein of the immunoglobulin superfamily [
14], was found and confirmed to play an important role in HCMV-triggered early antiviral signaling for the first time. Interestingly, we also found that CD147 expression was down-regulated at the late stages of HCMV infection, and that CD147 was a target gene for HCMV-encoded miR-US25-1-5p. Simultaneously, the expression and secretion of Cyclophilin A (sCyPA), which has been reported as a ligand for CD147 [
15,
16], were up-regulated in response to HCMV infection. CD147 is a multifunctional transmembrane glycoprotein that is involved in neural function, pro-inflammation, tumor invasion, and metastasis [
17]. Extracellular CypA has been reported to induce the production of proinflammatory cytokines through direct binding with the ectodomain of CD147, as seen in our previous work [
18]. The specific biological significance and mechanism of HCMV miR-US25-1-5p by targeting CD147/EMMPRIN, during the early antiviral response to HCMV infection, were further investigated and discussed in this study.
2. Materials and Methods
2.1. Cells and Viruses
Human foreskin fibroblasts (HFF cells, passages 9 to 14), Human astrocytoma U251 cells (U251 cells), and Human Embryonic Kidney 293 cells (293 cells) were maintained in Dulbecco’s modified Eagle medium (DMEM), containing 10% fetal bovine serum, as previously described [
19,
20]. Herpes simplex virus 1 (HSV-1) was provided by Fenyong Liu (University of California, Berkeley, CA, USA). To generate the U251-CD147 overexpressing (U251-CD147) and U251-Control (U251-C) cell lines, U251 MG cells were transfected with the pcDNA3.1-CD147 plasmid or the pcDNA3.1(+) (Invitrogen, Carlsbad, CA, USA) empty vector control plasmid using Lipofectamine
TM 2000, as prescribed by the manufacturer (Life Technologies, Waltham, MA, USA). After a 48-h incubation, transfected cells were selected for resistance in cell culture medium containing 600 μg/mL G418 (Life Technologies). After two to three weeks of selection, single clones were randomly isolated, and each was plated separately. The CD147 levels in individual selected clones were determined by Western blot analysis. The maintenance medium for cell lines was supplemented with 200 μg/mL G418. The bacterial artificial chromosome (BAC)-derived clinical strain NR-1 with enhanced green fluorescent protein (eGFP) expression cassette, provided by Dr. Ke Zen (Nanjing University, Nanjing, China), was used in this study [
21]. HCMV mutant (mUS25-1-5p) and the revertant wild type (RvWT) was generated using a bacterial recombineering method, as previously described [
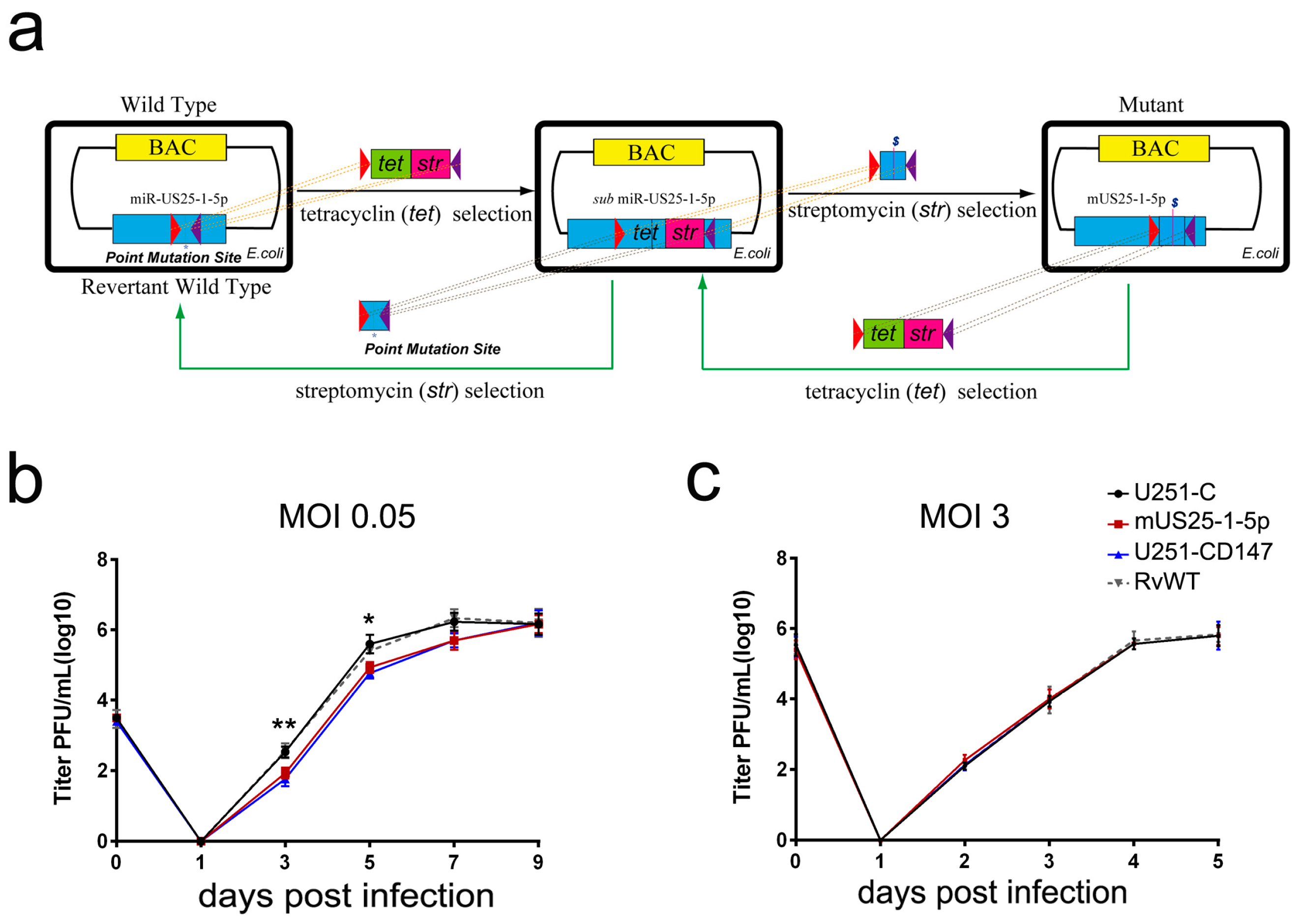
22]. In brief, approximately 300 bp of the miR-US25-1 flanking region was cloned into the pcDNA3.1(+) vector. The three nucleotides in the seed region of miR-US25-1-5p were mutated using site-directed polymerase chain reaction (PCR). Then, the PCR-amplified products containing point mutations of miR-US25-1-5p were used in the second round of recombination to replace the Tet/Str cassette that inserted in the region of miR-US25-1-5p at first. The amplified DNA fragments were transformed into
E. coli EL350 cells by electroporation (Bio-Rad, Hercules, CA, USA). The bacteria harboring the rescued BACs were selected in the presence of streptomycin. Mutants were confirmed using BAC DNA sequencing. Similarly, the BAC of RvWT was generated based on the newly constructed mUS25-1-5p, with two rounds of recombination. HCMV WT, mUS25-1-5p, and RvWT were propagated in HFF cells, and virus stocks were stored in DMEM supplemented with 10% fetal bovine serum (FBS) and 1.5% bovine serum albumin (BSA) at −80 °C.
2.2. Reagents and Antibodies
Cyclosporin A (CsA) reagent was obtained from Sigma-Aldrich (St. Louis, MO, USA). CD147 antibodies (HAb 18, IgG1) were prepared in our laboratory [
23]. Dylight 594-conjugated secondary antibody, used for immunofluorescence, was from Life Technology (San Jose, CA, USA). We also used anti-HCMV IE1/2 mouse mAb (ab53495, Abcam, Cambridge, UK), rabbit anti-human CyPA mAb (ab3563, Abcam), phospho-MEK1/2 (Ser217/221) rabbit mAb (#9154, Cell Signaling Technology (CST), Danvers, MA, USA), Erk1/2 rabbit mAb (#4695, CST), IRF-3 rabbit mAb (#11904, CST), phospho-IRF-3 (Ser396) rabbit mAb (#29047, CST), NF-κB p65 rabbit mAb (#8242, CST), phospho-NF-κB p65 (Ser536) rabbit mAb (#3033, CST), and anti-GAPDH mouse mAb (60004-1-Ig, Proteintech, Rosemont, IL, USA).
2.3. Construction of Plasmids
The following plasmids were used: CD147 pLKO.1 lentiviral shRNA (A6) and non-target shRNA control plasmid (pLKO.1-NTC) were purchased from Open Biosystems (GE Healthcare, Little Chalfont, UK). HCMV-encoded miR-US25-1-5p pLKO.1 lentiviral shRNA (pLKO.1-US25-1-5p) was constructed in this study. pcDNA3.1(+) empty vector was obtained from Invitrogen (Carlsbad, CA, USA). Full-length CD147-expressing plasmid pcDNA3.1-CD147 was constructed in our laboratory [
24]. Then, the extracellular domain (residues 1–185 of CD147) or intracellular domain (residues 230–269 of CD147) were deleted to generate pcDNA3.1-CD147-dECD and pcDNA3.1-CD147-dICD, respectively. The NF-κB-response promoter reporter plasmid (pNF-κB-Luc) and IFN-β promoter reporter plasmid (pIFN-β-Luc) were obtained from Beyotime (Shanghai, China). Dual luciferase miRNA target expression vector (pmirGLO) and the Renilla luciferase control reporter plasmid (pRL-TK) were purchased from Promega (Madison, WI, USA). The pmirGLO-CD147
3′UTR plasmid was made by inserting the 3′ UTR of the human CD147 gene into the pmirGLO empty vector using the primers as follows: (forward) 5′-AAGCTAGCGGCAGGTGGCCCGAGGACGCTCCCTG-3′ and (reverse) 5′-AGTCTAGAGAGGGTGGAGGTGGGGGCGATC-3′. Site-directed mutagenesis was performed using a QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Stratagene, San Diego, CA, USA) on the pmirGLO-CD147
3′UTR to generate a pmirGLO-CD147
3′UTRm plasmid with the following primers: (forward) 5′-AGTCATGGCCGGGTAGACAGCACAGCCTTCT-3′ and (reverse) 5′-AGAAGGCTGTGCTGTCTACCCGGCCATGACT-3′.
2.4. Indirect Immunofluorescence Assay (IFA)
Cells that were grown on chambered cover slips were infected with HCMV strain NR-1 at a multiplicity of infection (MOI) of 5. At 6 h posttransfection, cells were fixed with 4% formaldehyde and blocked with 4% bovine serum albumin (BSA) in PBS and stained with primary mouse IE1/2 antibody (ab53495, Abcam, Cambridge, UK), and then incubated with the secondary antibody Dylight 594 anti-mouse IgG (Life Technology). Cell nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Images were captured with a Nikon Eclipse TE300 microscope (Diagnostic Instruments, Inc., Sterling Heights, MI, USA) [
20]. The digital images were subsequently merged using FV10 ASW V4.1 software (Olympus, Tokyo, Japan).
2.5. RNAi-Transduced Stable Cells
The 293 cells were co-transfected with the two packaging plasmids (psPAX2 and pMD2G), together with a control or RNAi pLKO.1 lentiviral plasmid using LipofectamineTM 2000 (Invitrogen). The sequence for CD147 shRNA was: 5′-CCCATCATACACTTCCTTCTT-3′ (siCD147); the sequence for HCMV-miR-US25-1-5p shRNA was: 5′-CCGCTCAGTGGCTCGGACC-3′ (miR-US25-1-5p). After 24 h, cells were incubated with fresh medium without antibiotics for another 24 h. The medium containing the recombinant virus was collected and filtered, and then added to HFF or U251 cells in the presence of 6 mg/mL polybrene. The infected cells were selected by adding puromycin (4–6 mg/mL) to the culture medium for 14 days before additional experiments. The silencing of expression was verified by qPCR and Western blot.
2.6. Reporter Gene Assays
Cells (1 × 105) were seeded on 12-well plates and the following day were transfected using LipofectamineTM 2000 (Invitrogen) and the indicated plasmids. For transfection efficiency normalization, 0.01 μg Renilla luciferase reporter plasmids (pRL-TK) were added to each transfection. The total amount of transfeced DNA was maintained at a consistent level by adding empty vector DNA. Then, 36-h post-transfection, cells were lysed with 200 μL passive lysis buffer (Promega). Supernatants clarified by centrifugation were used to perform Luciferase assays using the dual luciferase assay kit (Promega). The values of Firefly luciferase activities were normalized to Renilla luciferase activities. All of the reporter assays were repeated at least three times in triplicate. Data shown are average values ± standard deviation (SD) from one representative experiment.
2.7. Real-Time PCR Quantitation of HCMV DNA Level
Viral DNA was extracted from mock or HCMV-infected cells using a DNeasy tissue kit (Qiagen, Hilden, Germany). Intracellular viral DNAs were quantified using primers and a probe for amplifying the HCMV UL83 DNA region, as previously described [
19]. The reaction was performed in a 20-μL amplification mixture (2 μL of DNA extract, 10 μL of 2 × Premix Ex Taq
TM (TaKaRa, Dalian, China), 0.4 μL each of primer at 10 μM, 0.8 μL of the fluorogenic probe at 5 μM, 0.4 μL 50 × ROX Reference Dye II) using an ABI 7500 device (Applied Biosystems Inc., Foster City, CA, USA). PCR cycling conditions were as follows: 95 °C for 30 s, 45 cycles at 95 °C for 5 s and 60 °C for 34 s. Viral genome copies were normalized to cellular RNase P with thepreviously described primers and probe [
25]. Unknown sample values were determined with a standard curve of known copy numbers of UL83 (NR-1 BAC) and RNase P. Each set of assays was repeated three times in triplicate, and data that are shown are average values ± SD from one representative experiment.
2.8. RNA Extraction and RT-qPCR Assays
Total RNA was isolated with TRIzol reagent (Invitrogen) and cDNA was reverse transcribed using a PrimeScriptTM RT Reagent Kit (TaKaRa). SYBR-Green real-time PCR (RT-PCR) was performed with the ABI 7500 device (Applied Biosystems Inc.) using SYBR Premix Ex Taq II (2×) (TaKaRa). Cellular GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) was used to normalize the RNA inputs. Relative changes in gene expression were analyzed using the 2−ΔΔCt method. All primers were synthesized by Shanghai GenePharma Co., Ltd., and are listed as following:
CD147: (forward) 5′-TCGCGCTGCTGGGCACC-3′ and (reverse) 5′-TGGCGCTGTCATTCAAGGA-3′; Cyclophilin A: (forward) 5′-CATGGTCAACCCCACGTGTTCTT-3′ and (reverse) 5′-TAGATGGACTTGCCACCAGTGCCAT-3′; IFNB1: (forward) 5′-AGGACAGGATGAACTTTGAC-3′ and (reverse) 5′-TGATAGACATTAGCCAGGAG-3′; IL-6: (forward) 5′-GACAGCCACTCACCTCTTCA-3′ and (reverse) 5′-AGTGCCTCTTTGCTGCTTTC-3′; ISG15: (forward) 5′-CACAGCCATGGGCTGGGACCTG-3′ and (reverse) 5′-GCACGCCGATCTTCTGGGTGA-3′; TNF-α: (forward) 5′-ATGAGCACTGAAAGCATGATCCGG-3′ and (reverse) 5′-CTACAACATGGGCTACAGGCTTGT-3′; GAPDH: (forward) 5′-AGCAATGCCTCCTGCACCACCAAC-3′ and (reverse) 5′-CCGGAGGGGCCATCCACAGTCT-3′.
2.9. Transfection of siRNA into Cells
The chemically synthetic siRNA, targeting the CD147 mRNA CDS region (miR-CD147, 5′-GUUCUUCGUGAGUUCCUCtt-3′) [
26], HCMV miR-US25-1-5p (MirBase, MIMAT0001581), miR-US25-1-3p (MirBase, MIMAT0004755), and 13 other screened HCMV-encoded mature microRNAs (
http://www.mirbase.org/) were synthesized by Ribobio Co., Ltd. (Guangzhou, China), along with the silencer negative control siRNA (C-siRNA) molecules. The siRNAs were transfected into cells using Lipofectamine
TM 2000 (Invitrogen), according to the manufacturer’s instructions.
2.10. Western Blot Analysis
Cells (1 × 108) were washed with cold PBS and lysed with 1 mL NP40 lysis buffer with 1 mM β-mercaptoethanol and protease inhibitor Cocktail (Roche, Basel, Switzerland) for 30 min on ice. Cell lysates were centrifuged at 12,000 rpm for 30 min, and supernatants were collected. The protein concentration was determined by BCA assay. Proteins were separated by 8% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membrane (GE Healthcare). Block in 5% BSA was performed for 1 h in TBS with 0.1% Triton X-100 (TBST), followed by incubation with primary antibody solution overnight at 4 °C. Then, the membrane was rinsed 3–5 times for 5 min with TBST. After rinsing, the membrane was incubated with the horse radish peroxidase (HRP)-conjugated secondary antibody solution for 2 h at room temperature. After another rinsing, the membrane was subsequently reacted with the chemiluminescent substrate, according to the recommendation of the ECL Western blot detection reagent kits (Thermo Scientific, Waltham, MA, USA), and were quantitated chemiluminescent signals using a CCD camera-based imager (Bio-Rad, Hercules, CA, USA). Quantitation was performed in the linear range of protein detection and analyzed by Image Lab 4.0 software (Bio-Rad). The experiments were repeated at least three times in triplicate, and a representative result is shown.
2.11. Viral Infection and Growth Curves
Cells (1 × 10
6) were either mock or HCMV infected at a MOI of 0.05 to 5 in 1.5 mL DMEM with 1% fetal bovine serum. The medium was replaced with 10% fetal bovine serum containing DMEM after 2 h of incubation. Protein extracts prepared from the infected cells at 3 to 12 days post-infection, were analyzed by Western blot analysis. To determine viral growth level, cells (1 × 10
5) were infected with HCMV at an MOI of 0.05 or 3. Viral stocks were prepared with the total cells and the medium was harvested 3 to 11 days post-infection by adding an equal volume of 10% (
w/
v) skim milk, followed by ultrasonication. The titers of the viral stocks were determined in 1 × 10
5 HFF cells, with standard virus plaque assay and the number of plaques were counted 10 to 14 days after viral infection. The values obtained were averages ± SD from triplicate experiments [
19].
2.12. Statistical Analysis
All of the statistical analyses were performed with GraphPad Prism version 6.00 (GraphPad Software, San Diego, CA, USA). Experiments were repeated at least three times in triplicate. Data are expressed as mean ± standard deviation. Means between two groups were compared by using unpaired t-test or one-way analysis of variance (ANOVA) with post hoc Bonferroni t-test. Differences were considered statistically significant at p < 0.05.
4. Discussion
Numerous cell surface glycoproteins, such as β2 microglobulin, annexin II, CD13, platelet-derived growth factor receptor-α (PDGFR-α), epithelial growth factor receptor (EGFR), and β1 integrins, have been suggested to act as receptors facilitating HCMV entry [
30]. However, none of the suggested glycoproteins has been validated as a bona fide receptor that is necessary for HCMV infection of all the susceptible cell types. The in vivo broad tissue tropism of HCMV infection and differences in permissiveness for different cell types suggest that HCMV likely uses distinct cell surface receptors depending on the target cells. Thus, identifying the specific HCMV cellular receptors is important for understanding the pathogenesis of HCMV. CD147/EMMPRIN is a transmembrane receptor glycoprotein that is belonging to the immunoglobulin superfamily and expressed at varying levels in many tissue cells. Notably, several bacterial pathogens, such as Neisseria meningitidis and Plasmodium falciparum, as well as many viruses, including HIV-1, HBV, SARS-CoV, and measles virus (Mev), have been previously found to infect target cells via CD147, indicating that it might constitute an evolutionarily conserved pathogen receptor for infection and spread within tissues and organisms [
31]. Unfortunately, neither the infection-blocking experiment using CD147-specfic antibody, nor the viral infection test with CD147-knockdown cells, showed any difference in facilitating HCMV entry (
Figure S2). Since a low passage clinical HCMV strain NR-1 was used in these experiments, we assumed that CD147 does not function as a broad-spectrum receptor for HCMV infection, at least in typical in vitro permissive cell lines.
In addition to the pathogen recognition receptor function, most transmembrane receptor proteins have been proposed to mediate signal transduction pathways immediately upon pathogenic particle attachment and penetration. CD147 belongs to the Type I membrane proteins and as a candidate tumor biomarker, its expression is up-regulated in breast cancer tissue, lung carcinoma tissue, and bladder cancer tissue, along with a tumor’s malignance, invasiveness, and metastasis [
32]. CD147 has been extensively studied since the discovery of its function in tumor progression and metastasis. Here, we have explored a previously unknown role for CD147 in the host innate immune defense against HCMV invasion. Cells detect HCMV infection as early as four to eight hours post-infection (hpi), responding by producing NF-κB-dependent cytokines and an early peak of antiviral IFN [
5,
33]. Our experiments indicate that endogenous CD147 is required for the maximum activation of NF-κB and IFN-β-associated antiviral signaling that is triggered by HCMV infection, possibly having a redundant effect with other cell surface receptor glycoproteins (
Figure 1). Many tumor-related proteins, such as Programmed death 1 (PD-1) and PTEN, are known to play key roles in regulating the innate immunity against viral and bacterial infection [
34,
35]. However, the involvement of CD147 in early antiviral immunity appears to be specific to HCMV infection, since it does not extend to other viruses, like HSV-1. Since the signaling pathways of CD147 remain to be well defined, we next demonstrated that the phosphorylation of ERK1/2 and then nuclear translocation of NF-κB might play a leading role in CD147-mediated HCMV-triggered antiviral signal transduction, with a subsequent activation of the IRF3 phosphorylation and the orchestrated NF-κB-dependent cytokines, IRF3 plus NF-κB-dependent interferon responses (
Figure 6). This finding can be supported by the fact that NF-κB is a key transcriptional enhancesome-component of the IFN-β, according to previous research [
36,
37].
Research has increasingly suggested that the modulation of the host immune microenvironment is not only important for tumor virus-induced inflammatory carcinogenesis, but it is also critical for persistent and latent viral infection. For example, cmvIL-10, a viral mimic of human IL-10 acquired from the cellular genome, binds with a high affinity to the human IL-10 receptor, and limits the innate and adaptive immune responses even more efficiently than IL-10 itself [
38]. In this study, we have demonstrated that HCMV infection leads to the expression and secretion of a cellular chemokine, CyPA. Studies show that CypA, as an extracellular ligand for its receptor CD147, can be secreted by various cell types, including vascular smooth muscle cells, macrophages, and fibroblasts, and triggers a cascade of inflammatory responses [
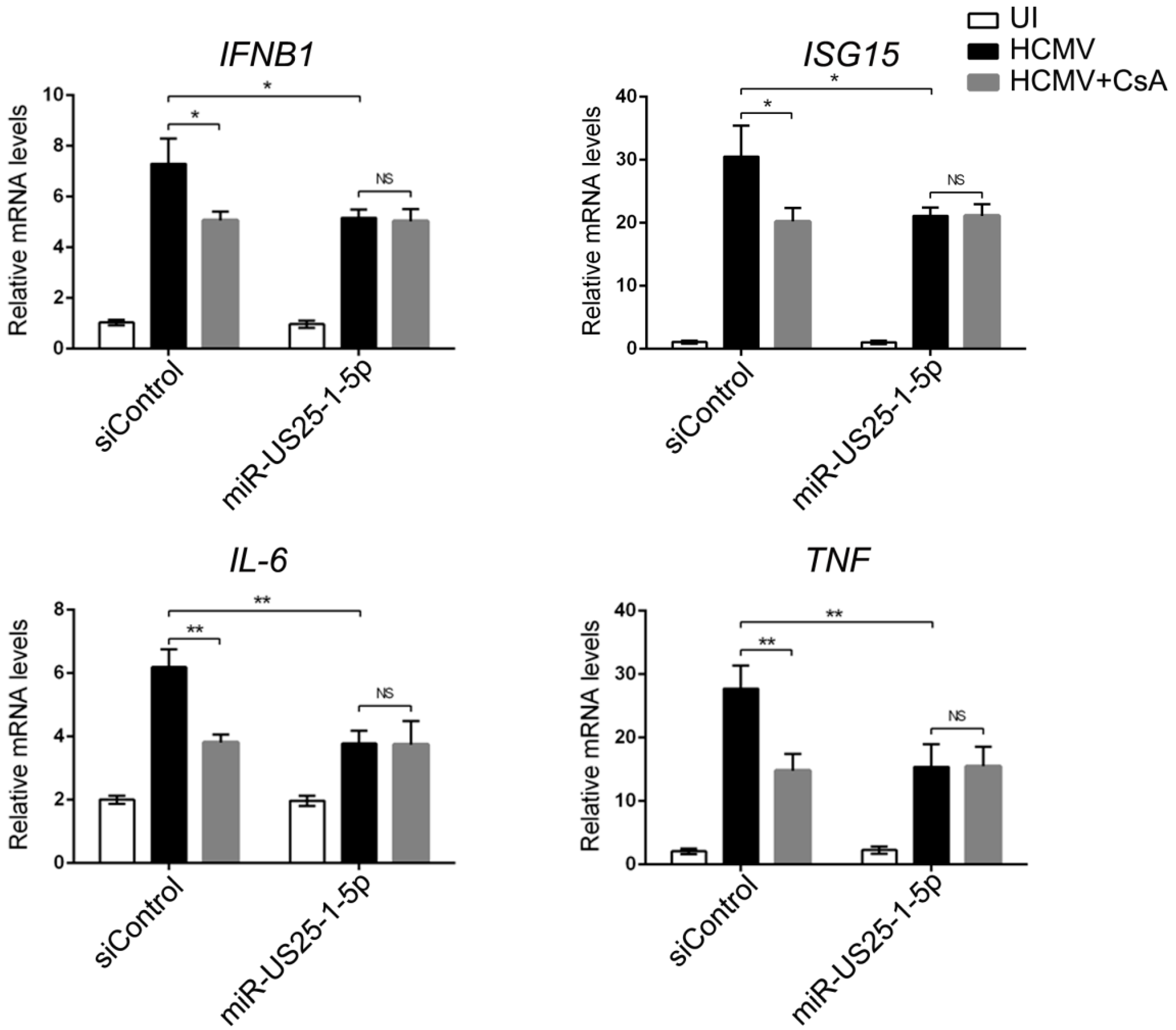
39]. We demonstrated that CD147-mediated HCMV-triggered phosphorylation of ERK1/2, and the activation of NF-κB depends on the release of CyPA from HCMV infected cells and its interaction with CD147 (
Figure 2). Although HCMV is highly species-specific with no animal model yet known to support its replication, we speculate that secreted extracellular CypA, triggered by HCMV infection, acts through a paracrine mechanism, to stimulate uninfected cells to defend against a second wave of infection by the progeny virus through the interaction with CD147.
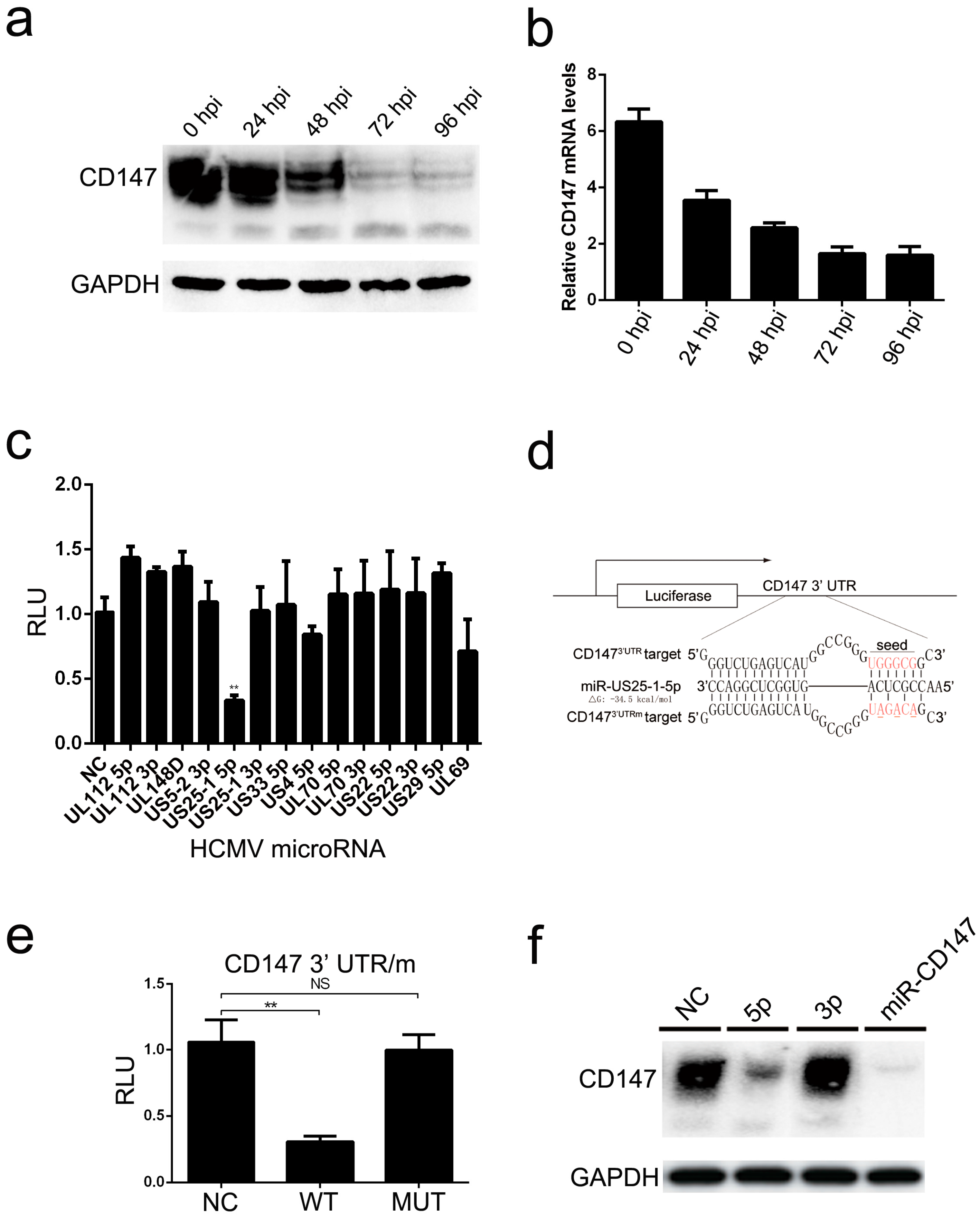
Moreover, we found that endogenous CD147 was targeted by HCMV-encoded miR-US25-1-5p at the 3′ UTR, which could replace CD147-specific siRNA mimics in inhibiting the CD147-mediated early innate immune response to HCMV infection (
Figure 4). Mutation of the HCMV miR-US25-1-5p seed sequence or ectopic expression of CD147 resulted in delayed multistep growth curves at low MOI, suggesting that miR-US25-1-5p targeting of CD147 is biologically significant (
Figure 5b). MicroRNAs (miRNAs) are small noncoding RNA molecules that generally target 3′ untranslated regions (3′ UTR) of the mRNAs. Since the discovery of HCMV-encoded miRNAs, at least 26 mature miRNA species encoded by 14 pre-miRNAs have been identified and reported to regulate multiple aspects of viral and cellular processes, including viral replication, immune evasion, formation of the virion, and eukaryotic translation [
40]. The pre-HCMV-miR-US25-1, from which the mature miR-US25-1-5p is derived, is encoded by the HCMV US24 and US26 intergenic regions [
41]. miR-US25-1-5p has been reported to be transcribed upon infection and highly expressed at 24 hpi, followed by a gradually increased expression, during the course of HCMV infection [
42]. One might expect that the cells would respond differently to infection with the mutant virus (HCMV-mUS25-1-5p), at late times of HCMV infection, when the expression levels of CD147 were obviously down-regulated by miR-US25-1-5p. However, we found no difference in the expression of ISGs after infection with the mutant virus. Based on previous studies and our overall findings, the reasons might be complex. At the late stage of infection, HCMV often uses multi-faceted and redundant strategies to regulate immune responses. Thus, the effect of natural miR-US25-1-5p may not be able to be detectable at late times of HCMV infection. We speculate that secreted extracellular CypA, which is triggered by HCMV infection, acts primarily to defend uninfected cells against progeny HCMV virus released from neighboring infected cells. As a result, the observed lack of differences in virus yield at the higher MOI, irrespective of the mutation of miR-US25-1-5p in the targeting of CD147 mRNA 3′ UTR or ectopic expression of CD147, are understandable (
Figure 5c).
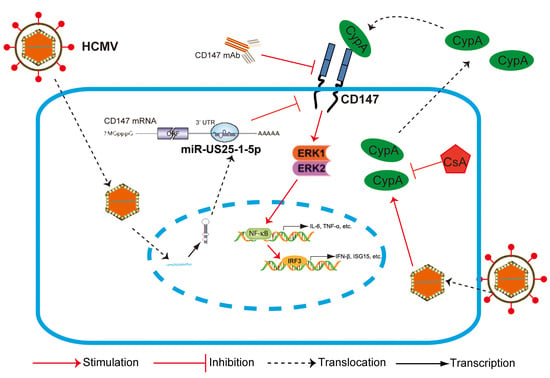
In summary, we have provided the first evidence that CD147, a non-HCMV receptor glycoprotein, is targeted by HCMV-encoded miR-US25-1-5p. This may be a tactic that is acquired by HCMV to antagonize the early innate immune response to benefit HCMV chronic infection. Additionally, HCMV-induced paracrine effects of the HCMV-induced expression of CypA could cause an immune microenvironment called “smoldering inflammation”, which could potentially contribute to the development of many HCMV inflammatory disorders.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





