
The Apis mellifera Filamentous Virus Genome



 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. AmFV DNA Isolation, Sequencing, and Assembly
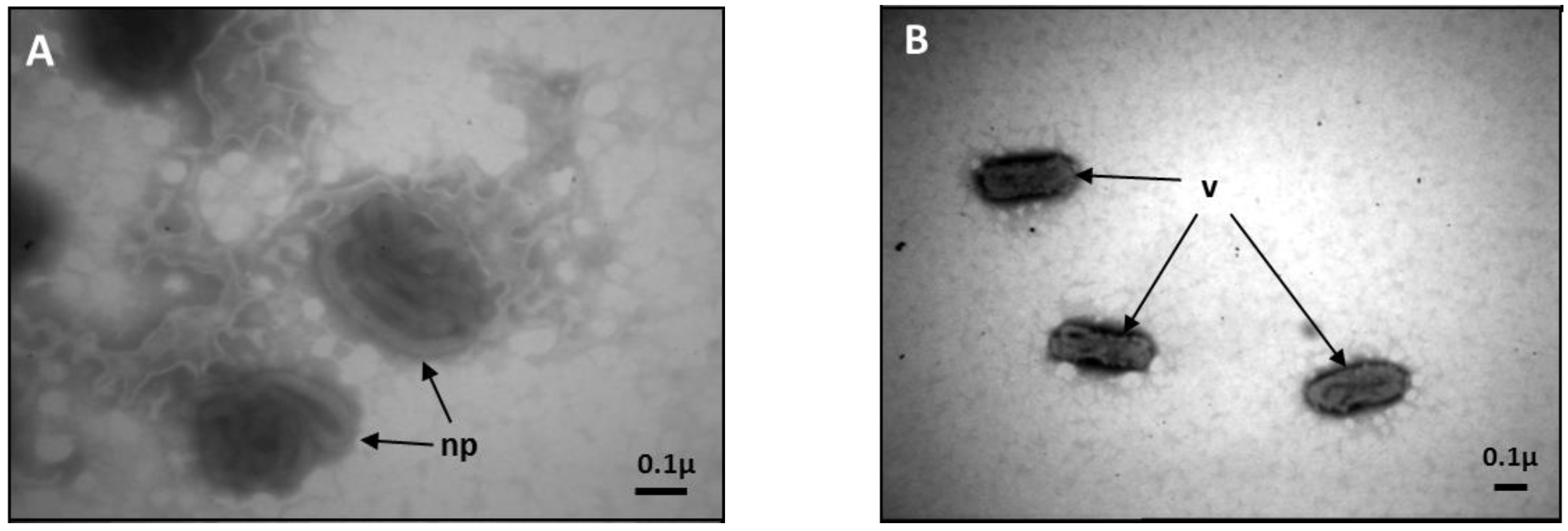
2.2. AmFV Sequence Analysis
2.3. Assembly of AmFV-Like Contigs from the USA
2.4. AmFV PCR Detection
2.5. Nucleotide Sequence Accession Number
3. Results and Discussion
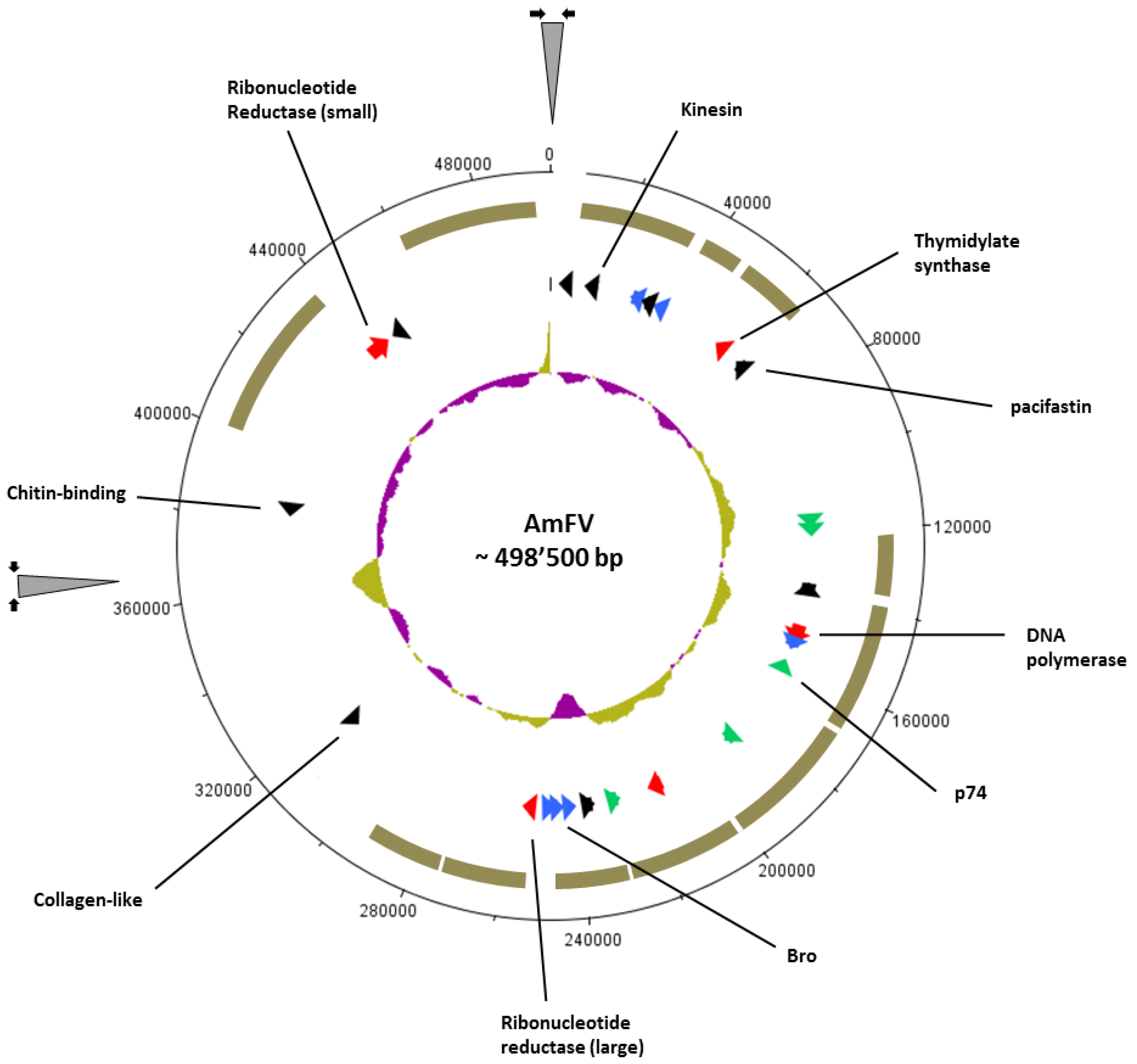
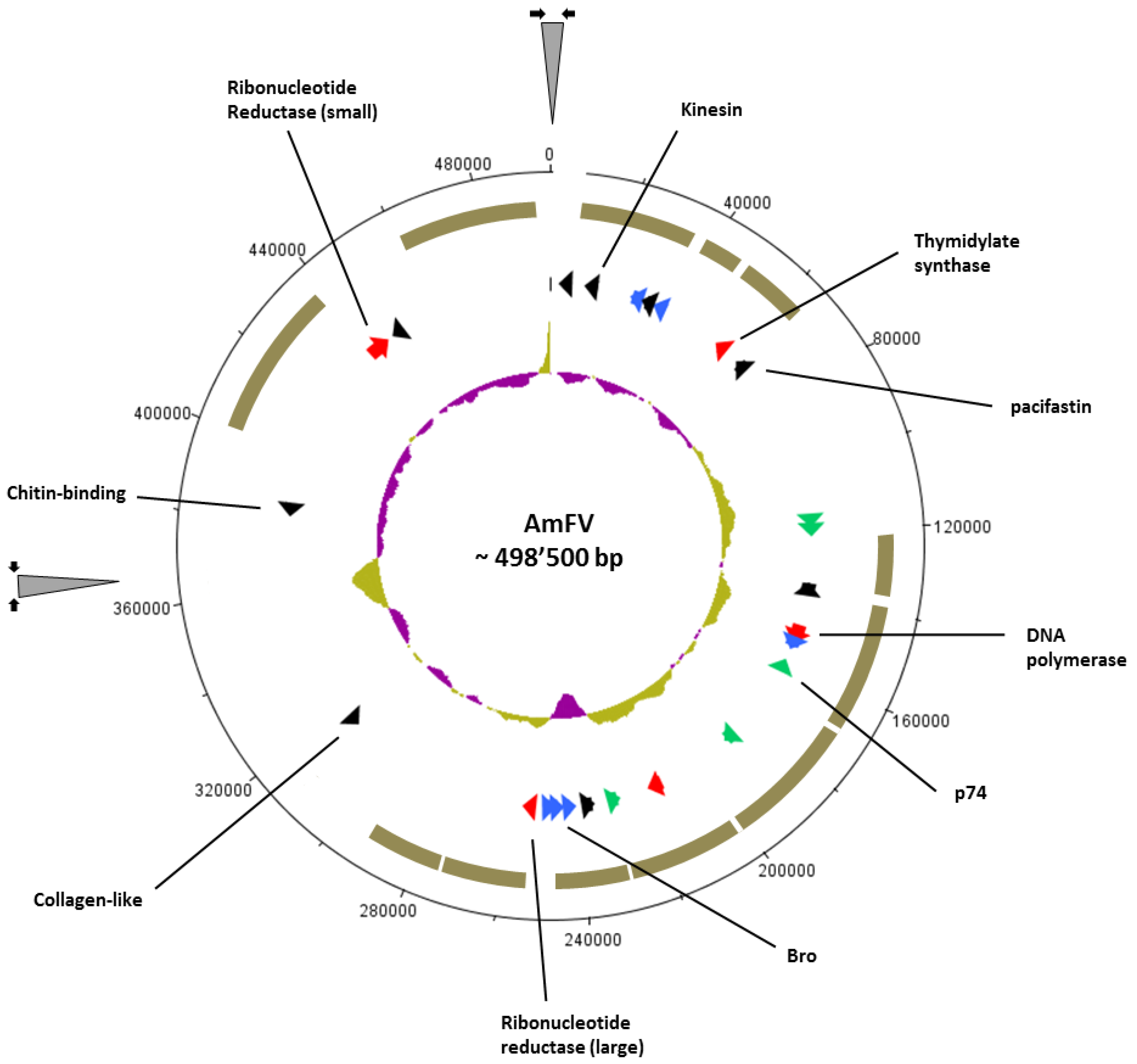
3.1. Assembly and Nucleotide Sequence Analysis of AmFV
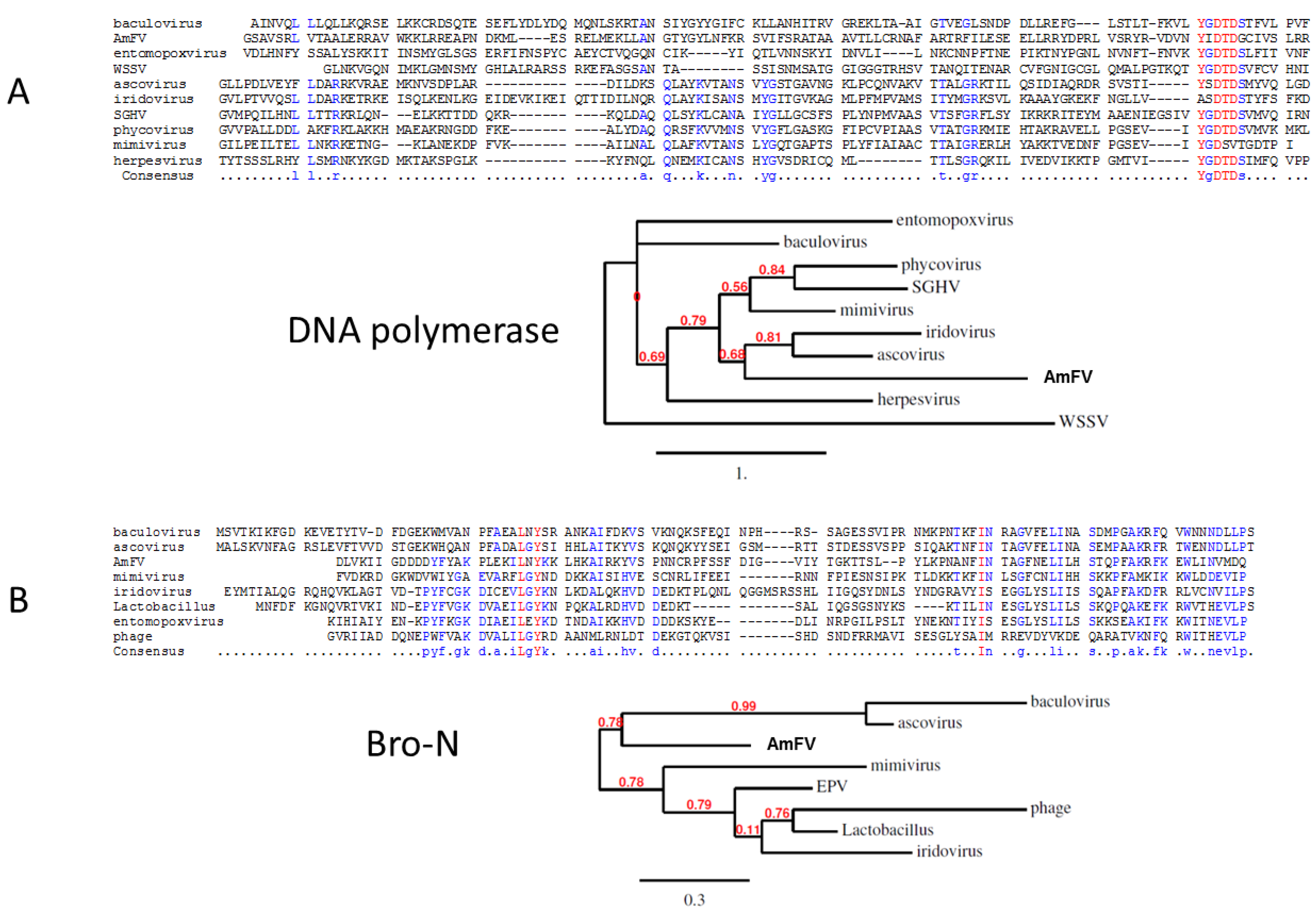
3.2. Gene Content Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Best Pfam-A database match with HMMer3 (0.1 cut off) | Best match with viral sequences database (BLASTP search taxid 10239, 1.0e-5 cut off) | AmFV (USA) % identity | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Putative function | AmFV ORF | Size (aa) | Pfam code domain | e-value | Pfam n° | CDS | e-value | Length / %similarity | Score | Accession number | |
| DNA replication and nucleotide metabolism | AmFV_76 | 1954 | DNA_pol_B | 6.5e-17 | PF00136.16 | - | - | - | - | - | 98.1 |
| AmFV_97 | 1602 | DNA ligase | 2.8e-08 | PF04675 | Hyp. protein [P. bursaria Chlorella virus AR158] | 4.0e-12 | 509 / 53% (61/115) | 72 | YP_001498814.1 | 93.7 | |
| AmFV_116 | 878 | Ribonuc_red_IgC | 5.9e-180 | PF02867.10 | RR1 [S. litura nucleopolyhedrovirus] | 0.0 | 770 / 42% (521/883) | 711 | NP_258291.1 | 99.7 | |
| AmFV_221 | 2332 | Ribonuc_red_sm | 3.4e-96 | PF00268.16 | RR2B [S. litura nucleopolyhedrovirus] | 7.e-56 | 333 / 72% (132/182) | 203 | NP_258331.1 | 97.8 | |
| AmFV_28 | 610 | Thymidylat_synt | 2.0e-86 | PF00303.14 | wsv067 [Shrimp white spot syndrome virus] | 3.0e-82 | 289 / 63% (188/294) | 265 | NP_477589.1 | 98.4 | |
| Virion structure and morphogenesis | AmFV_125 | 506 | Capsid_NCLDV | 1.5e-02 | PF04451.7 | Hyp. protein [Organic Lake phycodnavirus] | 5.0e-10 | 238 / 40% (116/283) | 65 | ADX05786.1 | 99.4 |
| AmFV_102 | 401 | Baculo_44 | 1.3e-16 | PF04631.7 | PIF-2 [G. bimaculatus nudivirus] | 2.0-15 | 378 / 48% (84/173) | 82 | YP_001111333.1 | 99.8 | |
| AmFV_90 | 279 | Pif-3 | 4.0e-06 | PF05006.7 | PIF3 [E. ello granulovirus] | 9.0e-06 | 188 / 39% (64/164) | 50 | YP_009091870.1 | 99.6 | |
| AmFV_61 | 1057 | Pif-1 | 1.6e-17 | PF05092.7 | PIF1 [S. littoralis nucleopolyhedrovirus] | 3.0-13 | 525 / 43% (99/230) | 77 | AGE89974.1 | 95.7 | |
| AmFV_62 | 829 | Pif-1 | 5.8e-25 | PF05092.7 | DekiORF31 [D. kikuchii nucleopolyhedrovirus] | 9.0e-19 | 536 / 48% (109/226) | 95 | AFS51909.1 | 98.6 | |
| AmFV_79 | 1196 | Baculo_p74 | 6.6e-10 | PF08404.5 | P74 [B. mori nucleopolyhedrovirus] | 1.0e-9 | 645 / 50% (72/144) | 68 | NP_047536.1 | 96.8 | |
| Unknown | AmFV_110 | 626 | Bro N | 1.82e-05 | PF02498 | BRO-C [M. configurata nucleopolyhedrovirus] | 8.0e-11 | 326 / 50% (110/219) | 69 | NP_689249.1 | 96.9 |
| AmFV_112 | 498 | Bro N | 1.5e-13 | PF02498.12 | BRO-B [C. chalcites nucleopolyhedrovirus] | 7.0e-17 | 628 / 48% (134/279) | 85 | YP_249673.1 | 97.8 | |
| AmFV_108 | 662 | Bro N | 1.7e-09 | PF02498.12 | BRO-M [L. xylina nucleopolyhedrovirus] | 2.0e-12 | 474 / 46% (95/204) | 75 | YP_003517887.1 | 99.2 | |
| AmFV_17 | 1313 | Bro N | 6.5e-09 | PF02498.12 | DekiORF51 [D. kikuchii nucleopolyhedrovirus] | 8.0e-08 | 480 / 49% (55/112) | 62 | AFS51929.1 | 90.9 | |
| AmFV_9 | 181 | ns | ns | ns | BRO-D [C. chalcites nucleopolyhedrovirus] | 7.0e-07 | 429 / 56% (42/75) | 53 | YP_249718.1 | 100 | |
| AmFV_77 | 432 | ns | ns | ns | BRO-6 [S. litura granulovirus] | 1.0e-05 | 485 / 42% (58/136) | 52 | YP_001257066.1 | 98.5 | |
| AmFV_197 | 898 | Chitin binding 3 | 6.8e-25 | PF03067.10 | - | - | - | - | - | 96.1 | |
| AmFV_104 | 596 | Collagen | 2.2e-07 | PF01391.13 | collagen repeat [Bacillus phage phBC6A52] | 9.0e-07 | 536 / 60% (46/76) | 57 | NP_852574.1 | 94.0 | |
| AmFV_69 | 422 | ns | ns | ns | collagen-like protein [A. polyphaga mimivirus] | 3.0e-07 | 1392 / 54% (54/99) | 58 | YP_003987190.1 | 95.9 | |
| AmFV_36 | 501 | Pacifastin I | 1.6e-10 | PF05375.8 | - | - | - | - | - | 94.3 | |
| AmFV_3 | 963 | Abhydrolase_3 | 1.3e-09 | PF07859.8 | - | - | - | - | - | 96.7 | |
| AmFV_6 | 1354 | Peptidase_M10 | 1.2e-10 | PF00413.19 | - | - | - | - | - | 92.2 | |
| AmFV_223 | 627 | Peptidase_M10 | 1.3e-10 | PF00413.19 | - | - | - | - | - | 97.9 | |
| AmFV_12 | 576 | Kinesin | 1.1e-67 | PF00225.18 | - | - | - | - | - | 99.8 | |
3.3. Sequence Variation between European and North American AmFV Isolates
3.4. AmFV Classification

3.5. AmFV Distribution in Honeybee Colonies

4. General Conclusions
Supplementary Information
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wille, H. Septikämien und Mischinfektionen. Schweiz. Bienenztg. 1962, 85, 222–226. [Google Scholar]
- Clark, T.B. A filamentous virus of the honey bee. J. Invertebr. Pathol. 1978, 32, 332–340. [Google Scholar] [CrossRef]
- Sitaropoulou, N.; Neophytou, E.P.; Thomopoulos, G.N. Structure of the Nucleocapsid of A Filamentous Virus of the Honey Bee (Apis-mellifera). J. Invertebr. Pathol. 1989, 53, 354–357. [Google Scholar] [CrossRef]
- Bailey, L.; Carpenter, J.M.; Woods, R.D. Properties of filamentous virus of the honey bee. Virology 1981, 114, 1–7. [Google Scholar] [CrossRef]
- Federici, B.A.; Bideshi, D.K.; Tan, Y.; Spears, T.; Bigot, Y. Ascoviruses: Superb Manipulators of Apoptosis for Viral Replication and Transmission. In Lesser Known Large DsDNA Viruses; Current Topics in Microbiology and Immunology; Van Etten, J.L., Ed.; Springer-Verlag: Berlin, Germany; Heidelberg, Germany, 2009; Volume 328, pp. 171–196. [Google Scholar]
- Allen, M.F.; Ball, B.V. The incidence and world distribution of honey bee viruses. Bee World 1996, 77, 141–162. [Google Scholar] [CrossRef]
- Bailey, L.; Ball, B.V. Honey Bee Pathology; Academic Press: London, UK, 1991. [Google Scholar]
- Varis, A.L.; Ball, B.V.; Allen, M. The Incidence of Pathogens in Honey-Bee (Apis mellifera L.) Colonies in Finland and Great-Britain. Apidologie 1992, 23, 133–137. [Google Scholar] [CrossRef]
- Bailey, L.; Ball, B.V.; Perry, J.N. Association of viruses with two protozoal pathogens of the honey bee. Ann. Appl. Biol. 1983, 103, 13–20. [Google Scholar] [CrossRef]
- Varaldi, J.; Ravallec, M.; Labrosse, C.; Lopez-Ferber, M.; Bouletreau, M.; Fleury, F. Artificial transfer and morphological description of virus particles associated with superparasitism behaviour in a parasitoid wasp. J. Insect Physiol. 2006, 52, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; UniProt Consortium. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genetics 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Bocs, S.; Cruveiller, S.; Vallenet, D.; Nuel, G.; Médigue, C. AMIGENE: Annotation of Microbial Genes. Nucleic Acids Res. 2003, 13, 3723–3726. [Google Scholar] [CrossRef]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Thomson, N.; Bleasby, A.; Berriman, M.; Parkhill, J. DNAPlotter: Circular and linear interactive genome visualization. Bioinformatics (Oxford, England) 2009, 25, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Cornman, R.S.; Schatz, M.C.; Spencer, J.; Chen, Y.P.; Pettis, J.; Hunt, G.; Bourgeois, L.; Elsik, C.; Anderson, D.; Grozinger, C.M.; et al. Genomic survey of the ectoparasitic mite Varroa destructor, a major pest of the honey bee Apis mellifera. BMC Genomics 2010, 11, e602. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.G.; Wetter, T.; Suhail, S. Using the miraEST Assembler for Reliable and Automated mRNA Transcript Assembly and SNP Detection in Sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Picard, A Set of Tools (in Java) for Working with Next Generation Sequencing Data in the BAM (http://samtools.sourceforge.net) Format. Available online: http://broadinstitute.github.io/picard/ (accessed on 7 July 2015).
- Carreck, N.L.; Andree, M.; Brent, C.S.; Cox-Foster, D.; Dade, H.A.; Ellis, J.D.; Hatjina, F.; van Engelsdorp, D. Standard methods for Apis mellifera anatomy and dissection. J. Apic. Res. 2013, 52. [Google Scholar] [CrossRef]
- Gauthier, L.; Ravallec, M.; Tournaire, M.; Cousserans, F.; Bergoin, M.; Dainat, B.; de Miranda, J.R. Viruses associated with ovarian degeneration in Apis mellifera L. queens. PLoS ONE 2011, 6, e16217. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M.; Abergel, C.; Ogata, H. Mimivirus. Curr. Top. Microbiol. Immunol. 2009, 328, 89–121. [Google Scholar] [PubMed]
- Van Hulten, M.C.W.; Witteveldt, J.; Peters, S.; Kloosterboer, N.; Tarchini, R.; Fiers, M.; Sandbrink, H.; Klein Lankhorst, R.; Vlak, J.M. The white spot syndrome virus DNA genome sequence. Virology 2001, 286, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Bawden, A.L.; Glassberg, K.J.; Diggans, J.; Shaw, R.; Farmerie, W.; Moyer, R.W. Complete Genomic Sequence of the Amsacta moorei Entomopoxvirus: Analysis and Comparison with Other Poxviruses. Virology 2000, 274, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Takatsuka, J.; Li, Z.; Gallais, J.; Doucet, D.; Arif, B.; Nakai, M.; Herniou, E.A. New insights into the evolution of Entomopoxvirinae from the complete genome sequences of four entomopoxviruses infecting Adoxophyes honmai, Choristoneura biennis, Choristoneura rosaceana, and Mythimna separata. J. Virol. 2013, 87, 7992–8003. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.; Eisen, J.A.; Nene, V. New evolutionary frontiers from unusual virus genomes. Genome Biol. 2005, 6, e212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herniou, E.A.; Arif, B.M.; Becnel, J.J.; Blissard, G.W.; Bonning, B.; Harrison, R.; Jehle, J.A.; Theilmann, D.A.; Vlak, J.M. Baculoviridae. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Oxford, UK, 2011; pp. 163–174. [Google Scholar]
- Wang, Y.; Jehle, J.A. Nudiviruses and other large, double-stranded circular DNA viruses of invertebrates: New insights on an old topic. J. Invertebr. Pathol. 2009, 101, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A. Nudiviruses: Their biology and genetics. In Insect Virology; Asgari, S., Johnson, K.N., Eds.; School of Biological Sciences, The University of Queensland: St Lucia QLD, Australia, 2010; pp. 153–170, 436. [Google Scholar]
- Lietze, V.U.; Abd-Alla, A.M.; Vreysen, M.J.; Geden, C.J.; Boucias, D.G. Salivary gland hypertrophy viruses: A novel group of insect pathogenic viruses. Annu. Rev. Entomol. 2011, 56, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Abd-Alla, A.M.; Wang, Y. Phylogeny and evolution of Hytrosaviridae. J. Invertebr. Pathol. 2013, 112, S62–S67. [Google Scholar] [CrossRef] [PubMed]
- Espagne, E.; Dupuy, C.; Huguet, E.; Cattolico, L.; Provost, B.; Martins, N.; Poirie, M.; Periquet, G.; Drezen, J.M. Genome sequence of a polydnavirus: Insights into symbiotic virus evolution. Science 2004, 306, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Bézier, A.; Herbinière, J.; Lanzrein, B.; Drezen, J.M. Polydnavirus hidden face: The genes producing virus particles of parasitic wasps. J. Invertebr. Pathol. 2009, 101, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Wetterwald, C.; Roth, T.; Kaeslin, M.; Annaheim, M.; Wespi, G.; Heller, M.; Mäser, P.; Roditi, I.; Pfister-Wilhelm, R.; Bézier, A.; et al. Identification of bracovirus particle proteins and analysis of their transcript levels at the stage of virion formation. J. Gen. Virol. 2010, 91, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Zhen, L.; Pavlik, L.; Arif, B. Entomopoxviruses. In Insect Virology; Asgari, S., Johnson, K.N., Eds.; School of Biological Sciences, The University of Queensland: St Lucia QLD, Australia, 2010; pp. 83–102, 436. [Google Scholar]
- Bideshi, D.K.; Renault, S.; Stasiak, K.; Federici, B.A.; Bigot, Y. Phylogenetic analysis and possible function of bro-like genes, a multigene family widespread among large double-stranded DNA viruses of invertebrates and bacteria. J. Gen. Virol. 2003, 84, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Takatsuka, J.; Nakai, M.; Arif, B.; Herniou, E.A. Gene acquisition convergence between entomopoxviruses and baculoviruses. Viruses 2015, 13, 1960–1974. [Google Scholar] [CrossRef] [PubMed]
- Ishimwe, E.; Hodgson, J.J.; Clem, R.J.; Passarelli, A.L. Reaching the melting point: Degradative enzymes and protease inhibitors involved in baculovirus infection and dissemination. Virology 2015, 479C–480C, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hukuhara, T. Enhanced infection of a nuclear polyhedrosis virus in larvae of the armyworm, Pseudaletia separata, by a factor in the spheroids of an entomopoxvirus. J. Invertebr. Pathol. 1992, 60, 259–264. [Google Scholar] [CrossRef]
- Mitsuhashi, W.; Kawakita, H.; Murakami, R.; Takemoto, Y.; Saiki, T.; Miyamoto, K.; Wada, S. Spindles of an entomopoxvirus facilitate its infection of the host insect by disrupting the peritrophic membrane. J. Virol. 2007, 81, 4235–4243. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalez, E.; Poppinga, L.; Funfhaus, A.; Hertlein, G.; Hedtke, K.; Jakubowska, A.; Genersch, E. Paenibacillus larvae Chitin-Degrading Protein PlCBP49 Is a Key Virulence Factor in American Foulbrood of Honey Bees. PLoS Pathog. 2014, 10, e7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cui, Z.; Shi, G.; Luo, D.; Wang, S.; Wang, C. PtPLC, a pacifastin-related inhibitor involved in antibacterial defense and prophenoloxidase cascade of the swimming crab Portunus trituberculatus. Fish Shellfish Immunol. 2015, 43, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Breugelmans, B.; Simonet, G.; van Hoef, V.; van Soest, S.; Vanden Broeck, J. Identification, distribution and molecular evolution of the pacifastin gene family in Metazoa. BMC Evol. Biol. 2009, 9, e97. [Google Scholar] [CrossRef] [PubMed]
- Antunez, K.; Arredondo, D.; Anido, M.; Zunino, P. Metalloprotease production by Paenibacillus larvae during the infection of honeybee larvae. Microbiology 2011, 157, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Way, M. A superhighway to virus infection. Cell 2006, 124, 741–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Bézier, A.; Louis, F.; Jancek, S.; Periquet, G.; Thézé, J.; Gyapay, G.; Musset, K.; Lesobre, J.; Lenoble, P.; Dupuy, C.; et al. Functional endogenous viral elements in the genome of the parasitoid wasp Cotesia congregata: Insights into the evolutionary dynamics of bracoviruses. Phil. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, e20130047. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K.; Ishido, S.; Jung, J.U. The collagen repeat sequence is a determinant of the degree of herpesvirus saimiri STP transforming activity. J. Virol. 2000, 74, 8102–8110. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Hülsmeier, A.J.; Hochhold, N.; Neidhart, M.; Gay, S.; Hennet, T. Exposure to mimivirus collagen promotes arthritis. J. Virol. 2014, 88, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.A.; Gundersen-Rindal, D.E.; Hostetler, J.B.; Tallon, L.J.; Fadrosh, D.W.; Fuester, R.W.; Pedroni, M.J.; Haas, B.J.; Schatz, M.C.; Jones, K.M.; et al. Comparative genomics of mutualistic viruses of Glyptapanteles parasitic wasps. Genome Biol. 2008, 9, eR183. [Google Scholar] [CrossRef] [PubMed]
- Drezen, J.M.; Bézier, A.; Lesobre, J.; Huguet, E.; Cattolico, L.; Periquet, G.; Dupuy, C. The few virus-like genes of Cotesia congregata bracovirus. Arch. Insect Biochem. Physiol. 2006, 61, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.L.; Tulman, E.R.; Lu, Z.; Oma, E.; Kutish, G.F.; Rock, D.L. The genome of Melanoplus sanguinipes entomopoxvirus. J. Virol. 1999, 73, 533–552. [Google Scholar] [PubMed]
- Stern, A.; Mayrose, I.; Penn, O.; Shaul, S.; Gophna, U.; Pupko, T. An evolutionary analysis of lateral gene transfer in thymidylate synthase enzymes. Syst. Biol. 2010, 59, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.K.; Gilbert, C.; Clark, M.S.; Feschotte, C. Repeated horizontal transfer of a DNA transposon in mammals and other tetrapods. Proc. Natl. Acad. Sci. USA 2008, 105, 17023–17028. [Google Scholar] [CrossRef] [PubMed]
- Filée, J. Route of NCLDV evolution: The genomic accordion. Curr. Opin. Virol. 2013, 3, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, U.; Forsgren, E.; Charrière, J.D.; Neumann, P.; Gauthier, L. Dynamics of Apis mellifera Filamentous Virus (AmFV) Infections in Honey Bees and Relationships with Other Parasites. Viruses 2015, 7, 2654–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Miranda, J.R.; Fries, I. Venereal and vertical transmission of deformed wing virus in honeybees (Apis mellifera L.). J. Invertebr. Pathol. 2008, 98, 184–189. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gauthier, L.; Cornman, S.; Hartmann, U.; Cousserans, F.; Evans, J.D.; De Miranda, J.R.; Neumann, P. The Apis mellifera Filamentous Virus Genome. Viruses 2015, 7, 3798-3815. https://doi.org/10.3390/v7072798
Gauthier L, Cornman S, Hartmann U, Cousserans F, Evans JD, De Miranda JR, Neumann P. The Apis mellifera Filamentous Virus Genome. Viruses. 2015; 7(7):3798-3815. https://doi.org/10.3390/v7072798
Chicago/Turabian StyleGauthier, Laurent, Scott Cornman, Ulrike Hartmann, François Cousserans, Jay D. Evans, Joachim R. De Miranda, and Peter Neumann. 2015. "The Apis mellifera Filamentous Virus Genome" Viruses 7, no. 7: 3798-3815. https://doi.org/10.3390/v7072798
APA StyleGauthier, L., Cornman, S., Hartmann, U., Cousserans, F., Evans, J. D., De Miranda, J. R., & Neumann, P. (2015). The Apis mellifera Filamentous Virus Genome. Viruses, 7(7), 3798-3815. https://doi.org/10.3390/v7072798