
Comparative Analysis of HaSNPV-AC53 and Derived Strains


Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Source and Passage
2.2. Test for Latent Virus
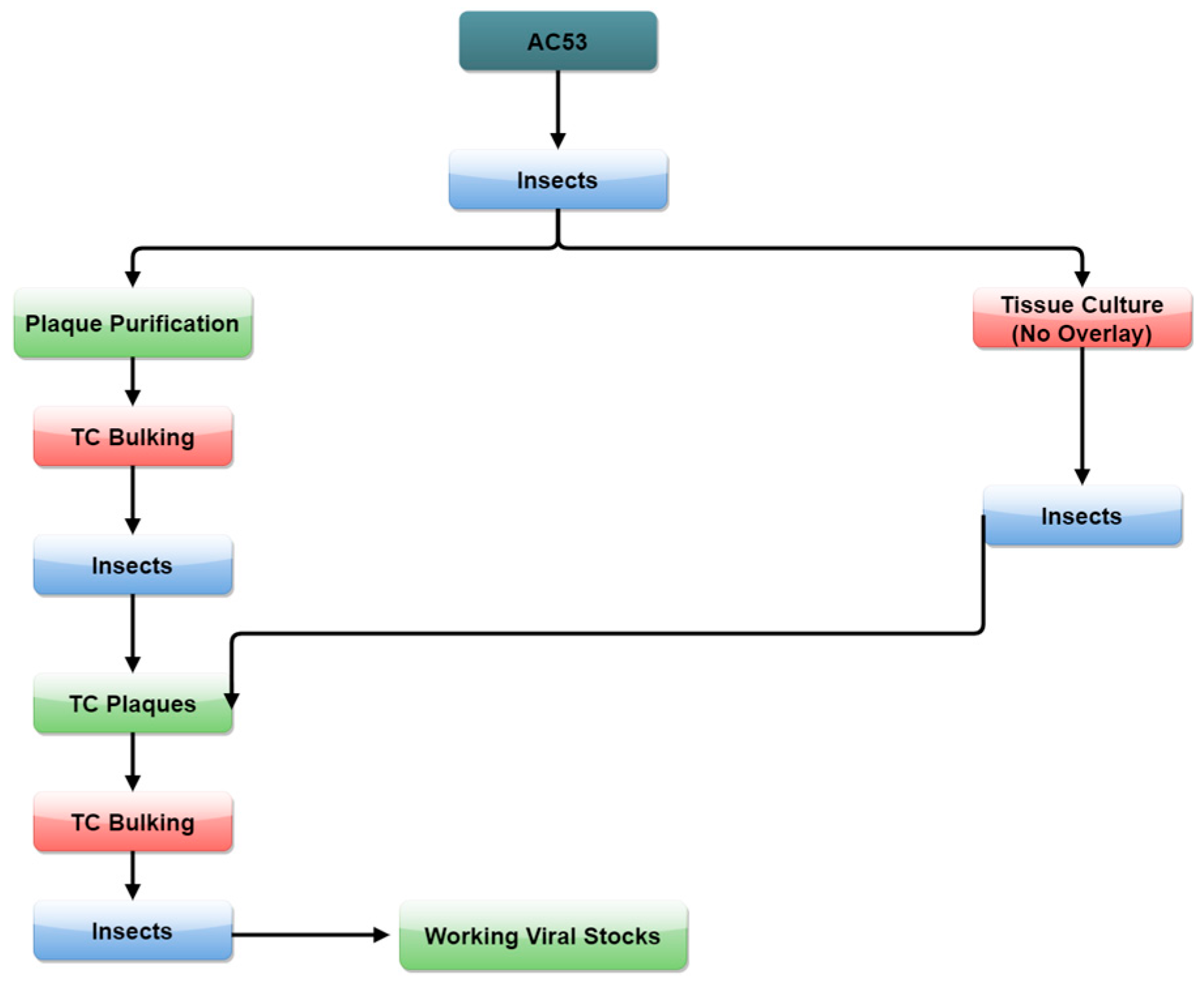
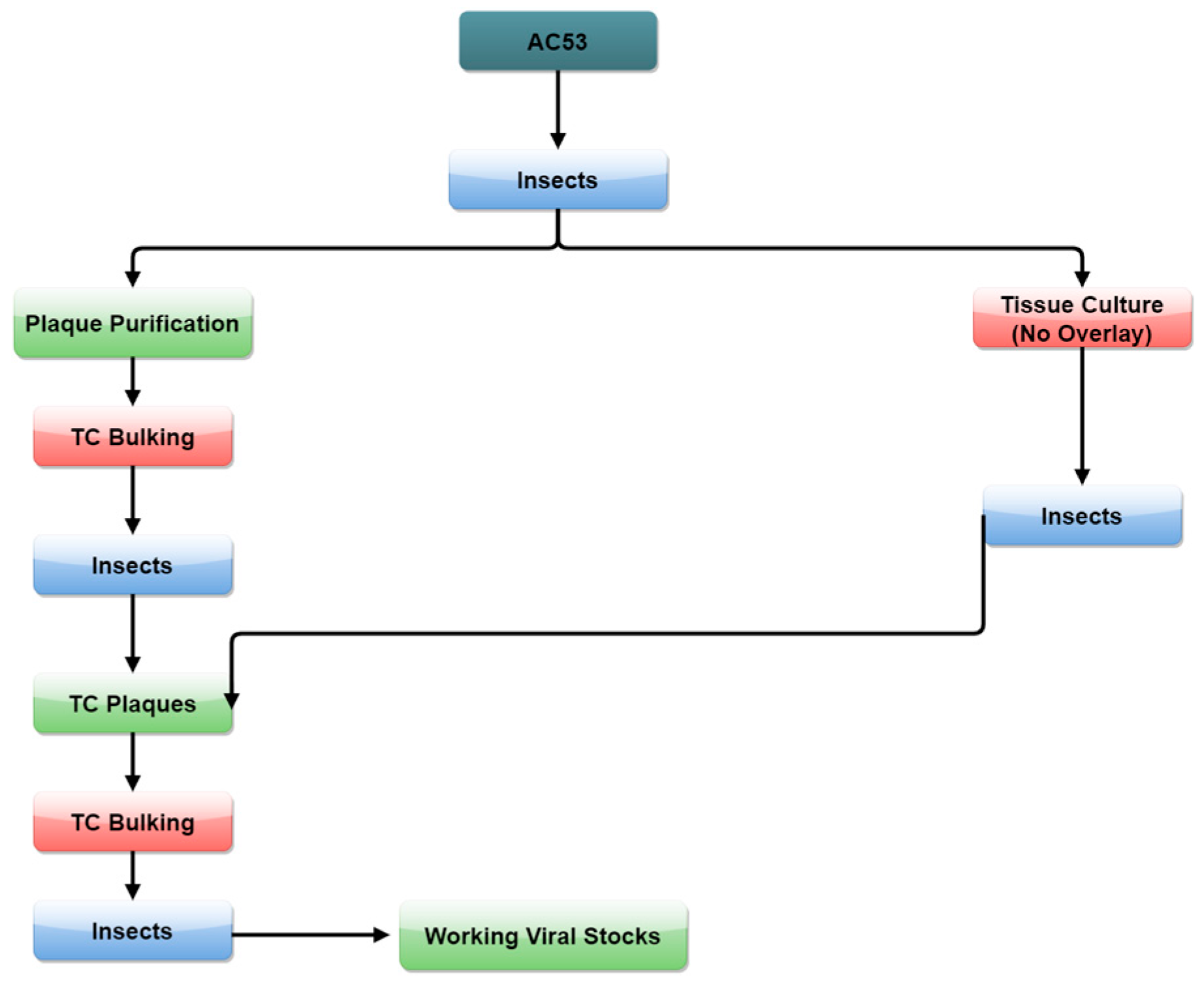
2.3. Strain Isolation by Passage and Selection in Tissue Culture
2.4. DNA Extraction, Next Generation Sequencing Library Preparation, Sequencing and Genome Assembly
2.5. Sequence Analysis and Maximum-Likelihood Estimation (MLE)
3. Results
3.1. Test for Latent Virus
3.2. Strain Isolation
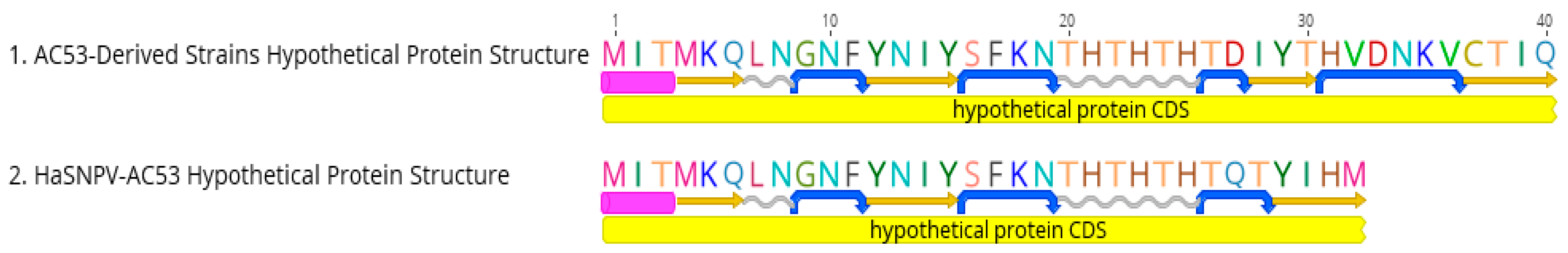
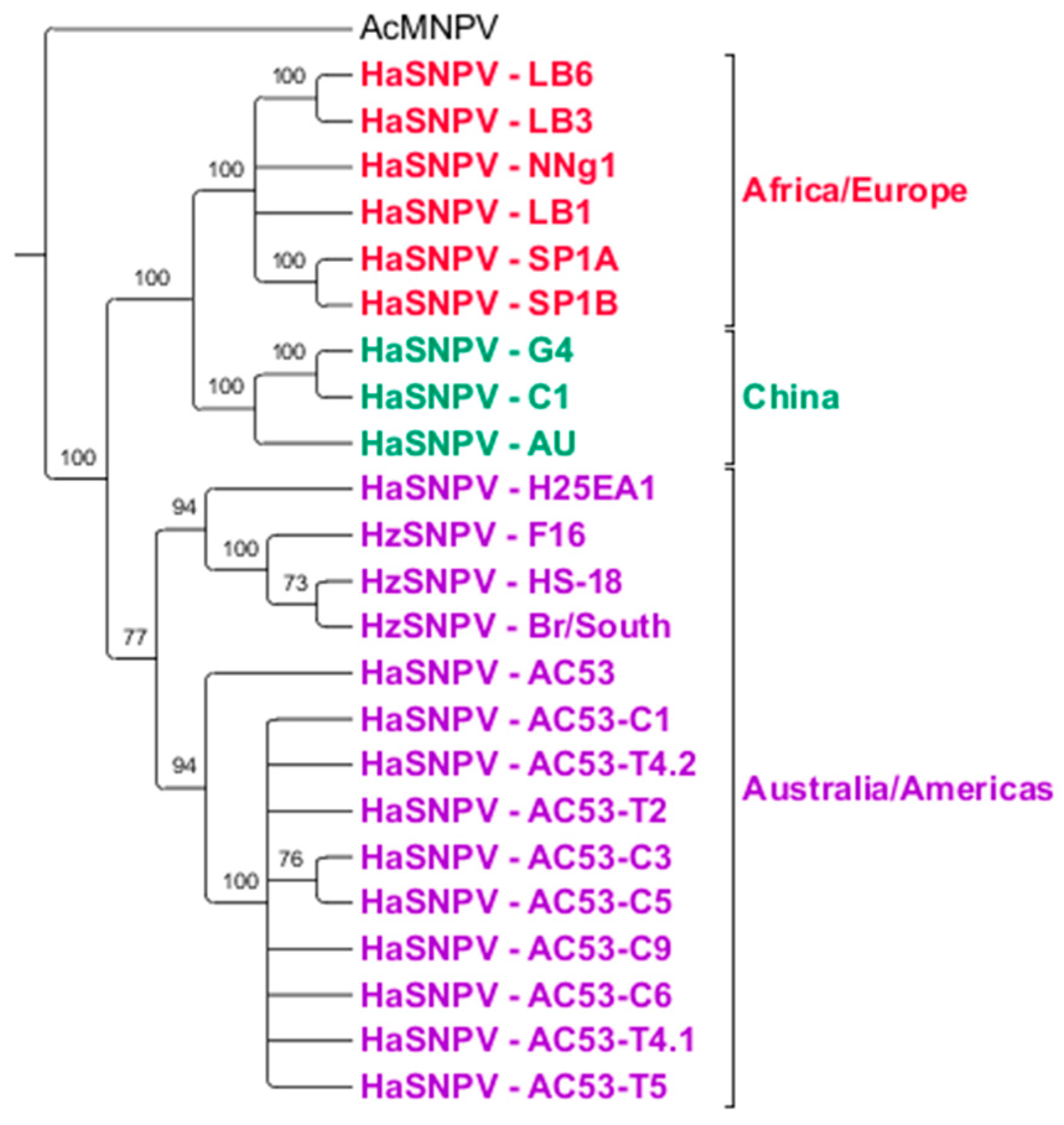
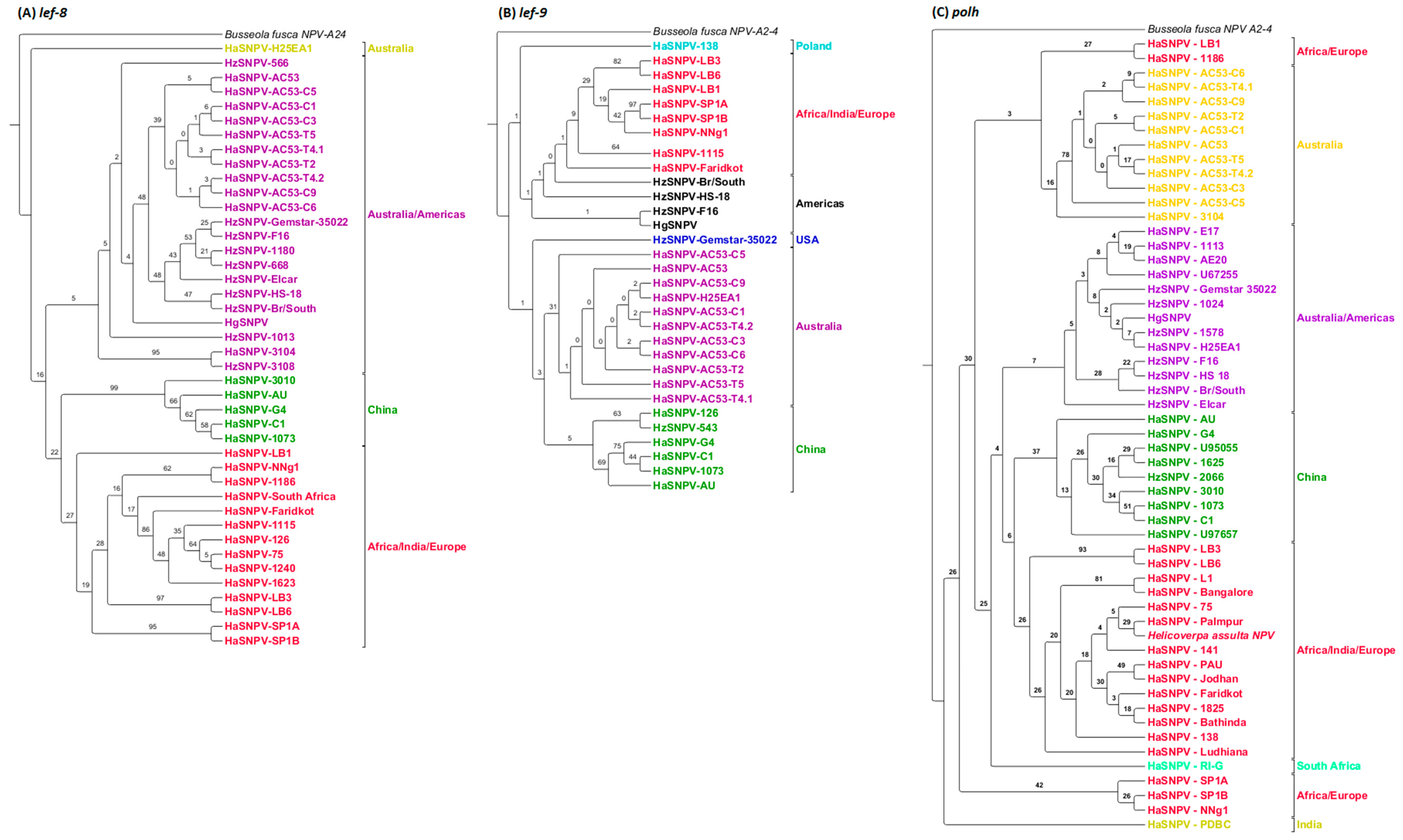
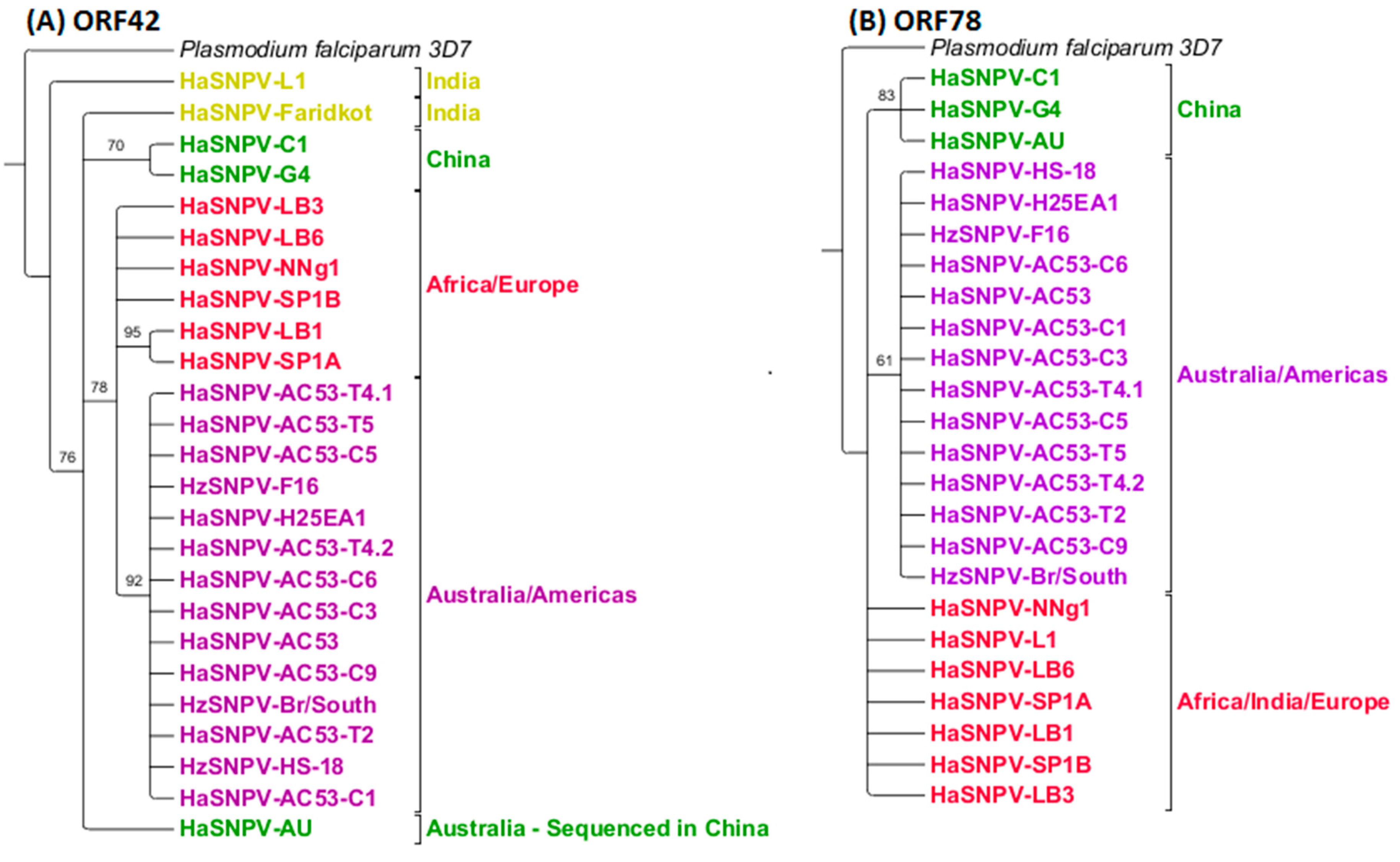
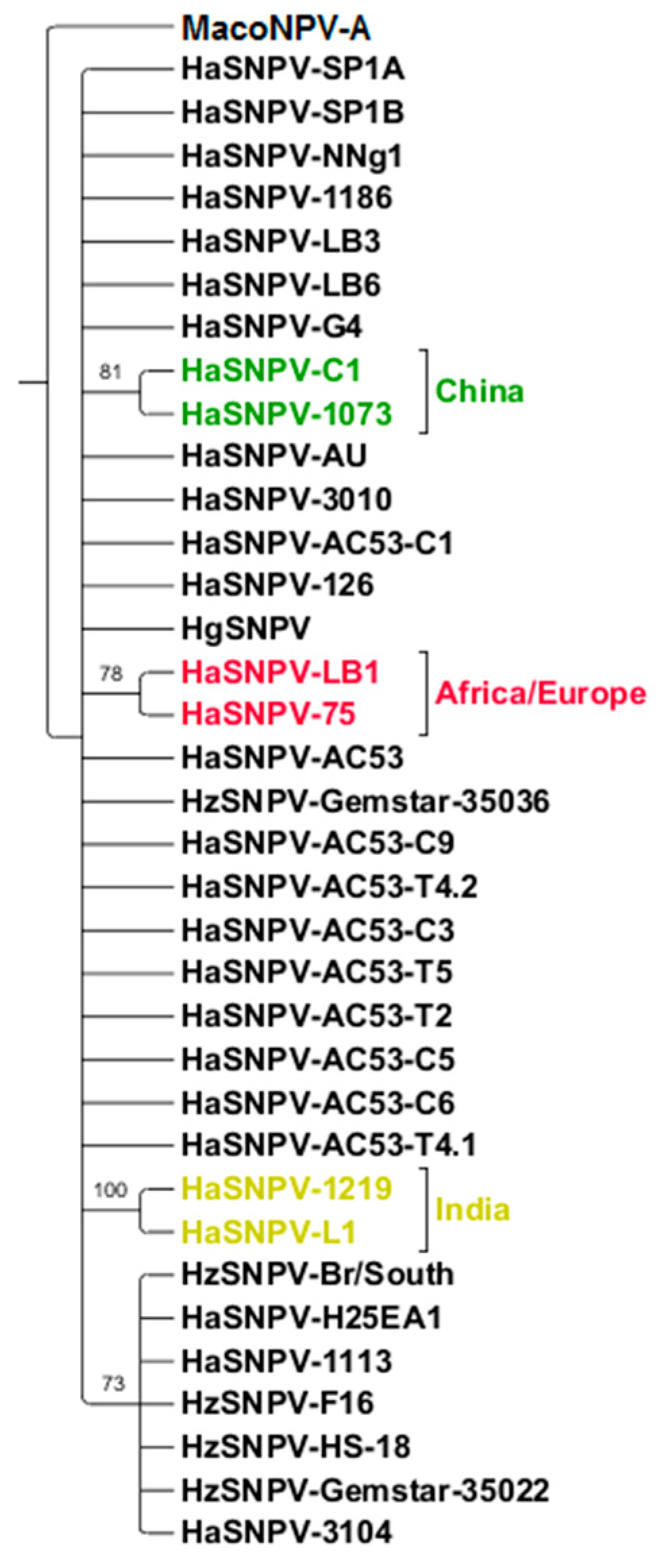
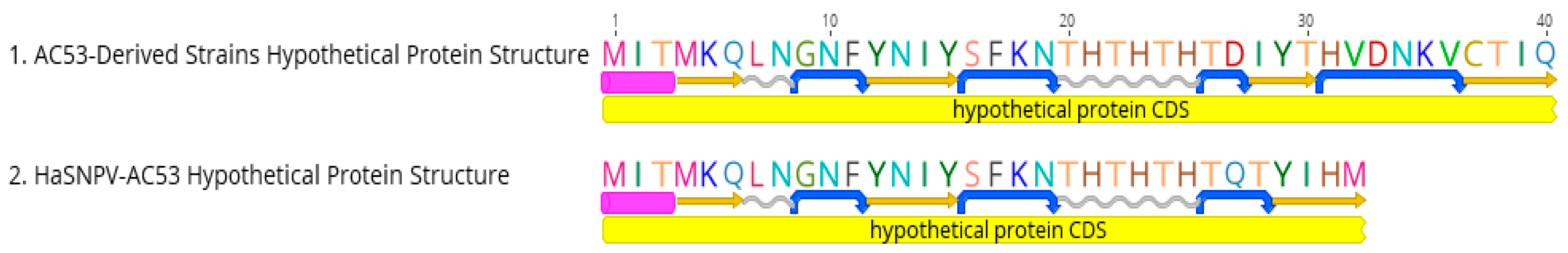
3.3. Sequence Analysis
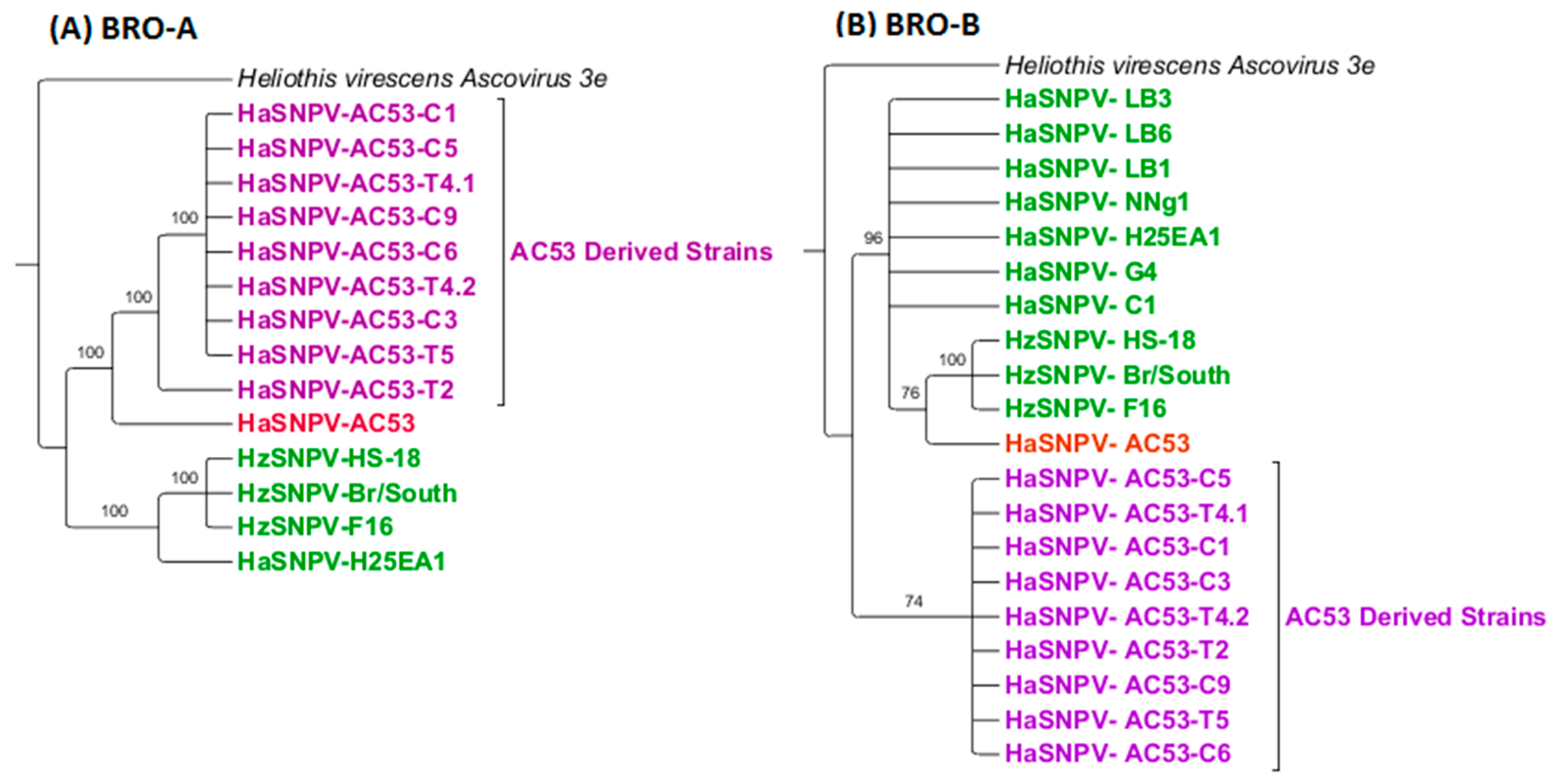
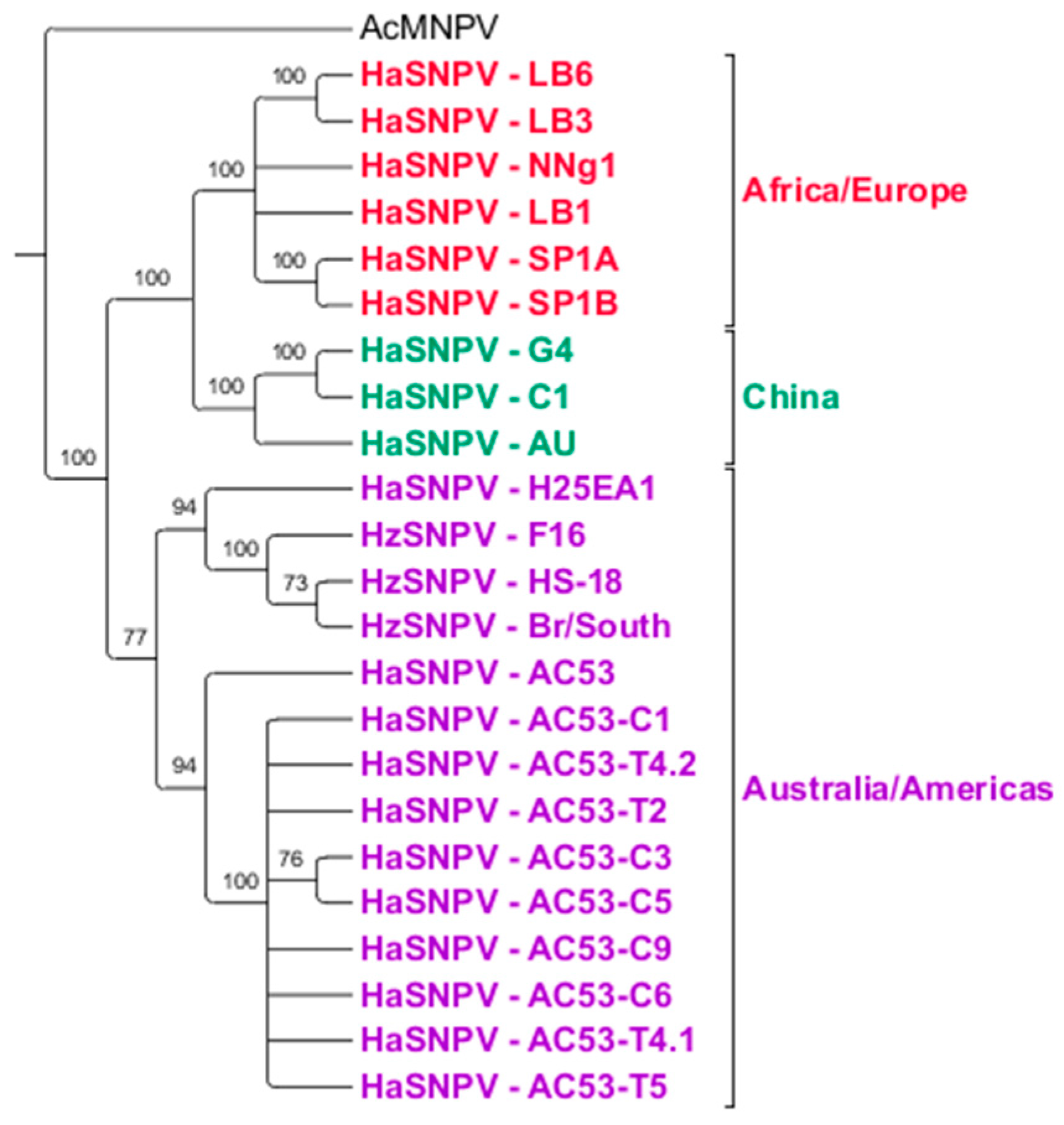
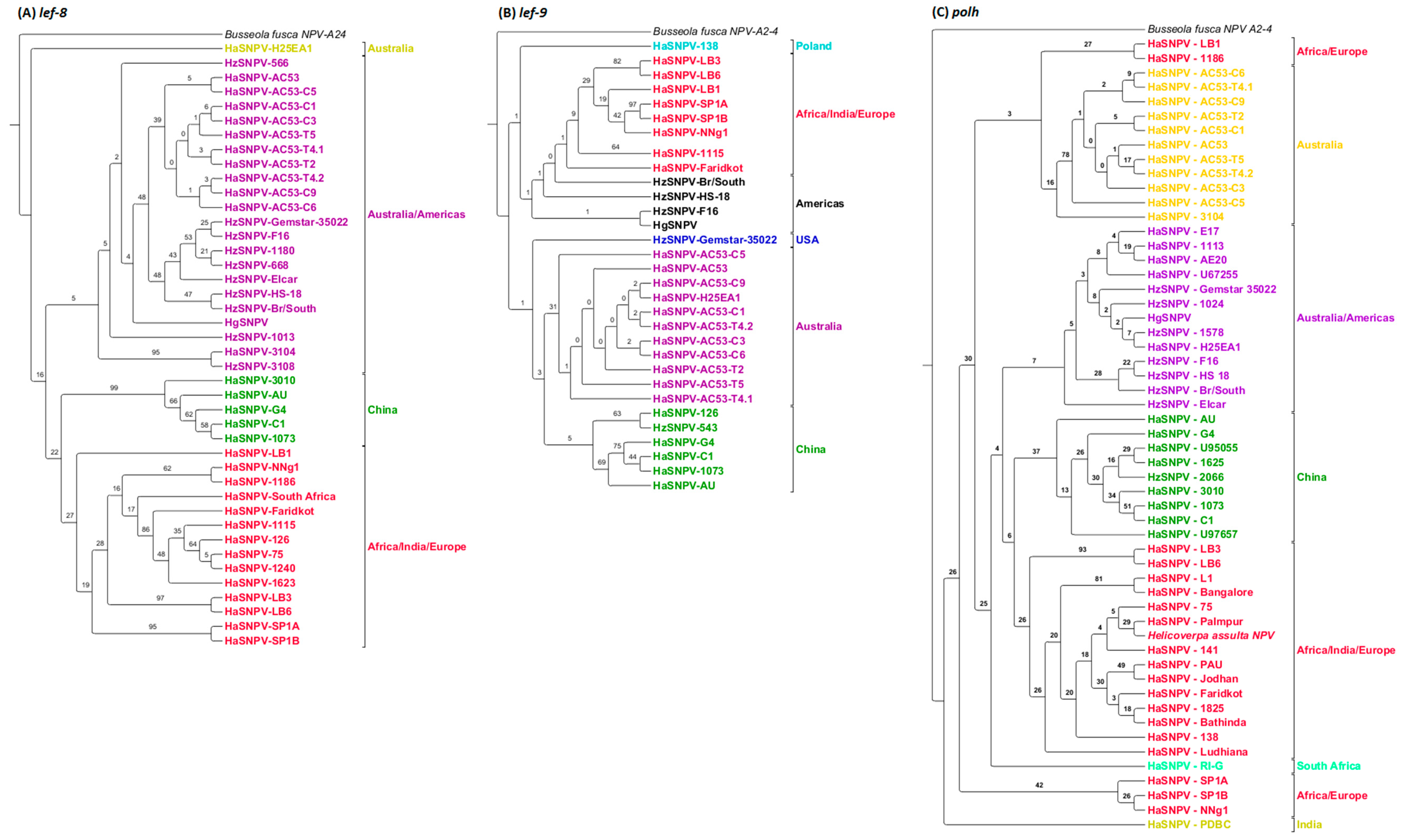
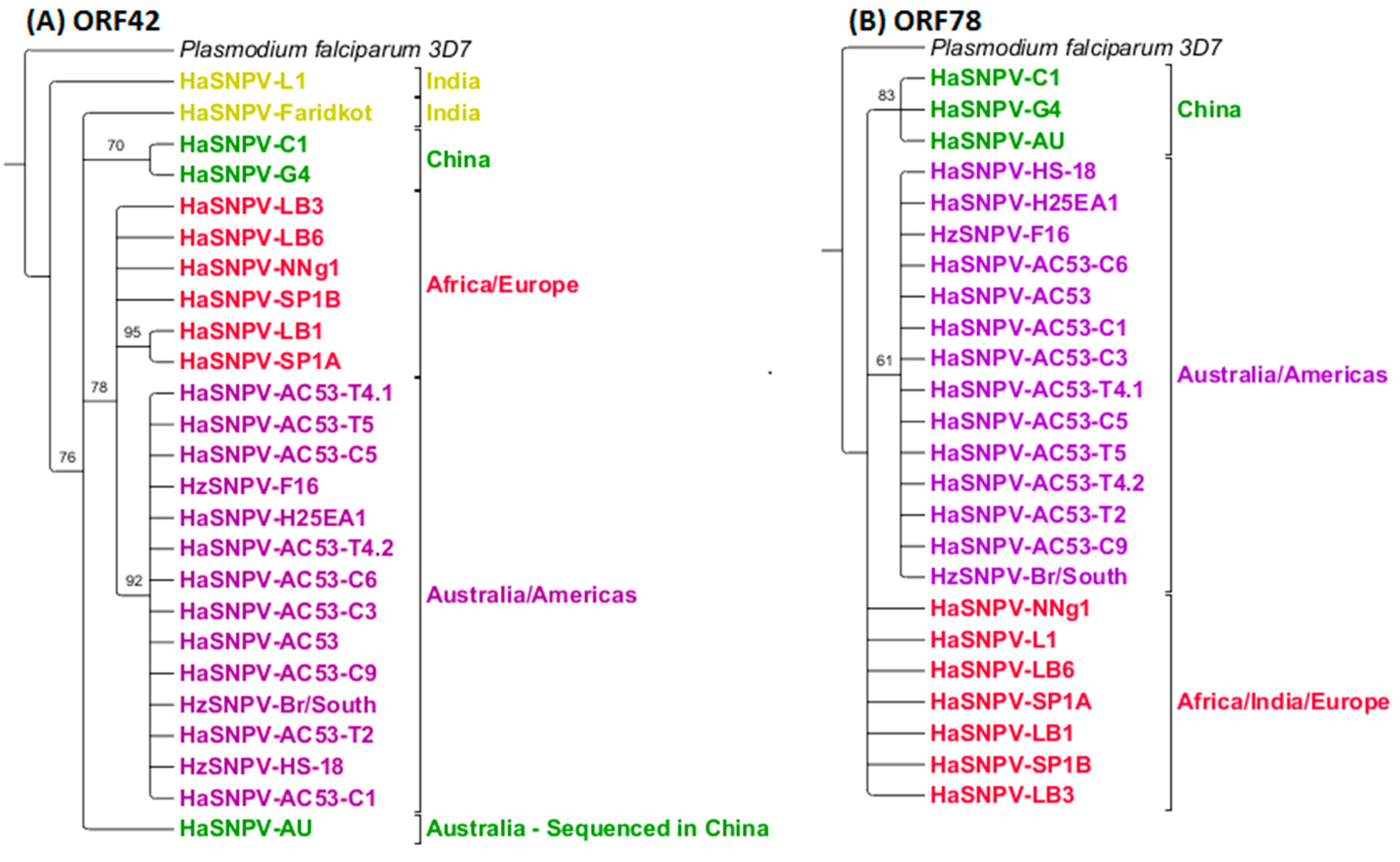
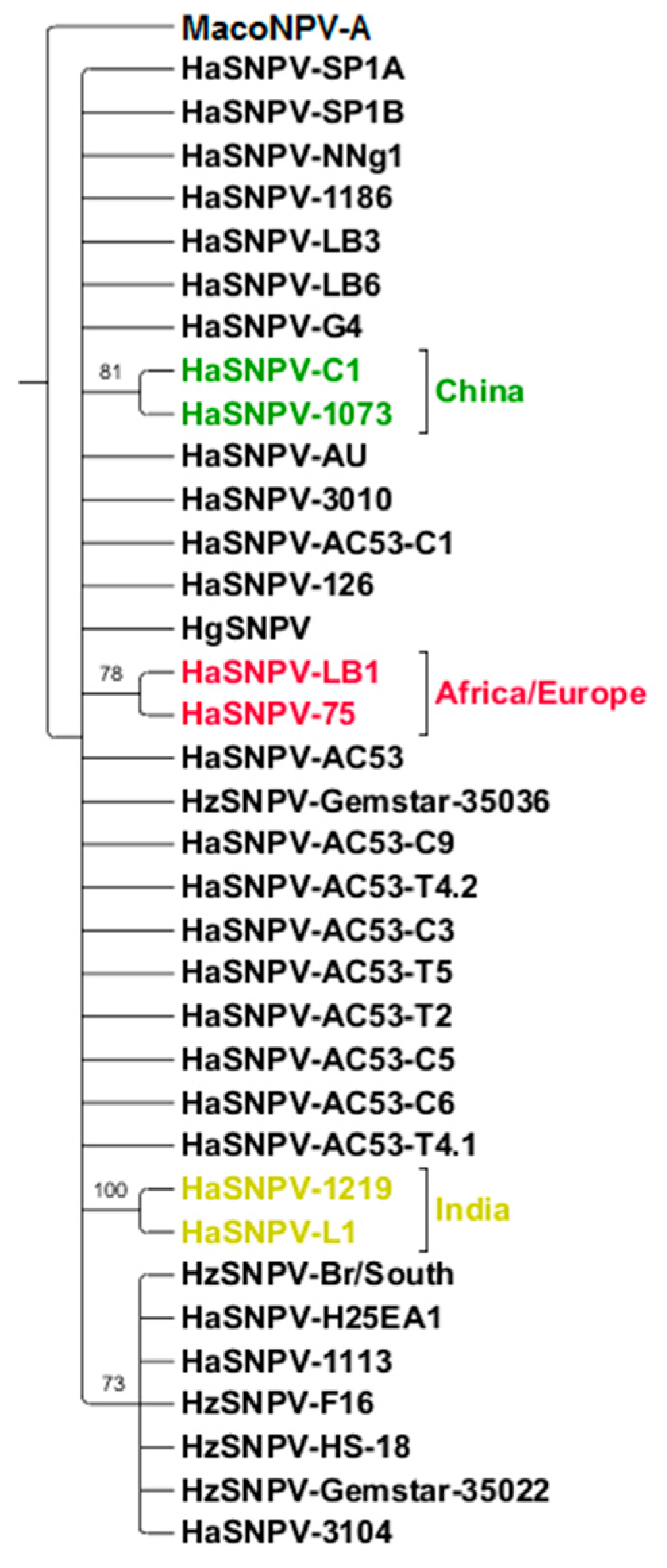
3.4. Maximum Likelihood Estimation
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Script Availability
References
- Rohrmann, G. Introduction to the baculoviruses and their taxonomy. In Baculovirus Molecular Biology, 2nd ed.National Center for Biotechnology Information: Bethesda, MD, USA, 2011. [Google Scholar]
- Blissard, G.W.; Rohrmann, G.F. Baculovirus diversity and molecular biology. Annu. Rev. Entomol. 1990, 35, 127–155. [Google Scholar] [CrossRef] [PubMed]
- Rowley, D.L.; Popham, H.J.R.; Harrison, R.L. Genetic variation and virulence of nucleopolyhedroviruses isolated worldwide from the heliothine pests Helicoverpa armigera, Helicoverpa zea, and Heliothis virescens. J. Invertebr. Pathol. 2011, 107, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Lange, M.; Wang, H.; Hu, Z.; Wang, Y.; Hauschild, R. Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology 2006, 346, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Blissard, G.W.; Bonning, B.C.; Cory, J.S.; Herniou, E.A.; Rohrmann, G.F.; Theilmann, D.A.; Thiem, S.M.; Vlak, J.M. On the classification and nomenclature of baculoviruses: A proposal for revision. Arch. Virol. 2006, 151, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Jehle, J.A. Baculovirus phylogeny and evolution. Curr. Drug Targets 2007, 8, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Wardhaugh, K.G.; Room, P.M.; Greenup, L.R. The incidence of Heliothis armigera (Hübner) and H. punctigera Wallengren (Lepidoptera: Noctuidae) on cotton and other host-plants in the Namoi Valley of New South Wales. Bull. Entomol. Res. 1980, 70, 113–131. [Google Scholar] [CrossRef]
- Daly, J.C.; Gregg, P. Genetic variation in Heliothis in Australia: Species identification and gene flow in the two pest species H. armigera (Hübner) and H. punctigera Wallengren (Lepidoptera: Noctuidae). Bull. Entomol. Res. 1985, 75, 169–184. [Google Scholar] [CrossRef]
- Zhang, G. Commercial viral insecticide Heliothis armigera viral insecticide in China. IPM Pract. 1989, 11, 13. [Google Scholar]
- Richards, A.R.; Christian, P.D. A rapid bioassay screen for quantifying nucleopolyhedroviruses (Baculoviridae) in the environment. J. Virol. Methods 1999, 82, 63–75. [Google Scholar] [CrossRef]
- Noune, C.; Hauxwell, C. Complete genome sequences of seven Helicoverpa armigera SNPV-AC53-derived strains. Genome Announc. 2016, 4, e00260-16. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L.; Herniou, E.A.; Theilmann, D.A.; Becnel, J.J.; Arif, B.; Jehle, J.A.; Burand, J.P.; Oers, M.V. Removal of Species Helicoverpa zea Single Nucleopolyhedrovirus from the Genus Alphabaculovirus; International Committee on Taxonomy of Viruses: Edinburgh, UK, 2013. [Google Scholar]
- Chen, X.; Zhang, W.-J.; Wong, J.; Chun, G.; Lu, A.; McCutchen, B.; Presnail, J.; Herrmann, R.; Dolan, M.; Tingey, S.; et al. Comparative analysis of the complete genome sequences of Helicoverpa zea and Helicoverpa armigera single-nucleocapsid nucleopolyhedroviruses. J. Gen. Virol. 2002, 83, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Buerger, P.; Hauxwell, C.; Murray, D. Nucleopolyhedrovirus introduction in Australia. Virol. Sin. 2007, 22, 173–179. [Google Scholar] [CrossRef]
- Nguyen, Q.; Qi, Y.M.; Wu, Y.; Chan, L.C.L.; Nielsen, L.K.; Reid, S. In vitro production of Helicoverpa baculovirus biopesticides—Automated selection of insect cell clones for manufacturing and systems biology studies. J. Virol. Methods 2011, 175, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Christian, P.D.; Gibb, N.; Kasprzak, A.B.; Richards, A. A rapid method for the identification and differentiation of Helicoverpa nucleopolyhedroviruses (NPV Baculoviridae) isolated from the environment. J. Virol. Methods 2001, 96, 51–65. [Google Scholar] [CrossRef]
- Noune, C.; Hauxwell, C. Complete genome sequences of Helicoverpa armigera single nucleopolyhedrovirus strains AC53 and H25EA1 from Australia. Genome Announc. 2015, 3, e01083-15. [Google Scholar] [CrossRef] [PubMed]
- Baillie, V.L.; Bouwer, G. High levels of genetic variation within Helicoverpa armigera nucleopolyhedrovirus populations in individual host insects. Arch. Virol. 2012, 157, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Erlandson, M.A. Genetic variation in field populations of baculoviruses: Mechanisms for generating variation and its potential role in baculovirus epizootiology. Virol. Sin. 2009, 24, 458–469. [Google Scholar] [CrossRef]
- Gettig, R.R.; McCarthy, W.J. Genotypic variation among wild isolates of Heliothis spp. nuclear polyhedrosis viruses from different geographical regions. Virology 1982, 117, 245–252. [Google Scholar] [CrossRef]
- Crawford, A.M.; Zelazny, B.; Alfiler, A.R. Genotypic variation in geographical isolates of oryctes baculovirus. J. Gen. Virol. 1986, 67, 949–952. [Google Scholar] [CrossRef]
- Corsaro, B.G.; Fraser, M.J. Characterization of genotypic and phenotypic variation in plaque-purified strains of HzSNPV elkar isolate. Intervirology 1987, 28, 185–198. [Google Scholar] [PubMed]
- Ogembo, J.G.; Caoili, B.L.; Shikata, M.; Chaeychomsri, S.; Kobayashi, M.; Ikeda, M. Comparative genomic sequence analysis of novel Helicoverpa armigera nucleopolyhedrovirus (NPV) isolated from Kenya and three other previously sequenced Helicoverpa spp. NPVs. Virus Genes 2009, 39, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Ogembo, J.G.; Chaeychomsri, S.; Kamiya, K.; Ishikawa, H.; Katou, Y.; Ikeda, M.; Kobayashi, M. Cloning and comparative characterization of nucleopolyhedroviruses isolated from African bollworm, Helicoverpa armigera, (Lepidoptera: Noctudiae) in different geographic regions. J. Insect Biotechnol. Sericol. 2007, 76, 39–49. [Google Scholar]
- Kabaluk, J.T.; Svircev, A.M.; Goettel, M.S.; Woo, S.G. (Eds.) The Use and Regulation of Microbial Pesticides in Representative Jurisdictions Worldwide; IOBC Global, 2010; p. 99. Available online: http://www.iobc-global.org/download/Microbial_Regulation_Book_Kabaluk_et_al_2010.pdf (accessed on 17 October 2016).
- Baillie, V.L.; Bouwer, G. Development of highly sensitive assays for detection of genetic variation in key Helicoverpa armigera nucleopolyhedrovirus genes. J. Virol. Methods 2011, 178, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Craveiro, S.R.; Inglis, P.W.; Togawa, R.C.; Grynberg, P.; Melo, F.L.; Ribeiro, Z.M.; Ribeiro, B.M.; Báo, S.N.; Castro, M.E. The genome sequence of Pseudoplusia includens single nucleopolyhedrovirus and an analysis of p26 gene evolution in the baculoviruses. BMC Genom. 2015, 16, 127. [Google Scholar] [CrossRef] [PubMed]
- Chateigner, A.; Bézier, A.; Labrousse, C.; Jiolle, D.; Barbe, V.; Herniou, E.A. Ultra deep sequencing of a baculovirus population reveals widespread genomic variations. Viruses 2015, 7, 3625–3646. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three Next Generation Sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L. Structural divergence among genomes of closely related baculoviruses and its implications for baculovirus evolution. J. Invertebr. Pathol. 2009, 101, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-X.; Ma, X.-C.; Guo, Z.-J. Comparison of the complete genome sequence between C1 and G4 isolates of the Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus. Virology 2005, 333, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, M.R.; Christian, P.; Nielsen, L.K.; Reid, S.; Chan, L.C. Importance of virus—Medium interactions on the biological activity of wild-type Heliothine nucleopolyhedroviruses propagated via suspension insect cell cultures. J. Virol. Methods 2006, 136, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.; Nielsen, L.K.; Reid, S. Genome scale transcriptomics of baculovirus-insect interactions. Viruses 2013, 5, 2721–2747. [Google Scholar] [CrossRef] [PubMed]
- Lua, L.H.; Reid, S. Virus morphogenesis of Helicoverpa armigera nucleopolyhedrovirus in Helicoverpa zea serum-free suspension culture. J. Gen. Virol. 2000, 81, 2531–2543. [Google Scholar] [CrossRef] [PubMed]
- Lua, L.H.; Pedrini, M.R.; Reid, S.; Robertson, A.; Tribe, D.E. Phenotypic and genotypic analysis of Helicoverpa armigera nucleopolyhedrovirus serially passaged in cell culture. J. Gen. Virol. 2002, 83, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.R.; Wood, H.A. A synchronous peroral technique for the bioassay of insect viruses. J. Invertebr. Pathol. 1981, 37, 154–159. [Google Scholar] [CrossRef]
- Hughes, D.S.; Possee, R.D.; King, L.A. Evidence for the presence of a low-level, persistent baculovirus infection of Mamestra brassicae insects. J. Gen. Virol. 1997, 78, 1801–1805. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.S.; Possee, R.D.; King, L.A. Activation and detection of a latent baculovirus resembling Mamestra brassicae nuclear polyhedrosis virus in M. brassicae insects. Virology 1993, 194, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Faulkner, P. Plaque assay of nuclear polyhedrosis viruses in cell culture. Appl. Environ. Microbiol. 1978, 36, 31–35. [Google Scholar] [PubMed]
- BDBiosciences. Plaque Assay. Available online: http://www.bdbiosciences.com/br/resources/baculovirus/protocols/plaque_assay.jsp (accessed on 17 September 2012).
- Hauxwell, I.C. Evaluation of Potential Baculovirus Insecticides: Studies of the Infection Process and Host Susceptibility; Imperial College London (University of London): London, UK, 1999. [Google Scholar]
- Matsuura, Y.; Possee, R.D.; Overton, H.A.; Bishop, D.H. Baculovirus expression vectors: The requirements for high level expression of proteins, including glycoproteins. J. Gen. Virol. 1987, 68, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Doyle, C.J.; Hirst, M.L.; Cory, J.S.; Entwistle, P.F. Risk assessment studies: Detailed host range testing of wild-type cabbage moth, Mamestra brassicae (Lepidoptera: Noctuidae), nuclear polyhedrosis virus. Appl. Environ. Microbiol. 1990, 56, 2704–2710. [Google Scholar] [PubMed]
- Zhang, H.; Yang, Q.; Qin, Q.L.; Zhu, W.; Zhang, Z.F.; Li, Y.N.; Zhang, N.; Zhang, J.H. Genomic sequence analysis of Helicoverpa armigera nucleopolyhedrovirus isolated from Australia. Arch. Virol. 2014, 159, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Arrizubieta, M.; Williams, T.; Caballero, P.; Simón, O. Selection of a nucleopolyhedrovirus isolate from Helicoverpa armigera as the basis for a biological insecticide. Pest Manag. Sci. 2014, 70, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Arrizubieta, M.; Simón, O.; Williams, T.; Caballero, P. A novel binary mixture of Helicoverpa armigera single nucleopolyhedrovirus genotypic variants has improved insecticidal characteristics for control of cotton bollworms. Appl. Environ. Microbiol. 2015, 81, 3984–3993. [Google Scholar] [CrossRef] [PubMed]
- Arrizubieta, M.; Simón, O.; Williams, T.; Caballero, P. Genomic sequences of five Helicoverpa armigera nucleopolyhedrovirus genotypes from Spain that differ in their insecticidal properties. Genome Announc. 2015, 3, e00548-15. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; IJkel, W.F.; Tarchini, R.; Sun, X.; Sandbrink, H.; Wang, H.; Peters, S.; Zuidema, D.; Lankhorst, R.K.; Vlak, J.M. The sequence of the Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus genome. J. Gen. Virol. 2001, 82, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Ardisson-Araújo, D.M.; Sosa-Gomez, D.R.; Melo, F.L.; Báo, S.N.; Ribeiro, B.M. Characterization of Helicoverpa zea single nucleopolyhedrovirus isolated in Brazil during the first old world bollworm (Noctuidae: Helicoverpa armigera) nationwide outbreak. Virus Rev. Res. 2015, 20, 2. [Google Scholar] [CrossRef]
- Ayres, M.D.; Howard, S.C.; Kuzio, J.; Lopez-Ferber, M.; Possee, R.D. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology 1994, 202, 586–605. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinf. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Wang, H.; Zhihong, H.; Jehle, J.A. Towards a molecular identification and classification system of lepidopteran-specific baculoviruses. Virology 2004, 325, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Ferrelli, M.; Taibo, C.; Fichetti, P.; Sciocco-Cap, A.; Arneodo, J. Characterization of a new Helicoverpa armigera nucleopolyhedrovirus variant causing epizootic on a previously unreported host, Helicoverpa gelotopoeon (Lepidoptera: Noctuidae). J. Invertebr. Pathol. 2015, 138, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-D.; Choi, J.Y.; Je, Y.H.; Jin, B.R. Characterization of the Helicoverpa assulta nucleopolyhedrovirus genome and sequence analysis of the polyhedrin gene region. J. Biosci. 2006, 31, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Donly, C.; Li, L.; Willis, L.G.; Theilmann, D.A.; Erlandson, M. Sequence and organization of the Mamestra configurata nucleopolyhedrovirus genome. Virology 2002, 294, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Erlandson, M.; Moody, D.; Gillott, C. A physical map of the Mamestra configurata nucleopolyhedrovirus genome and sequence analysis of the polyhedrin gene. J. Gen. Virol. 1997, 78 Pt 1, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Asgari, S.; Davis, J.; Wood, D.; Wilson, P.; McGrath, A. Sequence and organization of the Heliothis virescens ascovirus genome. J. Gen. Virol. 2007, 88, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schaffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Harrison, R.L. Genomic sequence analysis of the Illinois strain of the Agrotis ipsilon multiple nucleopolyhedrovirus. Virus Genes 2009, 38, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Simon, O.; Palma, L.; Beperet, I.; Munoz, D.; Lopez-Ferber, M.; Caballero, P.; Williams, T. Sequence comparison between three geographically distinct Spodoptera frugiperda multiple nucleopolyhedrovirus isolates: Detecting positively selected genes. J. Invertebr. Pathol. 2011, 107, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Parrish, J.T.; Peterson, F. Wind directions predicted from global circulation models and wind directions determined from eolian sandstones of the western United States—A comparison. Sediment. Geol. 1988, 56, 261–282. [Google Scholar] [CrossRef]
- Trenberth, K.E.; Large, W.G.; Olson, J.G. The mean annual cycle in global ocean wind stress. J. Phys. Oceanogr. 1990, 20, 1742–1760. [Google Scholar] [CrossRef]
- Parrish, J.T.; Curtis, R.L. Atmospheric circulation, upwelling, and organic-rich rocks in the Mesozoic and Cenozoic eras. Palaeogeogr. Palaeoclimatol. Palaeoecol. 1982, 40, 31–66. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Time (h) Post-Infection (pi) | First Round Isolation Method |
|---|---|---|
| AC53-C1 | 48 and 48 | Agar Overlay |
| AC53-C5 | 48 and 48 | Agar Overlay |
| AC53-C6 | 48 and 48 | Agar Overlay |
| AC53-C9 | 48 and 48 | Agar Overlay |
| AC53-C3 | 72 and 72 | Agar Overlay |
| AC53-T2 | 48 and 48 | Tissue Culture—No Overlay |
| AC53-T4.1 * | 72 and 96 | Tissue Culture—No Overlay |
| AC53-T4.2 * | 72 and 96 | Tissue Culture—No Overlay |
| AC53-T5 | 72 and 120 | Tissue Culture—No Overlay |
| Common Name | Genbank Accession | Sequence Length (bp) | Nucleotide Identity to AC53 (%) | Country/Region of Origin |
|---|---|---|---|---|
| HaSNPV-AC53-C1 | KU738896 | 130,460 | 99.624 | Australia |
| HaSNPV-AC53-C5 | KU738898 | 130,439 | 99.600 | Australia |
| HaSNPV-AC53-C6 | KU738899 | 130,435 | 99.601 | Australia |
| HaSNPV-AC53-T4.1 | KU738902 | 130,440 | 99.602 | Australia |
| HaSNPV-AC53-T5 | KU738904 | 130,442 | 99.603 | Australia |
| HaSNPV-AC53-C9 | KU738897 | 130,437 | 99.599 | Australia |
| HaSNPV-AC53-T2 | KU738901 | 130,440 | 99.596 | Australia |
| HaSNPV-AC53-T4.2 | KU738896 | 130,443 | 99.530 | Australia |
| HaSNPV-AC53-C3 | KU738897 | 130,437 | 99.595 | Australia |
| HaSNPV-H25EA1 | KJ922128 | 130,436 | 99.423 | Australia |
| HzSNPV-HS18 | KJ004000 | 130,890 | 99.220 | Unknown—Sequenced in Russia |
| HzSNPV-F16 (Elcar-derived) | AF334030 | 130,869 | 99.208 | USA—Sequenced in China |
| HzSNPV-Br/South | KM596835 | 129,694 | 98.277 | Brazil |
| HaSNPV-NNg1 | AP010907 | 132,425 | 96.203 | Kenya |
| HaSNPV-LB1 | KJ701029 | 131,966 | 96.012 | Iberia |
| HaSNPV-SP1A | KJ701032 | 132,481 | 95.961 | Iberia |
| HaSNPV-SP1B | KJ701033 | 132,265 | 95.810 | Iberia |
| HaSNPV-LB3 | KJ701030 | 130,949 | 95.799 | Iberia |
| HaSNPV-LB6 | KJ701031 | 130,992 | 95.798 | Iberia |
| HaSNPV-C1 | AF303045 | 130,759 | 95.353 | China |
| HaSNPV-AU | JN584482 | 130,992 | 94.860 | Australia—Sequenced in China |
| HaSNPV-G4 | AF271059 | 131,405 | 94.442 | China |
| ORF | Protein | AC53-C1 | AC53-C3 | AC53-C5 | AC53-C6 | AC53-C9 | AC53-T2 | AC53-T4.1 | AC53-T4.2 | AC53-T5 | Notes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AA | N | AA | N | AA | N | AA | N | AA | N | AA | N | AA | N | AA | N | AA | N | |||
| 4 | HOAR | 95.6 | 95.7 | 95.8 | 96.1 | 96.9 | 97.1 | 96.8 | 96.9 | 96.9 | 96.9 | 96.7 | 96.9 | 96.7 | 96.9 | 96.9 | 96.9 | 96.7 | 96.9 | |
| 5 | 34.9 | 97.2 | 98.3 | 98.9 | 34.9 | 97.2 | 34.9 | 97.2 | 34.9 | 97.2 | 34.9 | 97.2 | 34.9 | 97.2 | 34.9 | 97.2 | 34.9 | 97.2 | AC53 and AC53-C3 have identical length | |
| 6 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | 99.3 | ||
| 7 | 94.1 | 99.3 | 94.1 | 99.3 | 94.1 | 99.3 | 94.1 | 99.3 | 94.1 | 99.3 | 94.1 | 99.3 | 94.1 | 99.3 | 94.1 | 99.3 | 94.1 | 99.3 | AC53 is 85 bp shorter | |
| Hr1 | N.A | 99.7 | N.A | 99.7 | N.A | 99.7 | N.A | 99.8 | N.A | 99.8 | N.A | 99.7 | N.A | 99.7 | N.A | 99.8 | N.A | 99.7 | ||
| Hr2 | N.A | 95.5 | N.A | 95.2 | N.A | 95.2 | N.A | 95.2 | N.A | 95.3 | N.A | 95.2 | N.A | 95.2 | N.A | 95.2 | N.A | 95.2 | ||
| Hypothetical ORF | Hypothetical Protein | 70.7 | 98.0 | 70.7 | 98.0 | 70.7 | 98.0 | 70.7 | 98.0 | 70.7 | 98.0 | 70.7 | 98.0 | 70.7 | 98.0 | 70.7 | 98.0 | 70.7 | 98.0 | AC53 is 24 bp shorter |
| 59 | BRO-A | 90.9 | 92.1 | 90.9 | 92.1 | 90.9 | 92.1 | 90.9 | 92.1 | 90.9 | 92.1 | 91.3 | 92.1 | 90.9 | 92.1 | 90.9 | 92.1 | 90.9 | 92.1 | |
| 60 | BRO-B | 90.4 | 94.0 | 90.4 | 94.0 | 90.4 | 94.0 | 90.4 | 94.0 | 90.4 | 94.0 | 90.4 | 94.0 | 90.4 | 94.0 | 90.4 | 94.0 | 90.4 | 94.0 | |
| Hr3 | N.A | 99.6 | N.A | 99.6 | N.A | 99.6 | N.A | 99.6 | N.A | 99.6 | N.A | 99.69 | N.A | 99.6 | N.A | 99.8 | N.A | 99.6 | ||
| 61 | 80.0 | 86.9 | 86.8 | 86.9 | 80.0 | 86.9 | 80.0 | 86.9 | 80.0 | 86.9 | 80.0 | 86.9 | 80.0 | 86.9 | 80.0 | 86.9 | 80.0 | 86.9 | AC53 is 41 bp longer | |
| 68 | DNA polymerase | 100 | 100 | 99.9 | 99.9 | 99.9 | 99.9 | 100 | 100 | 100 | 100 | 99.9 | 99.9 | 100 | 100 | 100 | 100 | 100 | 100 | |
| 78a/78b (ORF78 in all other strains) | 100 | 100 | 100 | 100 | 100 | 100 | 76.3 | 99.4 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | Split in two with AC53-C6 | |
| Hr4 | N.A | 99.5 | N.A | 99.0 | N.A | 99.0 | N.A | 99.0 | N.A | 99.0 | N.A | 99.0 | N.A | 99.0 | N.A | 99.0 | N.A | 99.0 | ||
| Hr5 | N.A | 99.3 | N.A | 99.1 | N.A | 99.1 | N.A | 99.5 | N.A | 99.2 | N.A | 99.1 | N.A | 99.4 | N.A | 99.1 | N.A | 99.4 | ||
| 126 | 38.7K | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 99.7 | 99.4 | 100 | 100 | 100 | 99.8 | 100 | 100 | |
| 128a/128b (ORF128 in all other strains) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 23.2 | 87.6 | 100 | 100 | Split in two with AC53-T4.2 | |
| 133 | PKIP-1 | 100 | 100 | 100 | 99.8 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |
| 137 | 97.2 | 99.3 | 97.2 | 99.3 | 97.2 | 99.3 | 97.2 | 99.3 | 97.2 | 99.3 | 97.2 | 99.3 | 97.2 | 99.3 | 97.2 | 99.3 | 97.2 | 99.3 | ||
| Total regions with sequence polymorphisms | 9 | 14 | 10 | 16 | 10 | 15 | 10 | 15 | 8 | 14 | 11 | 16 | 9 | 14 | 10 | 16 | 9 | 14 | ||
| ORF/Region | Nucleotide Similarity and Clusters within Derived Strains | Amino Acid Similarity and Clusters within Derived Strains |
|---|---|---|
| HOAR | - AC53-T4.1, AC53-T5 = 100% | - AC53-T4.1 and AC53-T5 = 100% |
| - AC53-C6, AC53-C9, AC53-T4.2 = 100% | - AC53-C6, AC53-C9 ,AC53-T4.2 = 100% | |
| - Remaining 4 strains all different at 96.7% to 99.9% | - Remaining 4 strains all different at 95.8% to 99.8% | |
| ORF5 * | - AC53-C3 = 96.1% | - AC53-C3 = 33.3% |
| - Remaining strains all identical | - Remaining strains all identical | |
| BRO-A | - AC53-T2 = 99.9% | - AC53-T2 = 99.5% |
| - Remaining strains all identical | - Remaining strains all identical | |
| DNA-Polymerase | - AC53-T5, AC53-T4.2, AC53-T4.1, AC53-C9, AC53-C6, AC53-C1 = 100% | - AC53-C3, AC53-C5, AC53-T2 = 100% |
| - AC53-C5, AC53-C3 = 100% | - AC53-T5, AC53-T4.2, AC53-T4.1, AC53-C9, AC53-C6 | |
| - AC53-T2 = 99.9% | - AC53-C1 = 100% | |
| ORF78/ORF78a and 78b in AC53-C6 | - AC53-C6 = 99.4% | - AC53-C6 = 77.9% |
| - Remaining strains all identical | - Remaining strains all identical | |
| 38.7K Protein | - AC53-T2 = 99.4% to other 7 strains and 99.6% to AC53-T4.2 | - AC53-T2 = 99.7% |
| - AC53-T4.2 = 99.8% to other 7 strains | - Remaining 8 strains all identical | |
| - Remaining 7 strains all identical | ||
| ORF128/ORF128a and 128b in AC53-T4.2 | - AC53-T4.2 = 87.6% | - AC53-T4.2 = 23.2% |
| - Remaining strains all identical | - Remaining strains all identical | |
| PKIP-1 | - AC53-C3 = 99.8% | - All strains = 100% |
| - Remaining strains all identical | ||
| Hr1 | - AC53-T4.2, AC53-C9, AC53-C6 = 100% | - Not Applicable |
| - AC53-T5, AC53-T4.1, AC53-T2, AC53-C5, AC53-C3, AC53-C1 = 100% | ||
| - 99.9% when both groups compared | ||
| Hr2 | - AC53-C6, AC53-T4.1 = 100% | - Not Applicable |
| - Remaining strains all identical | ||
| Hr3 | - AC53-T4.2 = 99.8% | - Not Applicable |
| - Remaining strains all identical | ||
| Hr4 | - AC53-C1 = 99.2% | - Not Applicable |
| - Remaining strains all identical | ||
| Hr5 | - AC53-T2, AC53-C5, AC53-C3 = 100% | - Not Applicable |
| - Remaining strains all identical |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noune, C.; Hauxwell, C. Comparative Analysis of HaSNPV-AC53 and Derived Strains. Viruses 2016, 8, 280. https://doi.org/10.3390/v8110280
Noune C, Hauxwell C. Comparative Analysis of HaSNPV-AC53 and Derived Strains. Viruses. 2016; 8(11):280. https://doi.org/10.3390/v8110280
Chicago/Turabian StyleNoune, Christopher, and Caroline Hauxwell. 2016. "Comparative Analysis of HaSNPV-AC53 and Derived Strains" Viruses 8, no. 11: 280. https://doi.org/10.3390/v8110280
APA StyleNoune, C., & Hauxwell, C. (2016). Comparative Analysis of HaSNPV-AC53 and Derived Strains. Viruses, 8(11), 280. https://doi.org/10.3390/v8110280