
A Tale of Two RNAs during Viral Infection: How Viruses Antagonize mRNAs and Small Non-Coding RNAs in The Host Cell


Abstract
1. Introduction
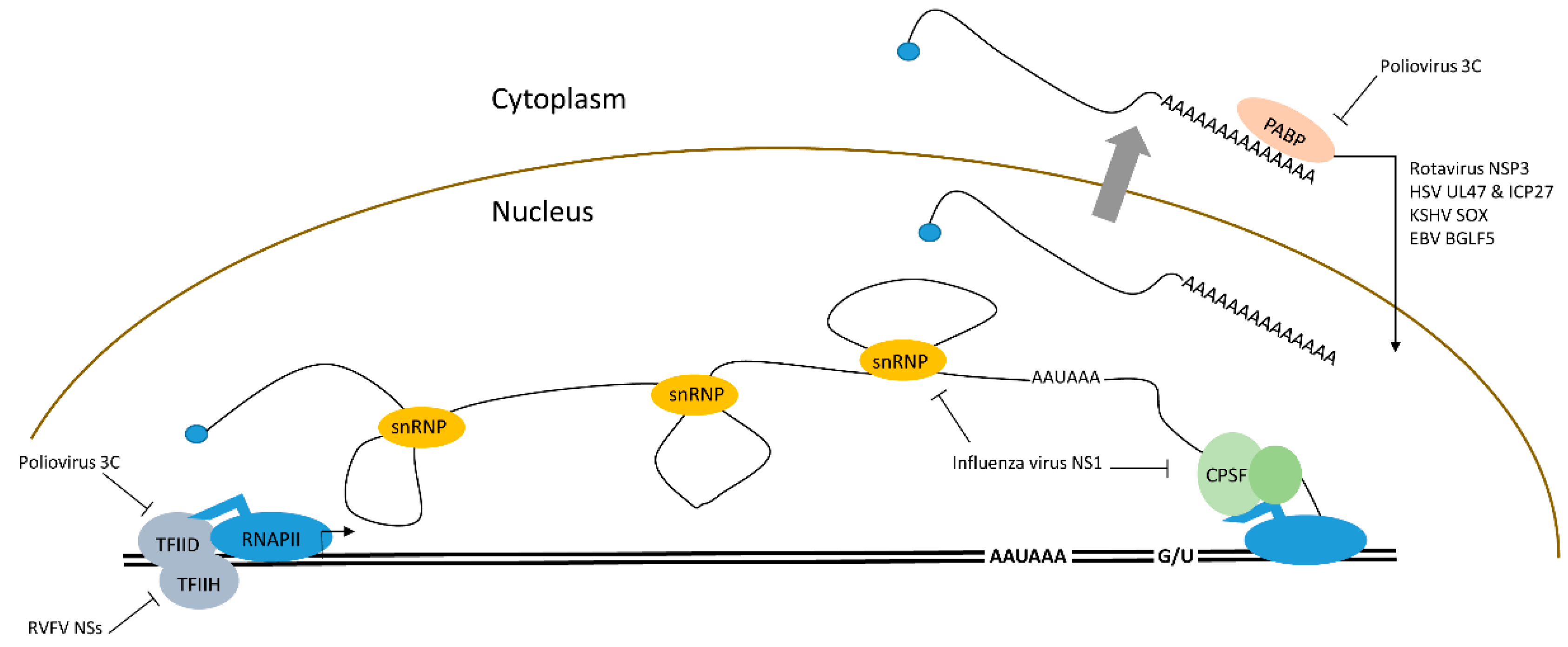
2. Shutting down Cellular mRNA
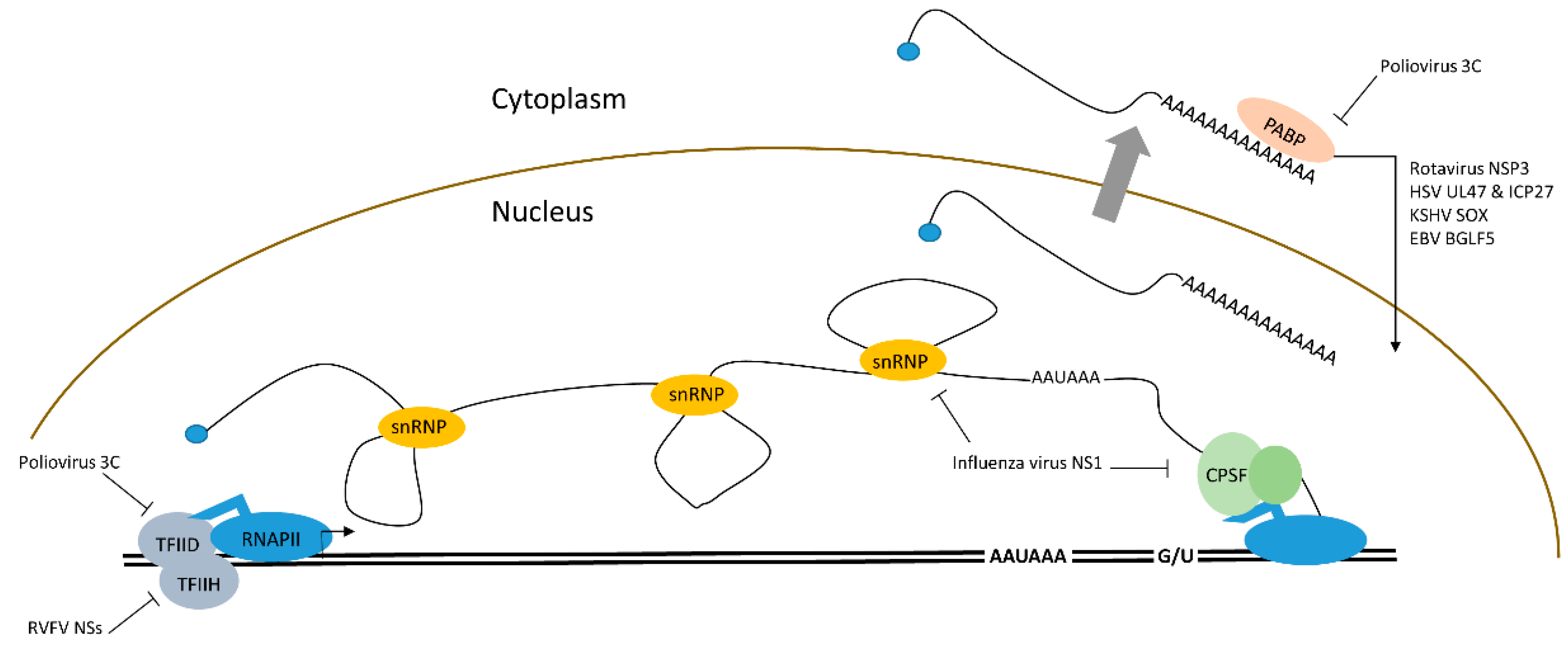
2.1. Transcriptional Down-regulation of mRNAs by Viral Proteins
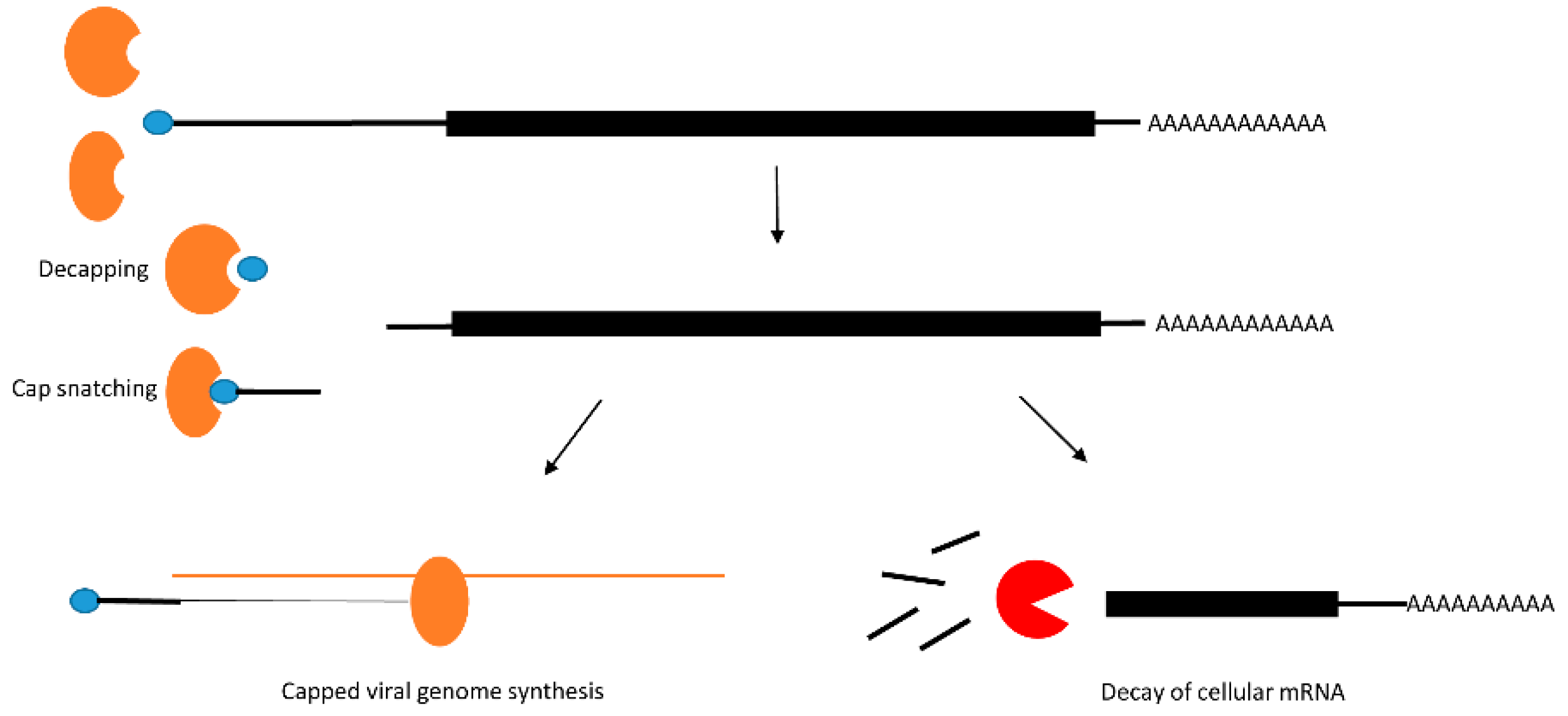
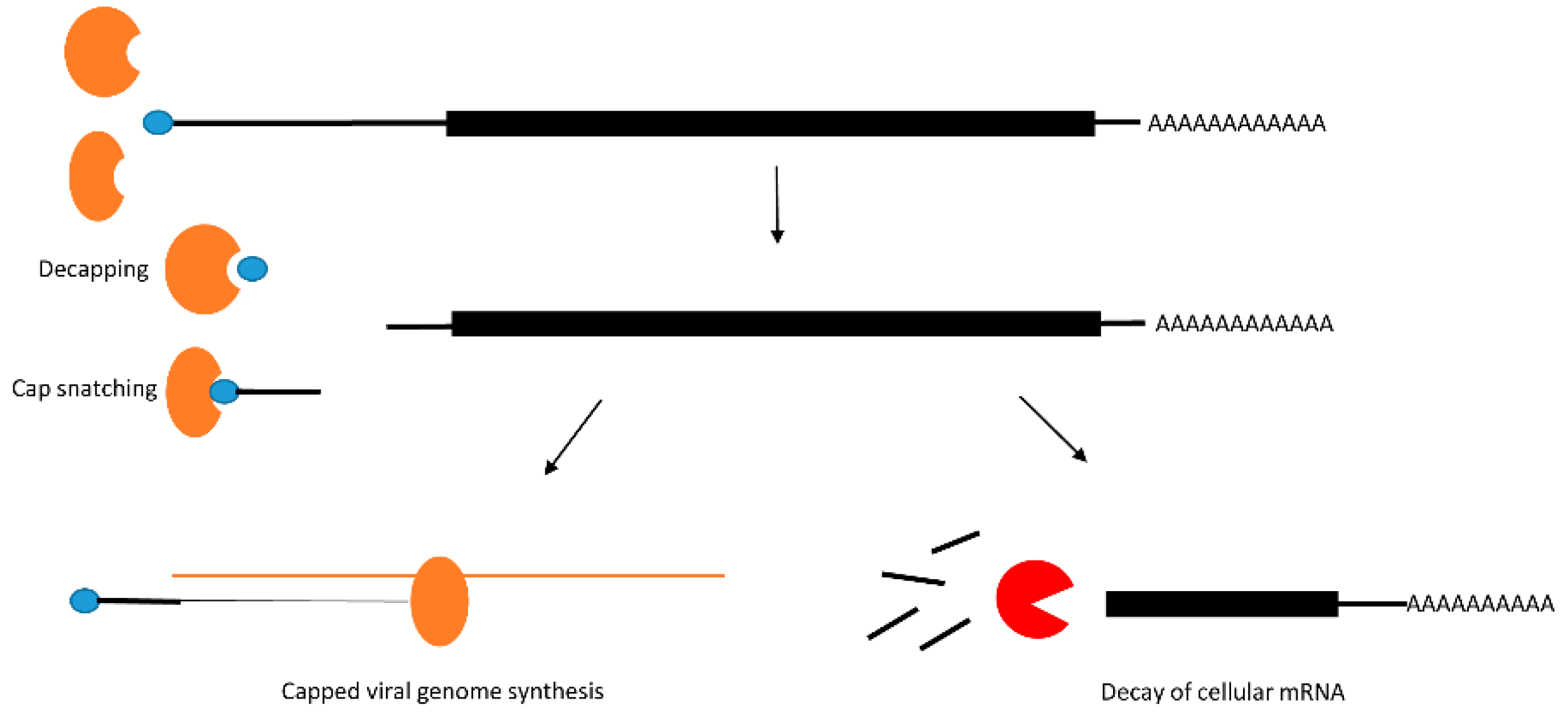
2.2. Post-Transcriptional Modification: Decay of mRNA by Decapping
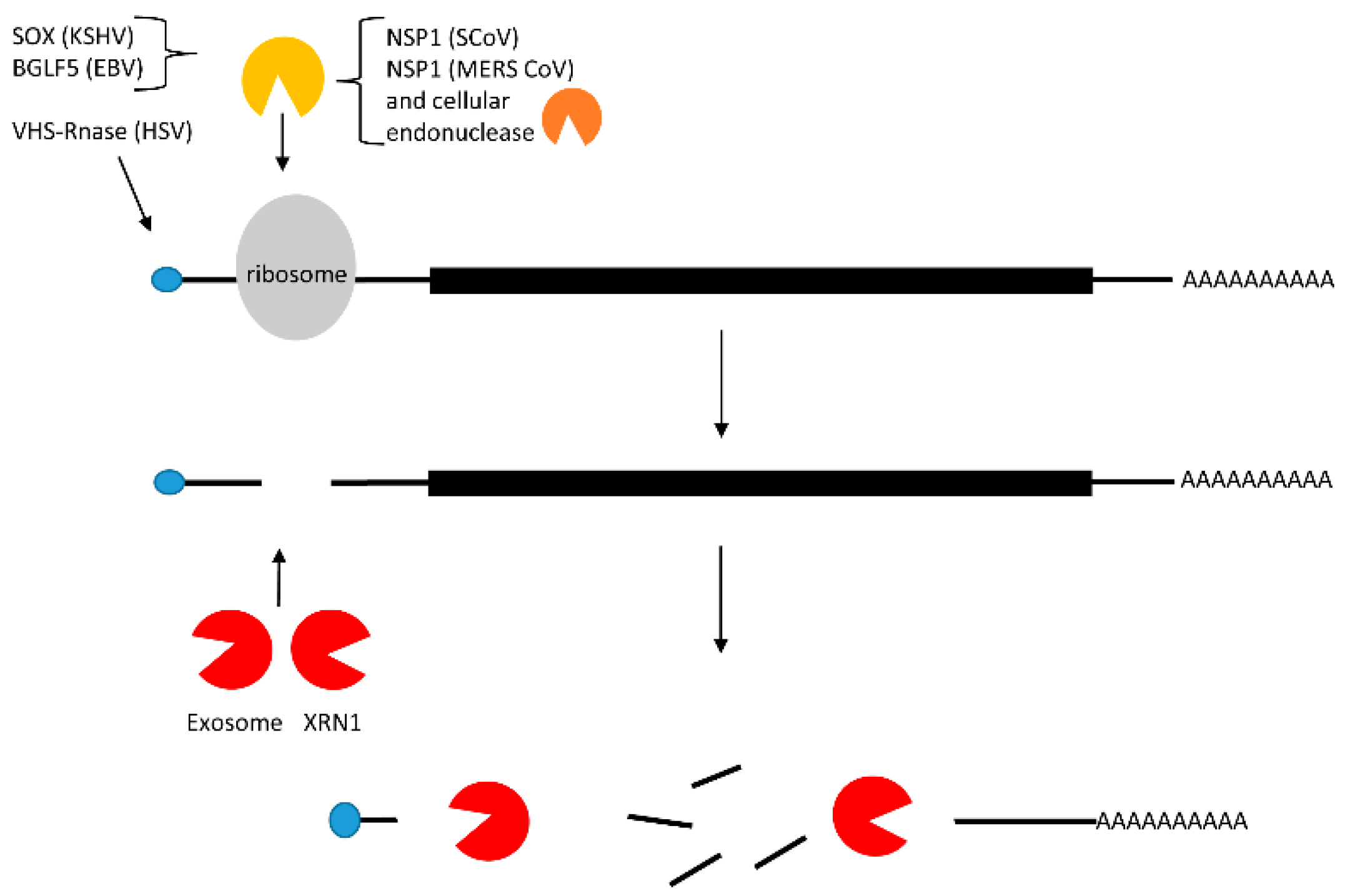
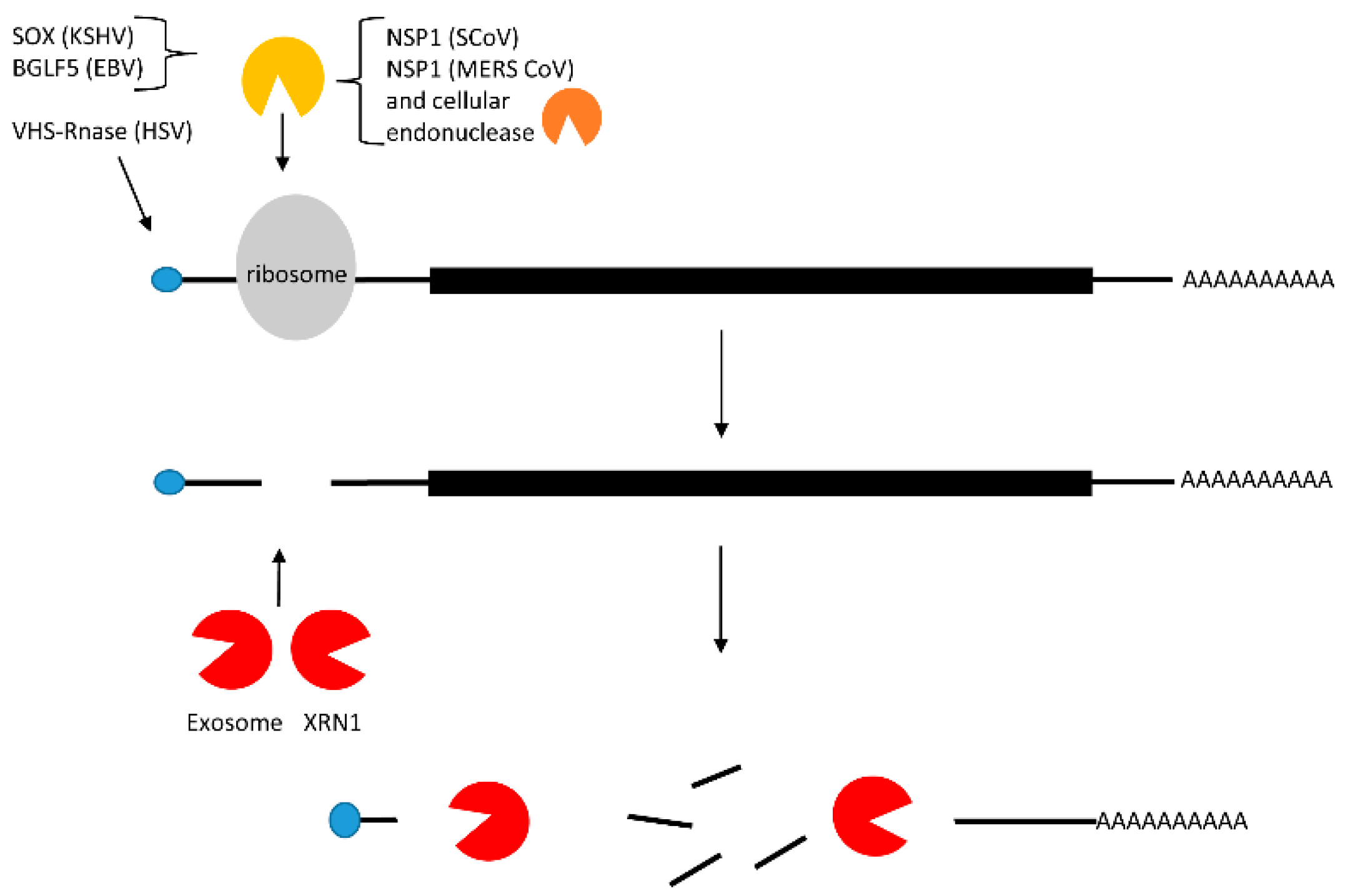
2.3. Translational Inhibition with and without mRNA Cleavage
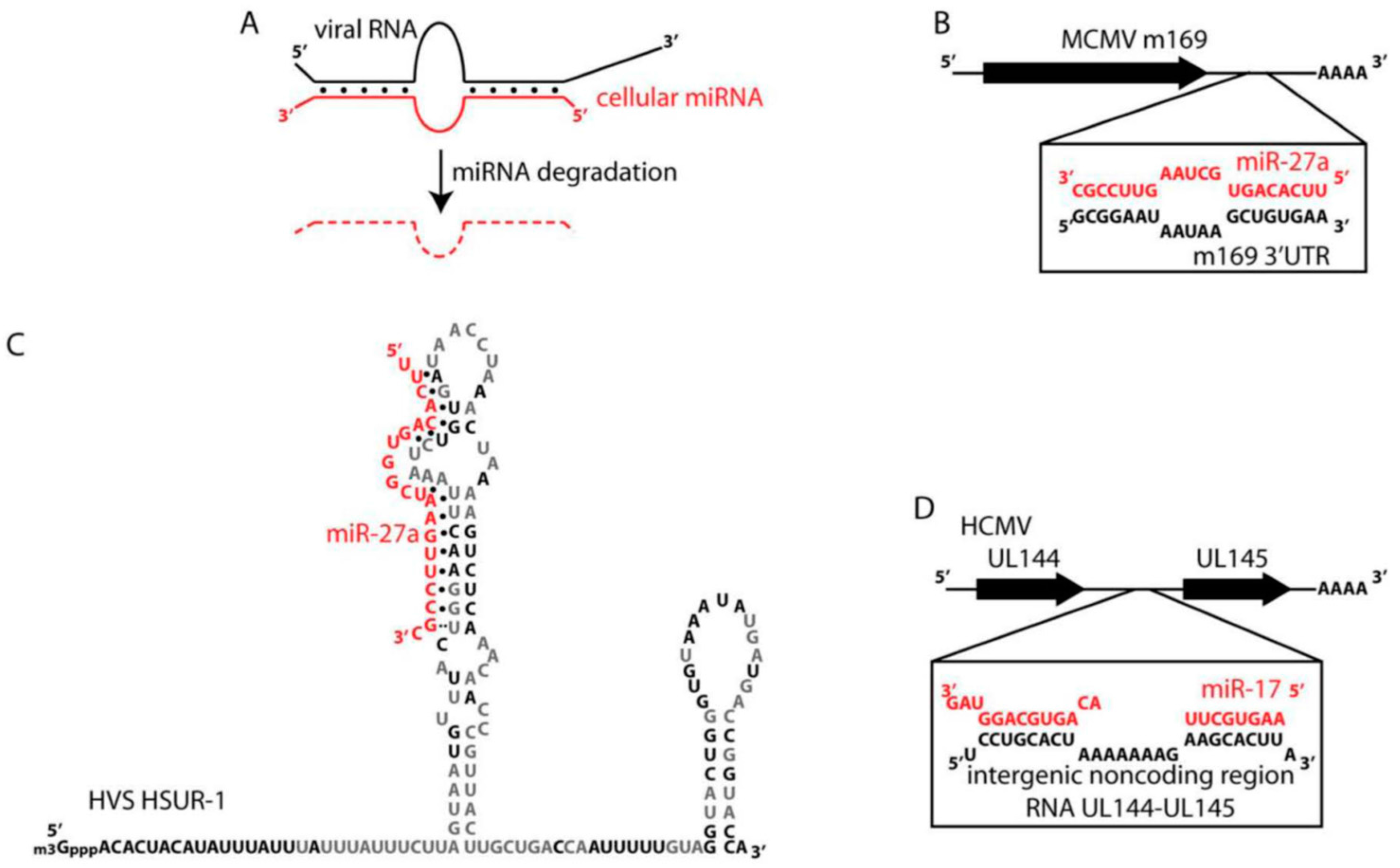
3. Shutting-Down Cellular Small Non-Coding RNAs
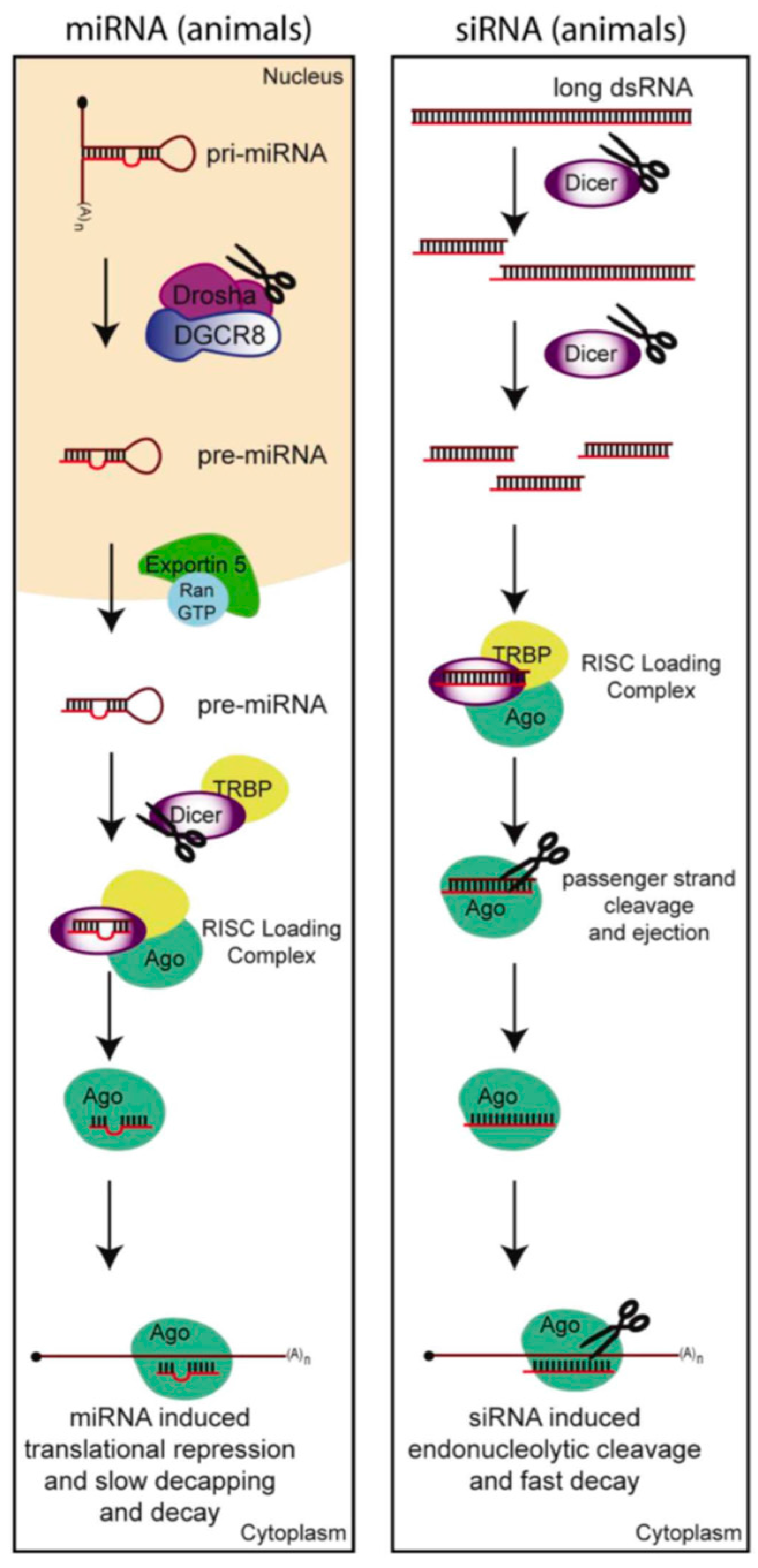
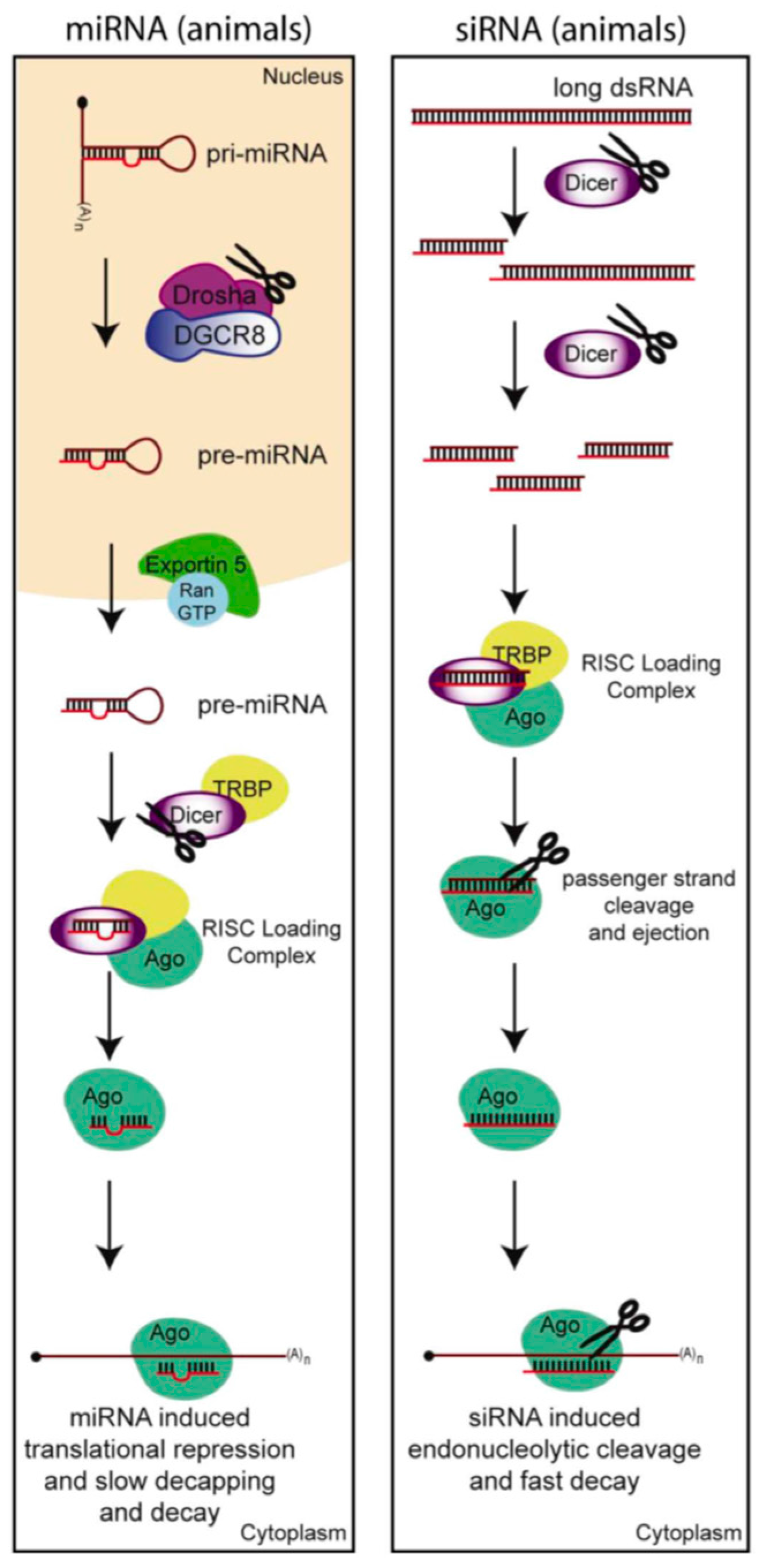
3.1. The Anti-Viral RNA Interference Pathway
3.2. Proposed Anti-Viral Role of Small RNAs in Mammals
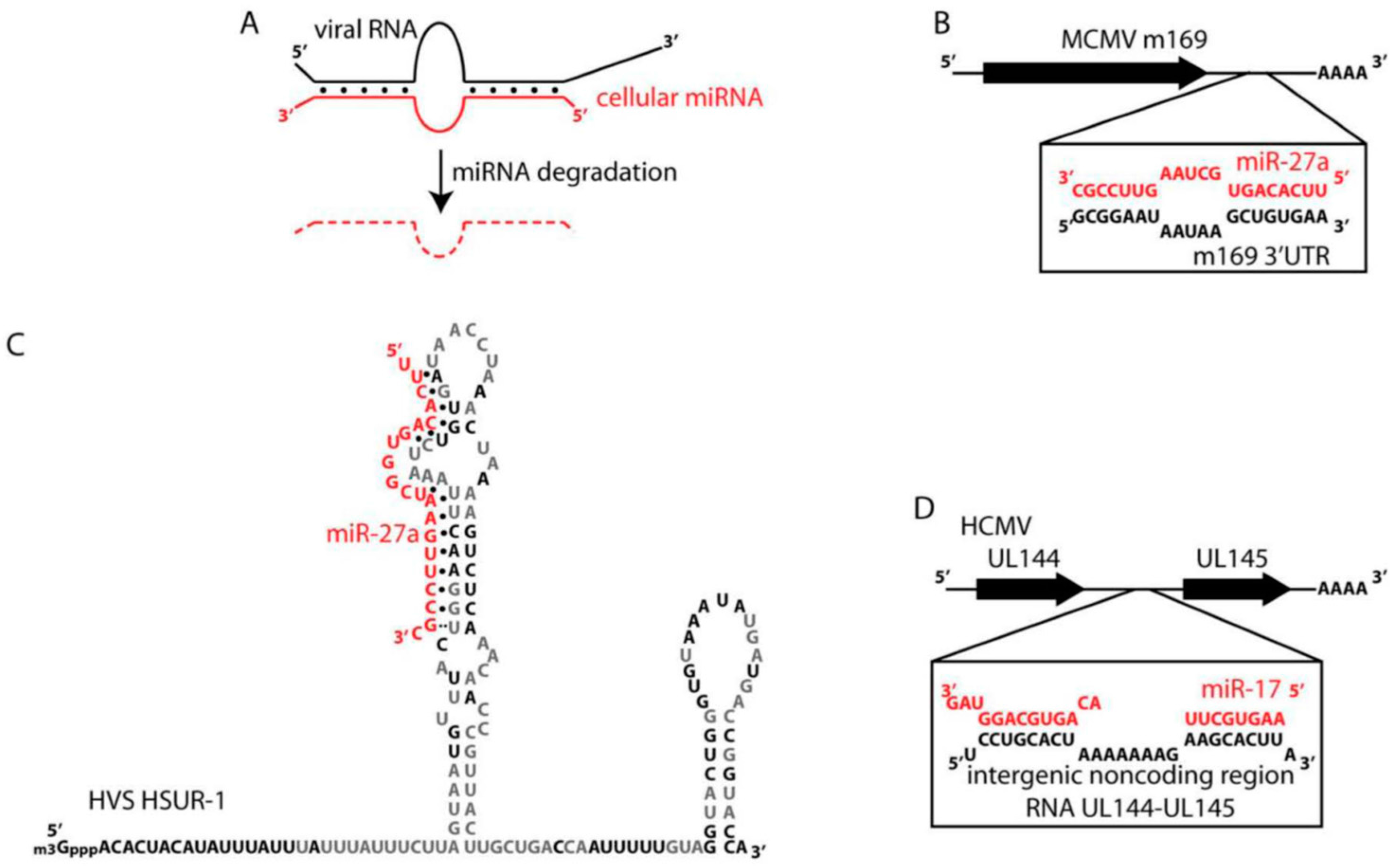
3.3. Suppression of Selected miRNAs by Mammalian Viruses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Benferhat, R.; Josse, T.; Albaud, B.; Gentien, D.; Mansuroglu, Z.; Marcato, V.; Souès, S.; Le Bonniec, B.; Bouloy, M.; Bonnefoy, E. Large-scale chromatin immunoprecipitation with promoter sequence microarray analysis of the interaction of the NSs protein of Rift Valley fever virus with regulatory DNA regions of the host genome. J. Virol. 2012, 86, 11333–11344. [Google Scholar] [CrossRef] [PubMed]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Covarrubias, S.; Wong, W.; Glaunsinger, B.A. A common strategy for host RNA degradation by divergent viruses. J. Virol. 2012, 86, 9527–9530. [Google Scholar] [CrossRef] [PubMed]
- Lyles, D.S. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol. Mol. Biol. Rev. 2000, 64, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Raychaudhuri, S.; Dasgupta, A. Nuclear entry of poliovirus protease-polymerase precursor 3CD: Implications for host cell transcription shut-off. Virology 2004, 320, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Kieft, J.S.; Zhou, K.; Jubin, R.; Doudna, J.A. Mechanism of ribosome recruitment by hepatitis c ires RNA. RNA 2001, 7, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Puckett, S.; Lyles, D.S. Inhibition of host transcription by vesicular stomatitis virus involves a novel mechanism that is independent of phosphorylation of TATA-binding protein (TBP) or association of TBP with TBP-associated factor subunits. J. Virol. 2001, 75, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Dubaele, S.; Proietti De Santis, L.; Billecocq, A.; Bouloy, M.; Egly, J.M. TfIIh transcription factor, a target for the rift valley hemorrhagic fever virus. Cell 2004, 116, 541–550. [Google Scholar] [CrossRef]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the innate immune response: The old world alphavirus nsp2 protein induces rapid degradation of rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef]
- Nag, A.; Narsinh, K.; Martinson, H.G. The poly(a)-dependent transcriptional pause is mediated by CPSF acting on the body of the polymerase. Nat. Struct. Mol. Biol. 2007, 14, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza a virus NS1 protein targets poly(a)-binding protein II of the cellular 3'-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Krug, R.M. U6atac snRNA, the highly divergent counterpart of U6 snRNA, is the specific target that mediates inhibition of AT-AC splicing by the influenza virus NS1 protein. RNA 1998, 4, 55–64. [Google Scholar] [PubMed]
- Yang, Y.C.; Chang, L.K. Role of TAF4 in transcriptional activation by rta of epstein-barr virus. PLoS ONE 2013, 8, e54075. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M.; DeLuca, N.A. Temporal association of herpes simplex virus ICP4 with cellular complexes functioning at multiple steps in polII transcription. PLoS ONE 2013, 8, e78242. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Makino, S. Interplay between viruses and host mRNA degradation. Biochim. Biophys. Acta 2013, 1829, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, K.; Cherry, S. Bunyaviral cap-snatching vs. decapping: Recycling cell cycle mRNAs. Cell Cycle 2013, 12, 3711–3712. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Wartchow, C.; Shia, S.; Uehara, K.; Steffek, M.; Warne, R.; Sutton, J.; Muiru, G.T.; Leonard, V.H.; Bussiere, D.E.; et al. Molecular basis of mRNA cap recognition by influenza B polymerase PB2 subunit. J. Biol. Chem. 2016, 291, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.W.; Katsafanas, G.C.; Liu, R.; Wyatt, L.S.; Moss, B. Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses. Cell. Host Microbe 2015, 17, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Shors, T.; Keck, J.G.; Moss, B. Down regulation of gene expression by the vaccinia virus D10 protein. J. Virol 1999, 73, 791–796. [Google Scholar] [PubMed]
- Abernathy, E.; Glaunsinger, B. Emerging roles for RNA degradation in viral replication and antiviral defense. Virology 2015, 479–480, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, S.; Richner, J.M.; Clyde, K.; Lee, Y.J.; Glaunsinger, B.A. Host shutoff is a conserved phenotype of gammaherpesvirus infection and is orchestrated exclusively from the cytoplasm. J. Virol. 2009, 83, 9554–9566. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Chavez, L.; Ganem, D. The exonuclease and host shutoff functions of the SOX protein of kaposi's sarcoma-associated herpesvirus are genetically separable. J. Virol. 2005, 79, 7396–7401. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive epstein-barr virus infection is mediated by BG1F5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Taddeo, B.; Zhang, W.; Roizman, B. Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the ul47 tegument protein. Proc. Natl. Acad. Sci. USA 2013, 110, E1669–E1675. [Google Scholar] [CrossRef] [PubMed]
- Elgadi, M.M.; Hayes, C.E.; Smiley, J.R. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 1999, 73, 7153–7164. [Google Scholar] [PubMed]
- Huang, C.; Lokugamage, K.G.; Rozovics, J.M.; Narayanan, K.; Semler, B.L.; Makino, S. SARS coronavirus NSP1 protein induces template-dependent endonucleolytic cleavage of mRNAs: Viral mRNAs are resistant to NSP1-induced RNA cleavage. PLoS Pathog. 2011, 7, e1002433. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, W.; Narayanan, K.; Huang, C.; Lokugamage, K.; Ikegami, T.; Ito, N.; Kubo, H.; Makino, S. Severe acute respiratory syndrome coronavirus NSP1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 12885–12890. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, W.; Huang, C.; Narayanan, K.; Lokugamage, K.G.; Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus NSP1 protein. Nat. Struct. Mol. Biol. 2009, 16, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, K.G.; Narayanan, K.; Huang, C.; Makino, S. Severe acute respiratory syndrome coronavirus protein NSP1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol. 2012, 86, 13598–13608. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, K.G.; Narayanan, K.; Nakagawa, K.; Terasaki, K.; Ramirez, S.I.; Tseng, C.T.; Makino, S. Middle east respiratory syndrome coronavirus NSP1 inhibits host gene expression by selectively targeting mRNAs transcribed in the nucleus while sparing mRNAs of cytoplasmic origin. J. Virol. 2015, 89, 10970–10981. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Ramirez, S.I.; Lokugamage, K.G.; Makino, S. Coronavirus nonstructural protein 1: Common and distinct functions in the regulation of host and viral gene expression. Virus Res. 2015, 202, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, S.; Gaglia, M.M.; Kumar, G.R.; Wong, W.; Jackson, A.O.; Glaunsinger, B.A. Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the mammalian exonuclease XRN1. PLoS Pathog. 2011, 7, e1002339. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.R.; Glaunsinger, B.A. Nuclear import of cytoplasmic poly(a) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol. Cell Biol. 2010, 30, 4996–5008. [Google Scholar] [CrossRef] [PubMed]
- Park, R.; El-Guindy, A.; Heston, L.; Lin, S.F.; Yu, K.P.; Nagy, M.; Borah, S.; Delecluse, H.J.; Steitz, J.; Miller, G. Nuclear translocation and regulation of intranuclear distribution of cytoplasmic poly(a)-binding protein are distinct processes mediated by two epstein barr virus proteins. PLoS ONE 2014, 9, e92593. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Darricarrère, N.; Darnell, A.; Myoung, J.; Steitz, J.A. A viral nuclear noncoding RNA binds re-localized poly(a) binding protein and is required for late kshv gene expression. PLoS Pathog. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.D.; Moon, S.L.; Emch, A.W.; Wilusz, C.J.; Wilusz, J. Changes in cellular mRNA stability, splicing, and polyadenylation through hur protein sequestration by a cytoplasmic RNA virus. Cell. Rep. 2013, 5, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Falk, S.; Weir, J.R.; Hentschel, J.; Reichelt, P.; Bonneau, F.; Conti, E. The molecular architecture of the tramp complex reveals the organization and interplay of its two catalytic activities. Mol. Cell 2014, 55, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Conrad, N.K.; Steitz, J.A. A kaposi’s sarcoma virus RNA element that increases the nuclear abundance of intronless transcripts. EMBO J. 2005, 24, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Conrad, N.K.; Mili, S.; Marshall, E.L.; Shu, M.D.; Steitz, J.A. Identification of a rapid mammalian deadenylation-dependent decay pathway and its inhibition by a viral RNA element. Mol. Cell 2006, 24, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Mitton-Fry, R.M.; DeGregorio, S.J.; Wang, J.; Steitz, T.A.; Steitz, J.A. Poly(a) tail recognition by a viral RNA element through assembly of a triple helix. Science 2010, 330, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Burmeister, W.P.; Boer, I.G.; van Leeuwen, D.; Buisson, M.; Gorbalenya, A.E.; Wiertz, E.J.; Ressing, M.E. The “Bridge” In the epstein-barr virus alkaline exonuclease protein BG1F5 contributes to shutoff activity during productive infection. J. Virol. 2012, 86, 9175–9187. [Google Scholar] [CrossRef] [PubMed]
- Clyde, K.; Glaunsinger, B.A. Deep sequencing reveals direct targets of gammaherpesvirus-induced mRNA decay and suggests that multiple mechanisms govern cellular transcript escape. PLoS ONE 2011, 6, e19655. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Taddeo, B.; Roizman, B. Tristetraprolin recruits the herpes simplex virion host shutoff RNase to AU-rich elements in stress response mRNAs to enable their cleavage. J. Virol. 2015, 89, 5643–5650. [Google Scholar] [CrossRef] [PubMed]
- Saffran, H.A.; Read, G.S.; Smiley, J.R. Evidence for translational regulation by the herpes simplex virus virion host shutoff protein. J. Virol. 2010, 84, 6041–6049. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.L.; Barnhart, M.D.; Wilusz, J. Inhibition and avoidance of mRNA degradation by RNA viruses. Curr. Opin. Microbiol. 2012, 15, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V.; van Boom, J.H.; Filippov, D.; Wimmer, E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature 1998, 393, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Leen, E.N.; Sorgeloos, F.; Correia, S.; Chaudhry, Y.; Cannac, F.; Pastore, C.; Xu, Y.; Graham, S.C.; Matthews, S.J.; Goodfellow, I.G.; et al. Correction: A conserved interaction between a C-terminal motif in norovirus VPg and the heat-1 domain of eIF4G is essential for translation initiation. PLoS Pathog. 2016, 12, e1005509. [Google Scholar] [CrossRef] [PubMed]
- Thibault, P.A.; Huys, A.; Amador-Cañizares, Y.; Gailius, J.E.; Pinel, D.E.; Wilson, J.A. Regulation of hepatitis C virus genome replication by XRN1, and microRNA-122 binding to individual sites in the 5′ UTR. J. Virol. 2015, 89, 6294–6311. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant. Cell. 1990, 2, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Romano, N.; Macino, G. Quelling: Transient inactivation of gene expression in Neurospora crassa by transformation with homologous sequences. Mol. Microbiol. 1992, 6, 3343–3353. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.M.; Andrewski, M.D.; Roossinck, M.J.; Keller, N.P. Aspergillus mycoviruses are targets and suppressors of RNA silencing. Eukaryot Cell. 2008, 7, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Segers, G.C.; Zhang, X.; Deng, F.; Sun, Q.; Nuss, D.L. Evidence that RNA silencing functions as an antiviral defense mechanism in fungi. Proc. Natl. Acad. Sci. USA 2007, 104, 12902–12906. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Choi, G.H.; Nuss, D.L. A single argonaute gene is required for induction of RNA silencing antiviral defense and promotes viral RNA recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 17927–17932. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, V.S.; Drake, A.; Chen, J. Virus-specific host miRNAs: Antiviral defenses or promoters of persistent infection? Trends Immunol. 2009, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, Y.R.; Pei, Y.; Lin, S.S.; Tuschl, T.; Patel, D.J.; Chua, N.H. Cucumber mosaic virus-encoded 2B suppressor inhibits arabidopsis Argonaute1 cleavage activity to counter plant defense. Genes Dev. 2006, 20, 3255–3268. [Google Scholar] [CrossRef] [PubMed]
- Csorba, T.; Lozsa, R.; Hutvagner, G.; Burgyan, J. Polerovirus protein P0 prevents the assembly of small RNA-containing risc complexes and leads to degradation of Argonaute1. Plant. J. 2010, 62, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, J.; Garcia, D.; Pontier, D.; Ohnesorge, S.; Yu, A.; Garcia, S.; Braun, L.; Bergdoll, M.; Hakimi, M.A.; Lagrange, T.; et al. Argonaute quenching and global changes in dicer homeostasis caused by a pathogen-encoded gw repeat protein. Genes Dev. 2010, 24, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Giner, A.; Lakatos, L.; Garcia-Chapa, M.; Lopez-Moya, J.J.; Burgyan, J. Viral protein inhibits risc activity by argonaute binding through conserved WG/GW motifs. PLoS Pathog. 2010, 6, e1000996. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Berry, B.; Tassetto, M.; Kunitomi, M.; Acevedo, A.; Deng, C.; Krutchinsky, A.; Gross, J.; Antoniewski, C.; Andino, R. Cricket paralysis virus antagonizes argonaute 2 to modulate antiviral defense in drosophila. Nat. Struct. Mol. Biol. 2010, 17, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Glick, E.; Zrachya, A.; Levy, Y.; Mett, A.; Gidoni, D.; Belausov, E.; Citovsky, V.; Gafni, Y. Interaction with host sgs3 is required for suppression of RNA silencing by tomato yellow leaf curl virus V2 protein. Proc. Natl. Acad. Sci. USA 2008, 105, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Pendon, J.A.; Li, F.; Li, W.X.; Ding, S.W. Suppression of antiviral silencing by cucumber mosaic virus 2B protein in arabidopsis is associated with drastically reduced accumulation of three classes of viral small interfering RNAs. Plant. Cell. 2007, 19, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Kobori, T.; Kosaka, Y.; Natsuaki, T.; Masuta, C. Characterization of silencing suppressor 2B of cucumber mosaic virus based on examination of its small RNA-binding abilities. Plant. Cell. Physiol. 2007, 48, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, X.; Ding, S.W. Viral suppressors of RNA-based viral immunity: Host targets. Cell. Host Microbe 2010, 8, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, L.; Csorba, T.; Pantaleo, V.; Chapman, E.J.; Carrington, J.C.; Liu, Y.P.; Dolja, V.V.; Calvino, L.F.; Lopez-Moya, J.J.; Burgyan, J. Small RNA binding is a common strategy to suppress RNA silencing by several viral suppressors. EMBO J. 2006, 25, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, W.J.; Kreuze, J.F.; Rajamaki, M.L.; Cruzado, K.R.; Untiveros, M.; Valkonen, J.P. Elimination of antiviral defense by viral RNase III. Proc. Natl. Acad. Sci. USA 2009, 106, 10354–10358. [Google Scholar] [CrossRef] [PubMed]
- Aliyari, R.; Wu, Q.; Li, H.W.; Wang, X.H.; Li, F.; Green, L.D.; Han, C.S.; Li, W.X.; Ding, S.W. Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in drosophila. Cell. Host Microbe 2008, 4, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Segers, G.C.; van Wezel, R.; Zhang, X.; Hong, Y.; Nuss, D.L. Hypovirus papain-like protease p29 suppresses RNA silencing in the natural fungal host and in a heterologous plant system. Eukaryot Cell. 2006, 5, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Zhang, Z.; Liu, Y. RNA interference pathways in fungi: Mechanisms and functions. Annu. Rev. Microbiol. 2012, 66, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Paddison, P.J.; Caudy, A.A.; Hannon, G.J. Stable suppression of gene expression by RNAi in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Billy, E.; Brondani, V.; Zhang, H.; Muller, U.; Filipowicz, W. Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc. Natl. Acad. Sci. USA 2001, 98, 14428–14433. [Google Scholar] [CrossRef] [PubMed]
- Tam, O.H.; Aravin, A.A.; Stein, P.; Girard, A.; Murchison, E.P.; Cheloufi, S.; Hodges, E.; Anger, M.; Sachidanandam, R.; Schultz, R.M.; et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 2008, 453, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Robalino, J.; Browdy, C.L.; Prior, S.; Metz, A.; Parnell, P.; Gross, P.; Warr, G. Induction of antiviral immunity by double-stranded RNA in a marine invertebrate. J. Virol. 2004, 78, 10442–10448. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R.; Cherry, S.; tenOever, B.R. Is RNA interference a physiologically relevant innate antiviral immune response in mammals? Cell. Host Microbe 2013, 14, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, S.; Sternberg, S.H.; Kellenberger, C.A.; Doudna, J.A. Substrate-specific kinetics of dicer-catalyzed RNA processing. J. Mol. Biol. 2010, 404, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Schafer, A.; Lu, S.; Bilello, J.P.; Desrosiers, R.C.; Edwards, R.; Raab-Traub, N.; Cullen, B.R. Epstein-barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006, 2, e23. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Cullen, B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.V.; Ciaudo, C.; Marchais, A.; Li, Y.; Jay, F.; Ding, S.W.; Voinnet, O. Antiviral RNA interference in mammalian cells. Science 2013, 342, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Tutton, S.; Pierce, E.; Yoon, K. Specific double-stranded RNA interference in undifferentiated mouse embryonic stem cells. Mol. Cell Biol. 2001, 21, 7807–7816. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.S.; Schmid, S.; Aguado, L.C.; Sabin, L.R.; Yasunaga, A.; Shim, J.V.; Sachs, D.; Cherry, S.; tenOever, B.R. Drosha as an interferon-independent antiviral factor. Proc. Natl. Acad. Sci. USA 2014, 111, 7108–7113. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.M.; Langlois, R.A.; TenOever, B.R. Replication in cells of hematopoietic origin is necessary for dengue virus dissemination. PLoS Pathog. 2012, 8, e1002465. [Google Scholar] [CrossRef] [PubMed]
- Langlois, R.A.; Varble, A.; Chua, M.A.; Garcia-Sastre, A.; tenOever, B.R. Hematopoietic-specific targeting of influenza A virus reveals replication requirements for induction of antiviral immune responses. Proc. Natl. Acad. Sci. USA 2012, 109, 12117–12122. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.T.; Pham, A.M.; Lorini, M.H.; Chua, M.A.; Steel, J.; tenOever, B.R. MicroRNA-mediated species-specific attenuation of influenza A virus. Nat. Biotechnol. 2009, 27, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Shapiro, J.S.; Sabin, L.R.; Pham, A.M.; Reyes, I.; Moss, B.; Cherry, S.; tenOever, B.R. Degradation of host microRNAs by poxvirus poly(a) polymerase reveals terminal RNA methylation as a protective antiviral mechanism. Cell. Host Microbe 2012, 12, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Langlois, R.A.; Schmid, S.; Varble, A.; Shim, J.V.; Sachs, D.; tenOever, B.R. The mammalian response to virus infection is independent of small RNA silencing. Cell. Rep. 2014, 8, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Kakumani, P.K.; Ponia, S.S.; S, R.K.; Sood, V.; Chinnappan, M.; Banerjea, A.C.; Medigeshi, G.R.; Malhotra, P.; Mukherjee, S.K.; Bhatnagar, R.K. Role of RNA interference (RNAi) in dengue virus replication and identification of ns4b as an RNAi suppressor. J. Virol. 2013, 87, 8870–8883. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding subgenomic flavivirus RNA: Multiple functions in west nile virus pathogenesis and modulation of host responses. Viruses 2014, 6, 404–427. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.L.; Anderson, J.R.; Kumagai, Y.; Wilusz, C.J.; Akira, S.; Khromykh, A.A.; Wilusz, J. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 2012, 18, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Schnettler, E.; Sterken, M.G.; Leung, J.Y.; Metz, S.W.; Geertsema, C.; Goldbach, R.W.; Vlak, J.M.; Kohl, A.; Khromykh, A.A.; Pijlman, G.P. Noncoding flavivirus RNA displays RNA interference suppressor activity in insect and mammalian cells. J. Virol. 2012, 86, 13486–13500. [Google Scholar] [CrossRef] [PubMed]
- Bennasser, Y.; Le, S.Y.; Benkirane, M.; Jeang, K.T. Evidence that hiv-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 2005, 22, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Li, H.; Lu, R.; Li, F.; Dus, M.; Atkinson, P.; Brydon, E.W.; Johnson, K.L.; Garcia-Sastre, A.; Ball, L.A.; et al. Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. USA 2004, 101, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Haasnoot, J.; de Vries, W.; Geutjes, E.J.; Prins, M.; de Haan, P.; Berkhout, B. The ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007, 3, e86. [Google Scholar] [CrossRef] [PubMed]
- Lecellier, C.H.; Dunoyer, P.; Arar, K.; Lehmann-Che, J.; Eyquem, S.; Himber, C.; Saib, A.; Voinnet, O. A cellular microRNA mediates antiviral defense in human cells. Science 2005, 308, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Sanghvi, V.R.; Steel, L.F. A re-examination of global suppression of RNA interference by hiv-1. PLoS ONE 2011, 6, e17246. [Google Scholar] [CrossRef] [PubMed]
- Lichner, Z.; Silhavy, D.; Burgyan, J. Double-stranded RNA-binding proteins could suppress RNA interference-mediated antiviral defences. J. Gen. Virol. 2003, 84, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza a virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Steitz, J.A. Virus meets host microRNA: The destroyer, the booster, the hijacker. Mol. Cell Biol. 2014, 34, 3780–3787. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.C.; Yu, S.L.; Chen, J.J.; Chang, S.Y.; Yan, B.S.; Hong, Q.S.; Singh, S.; Kao, C.L.; Chen, H.Y.; Su, K.Y.; et al. Enterovirus-induced MIR-141 contributes to shutoff of host protein translation by targeting the translation initiation factor EIF4E. Cell. Host Microbe 2011, 9, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as an orthologue of cellular MIR-155. Nature 2007, 450, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Sullivan, C.S. Virus-encoded microRNAs: An overview and a look to the future. PLoS Pathog. 2012, 8, e1003018. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Luna, J.M.; Liniger, M.; Nishiuchi, E.; Rozen-Gagnon, K.; Shlomai, A.; Auray, G.; Gerber, M.; Fak, J.; Keller, I.; et al. A broad RNA virus survey reveals both miRNA dependence and functional sequestration. Cell Host Microbe 2016, 19, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Scheel, T.K.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis c virus RNA functionally sequesters MIR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Buck, A.H.; Perot, J.; Chisholm, M.A.; Kumar, D.S.; Tuddenham, L.; Cognat, V.; Marcinowski, L.; Dolken, L.; Pfeffer, S. Post-transcriptional regulation of MIR-27 in murine cytomegalovirus infection. RNA 2010, 16, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Cazalla, D.; Yario, T.; Steitz, J.A. Down-regulation of a host microRNA by a herpesvirus saimiri noncoding RNA. Science 2010, 328, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Libri, V.; Helwak, A.; Miesen, P.; Santhakumar, D.; Borger, J.G.; Kudla, G.; Grey, F.; Tollervey, D.; Buck, A.H. Murine cytomegalovirus encodes a MIR-27 inhibitor disguised as a target. Proc. Natl. Acad. Sci. USA 2012, 109, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Song, J.; Kim, S.; Kim, J.; Hong, Y.; Kim, Y.; Kim, D.; Baek, D.; Ahn, K. Selective degradation of host microRNAs by an intergenic hcmv noncoding RNA accelerates virus production. Cell. Host Microbe 2013, 13, 678–690. [Google Scholar] [CrossRef] [PubMed]
- McCaskill, J.; Praihirunkit, P.; Sharp, P.M.; Buck, A.H. RNA-mediated degradation of microRNAs: A widespread viral strategy? RNA Biol. 2015, 12, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Marcinowski, L.; Tanguy, M.; Krmpotic, A.; Radle, B.; Lisnic, V.J.; Tuddenham, L.; Chane-Woon-Ming, B.; Ruzsics, Z.; Erhard, F.; Benkartek, C.; et al. Degradation of cellular MIR-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog. 2012, 8, e1002510. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.E.; Riley, K.J.; Iwasaki, A.; Steitz, J.A. Alternative capture of noncoding RNAs or protein-coding genes by herpesviruses to alter host T cell function. Mol. Cell 2014, 54, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Horwich, M.D.; Hung, J.H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification of Virus (Genome Type) | Family | Virus | Characteristics |
|---|---|---|---|
| I: Double-stranded DNA (dsDNA) | Herpesviridae | Epstein Barr Virus (EBV) | Linear dsDNA genome remains encased in enveloped viral capsid Viral DNA is replicated and transcribed in the cell’s nucleus Life cycle has separate latent and infectious lytic phase |
| Kaposi Sarcoma Herpes Virus (KSHV) | |||
| Herpes Simplex Virus 1 (HSV-1) | |||
| Herpes Virus Saimiri (HVS) | |||
| Cytomegalovirus (CMV) | |||
| Adenoviridae | Nonenveloped nuclear capsid encases dsDNA genome Nuclear replication Early and late phase life cycle | ||
| Poxviridae | Vaccinia virus (VACV) | Enveloped with dsDNA genome Replicates exclusively in the cytoplasm | |
| III: Double-stranded RNA (dsRNA) | Reoviridae | Rotavirus | Nonenveloped virion with linear dsRNA genome Replication is cytoplasmic |
| IV: Single-stranded (+) sense RNA ((+) ssRNA) | Coronaviridae | Human Coronavirus (HCoV) | Spiked, enveloped virion with nucleocapsid ssRNA is capped and polyadenylated Replication is cytoplasmic |
| Severe Acute Respiratory Syndrome Corona Virus (SARS CoV) | |||
| Middle East respiratory syndrome corona virus (MERS CoV) | |||
| Picornaviridae | Poliovirus | Nonenveloped virion with linear ssRNA genome mRNA is protected by 5’ linkage to VpG protein, polyadenylated | |
| Encephalomyocarditis virus (EMCV) | |||
| Caliciviridae | Nonenveloped virion with linear RNA genome mRNA is protected by 5’ linkage to VpG protein, polyadenylated Replication is cytoplasmic | ||
| Flaviviridae | Hepatitis C Virus (HCV) | Enveloped virion mRNA is without a poly(A) tail but can be capped (WNV and DENV) or uncapped (HCV) Replication is cytoplasmic | |
| Dengue Virus (DENV) | |||
| West Nile Virus (WNV) | |||
| Togaviridae | Sindbis Virus (SINV) | Enveloped virion mRNA is capped, polyadenylated Replication is cytoplasmic | |
| Nodaviridae | Nodamura Virus (NoV) | Non-enveloped virion with linear RNA genome mRNA is capped but not polyadenylated Replication is cytoplasmic | |
| V: Single-stranded (-) sense RNA ((-) RNA) | Bunyaviridae | Rift Valley Fever Virus (RVFV) | Negative stranded enveloped virus with RNA genome mRNA is capped by cap snatching but is not polyadenylated Replication is cytoplasmic |
| Orthomyxoviridae | Influenza Virus | Enveloped virion contains linear ssRNA genome mRNA is capped by cap snatching and polyadenylated Replication is nuclear | |
| Rhabdoviridae | Vesicular Somatic Virus (VSV) | Enveloped virus with linear ssRNA genome mRNA is capped and polyadenylated Replication is cytoplasmic | |
| Filoviridae | Ebola virus | Filamentous virus mRNA is capped and polyadenylated Replication is cytoplasmic | |
| VI: Single-stranded (+) sense RNA with DNA intermediate in life-cycle | Retroviridae | HIV-1 (human immunodeficiency virus type 1) | Enveloped virion contains two ssRNAs that are capped and polyadenylated Replication is nuclear |
| Primate foamy virus |
| Type of mRNA Suppression | Virus | Family | Viral protein and mechanism |
|---|---|---|---|
| mRNA Transcription | poliovirus | Picornaviridae | Protease 3C mediated cleavage of TFIID |
| VSV | Rhabdoviridae | Protein M mediated inhibition of TFIID | |
| RVFV | Bunyaviridae | NSs mediated inhibition of TFIIH | |
| Influenza | Orthomyxoviridae | NS1 blocks RNA processing by CPSF, PAB, U6 RNA | |
| Post-transcription suppression of mRNA | RVFV | Bunyaviridae | Nucleocapsid protein mediated 5′-cap removal |
| Influenza | Orthomyxoviridae | PB2 mediated 5′-cap removal, PA mediated RNA cleavage | |
| Poxvirus SINV | Poxviridae Togaviridae | D9 and D10 mediated decapping of mRNA HuR capture by viral 3′UTR | |
| Translational inhibition with and without mRNA cleavage | KSHV | Herpesviridae | SOX mediated mRNA cleavage |
| EBV | Herpesviridae | BGLF5 mediated mRNA cleavage | |
| HSV | Herpesviridae | HSV-1 RNase mediated mRNA cleavage | |
| HCoV | Coronaviridae | NSP1 mediated ribosome stalling | |
| SARS CoV | Coronaviridae | NSP1 mediated ribosome stalling followed by cleavage by unknown cellular protein | |
| MERSCoV | Coronaviridae | NSP1 mediated ribosome stalling and cleavage by unknown nuclease |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbert, K.M.; Nag, A. A Tale of Two RNAs during Viral Infection: How Viruses Antagonize mRNAs and Small Non-Coding RNAs in The Host Cell. Viruses 2016, 8, 154. https://doi.org/10.3390/v8060154
Herbert KM, Nag A. A Tale of Two RNAs during Viral Infection: How Viruses Antagonize mRNAs and Small Non-Coding RNAs in The Host Cell. Viruses. 2016; 8(6):154. https://doi.org/10.3390/v8060154
Chicago/Turabian StyleHerbert, Kristina M., and Anita Nag. 2016. "A Tale of Two RNAs during Viral Infection: How Viruses Antagonize mRNAs and Small Non-Coding RNAs in The Host Cell" Viruses 8, no. 6: 154. https://doi.org/10.3390/v8060154
APA StyleHerbert, K. M., & Nag, A. (2016). A Tale of Two RNAs during Viral Infection: How Viruses Antagonize mRNAs and Small Non-Coding RNAs in The Host Cell. Viruses, 8(6), 154. https://doi.org/10.3390/v8060154