
Fluorescent Protein Approaches in Alpha Herpesvirus Research

Abstract
:1. Introduction
1.1. A Brief History of Fluorescent Proteins
1.2. Alpha Herpesviruses Strains Expressing Fluorescent Proteins and Biosensors
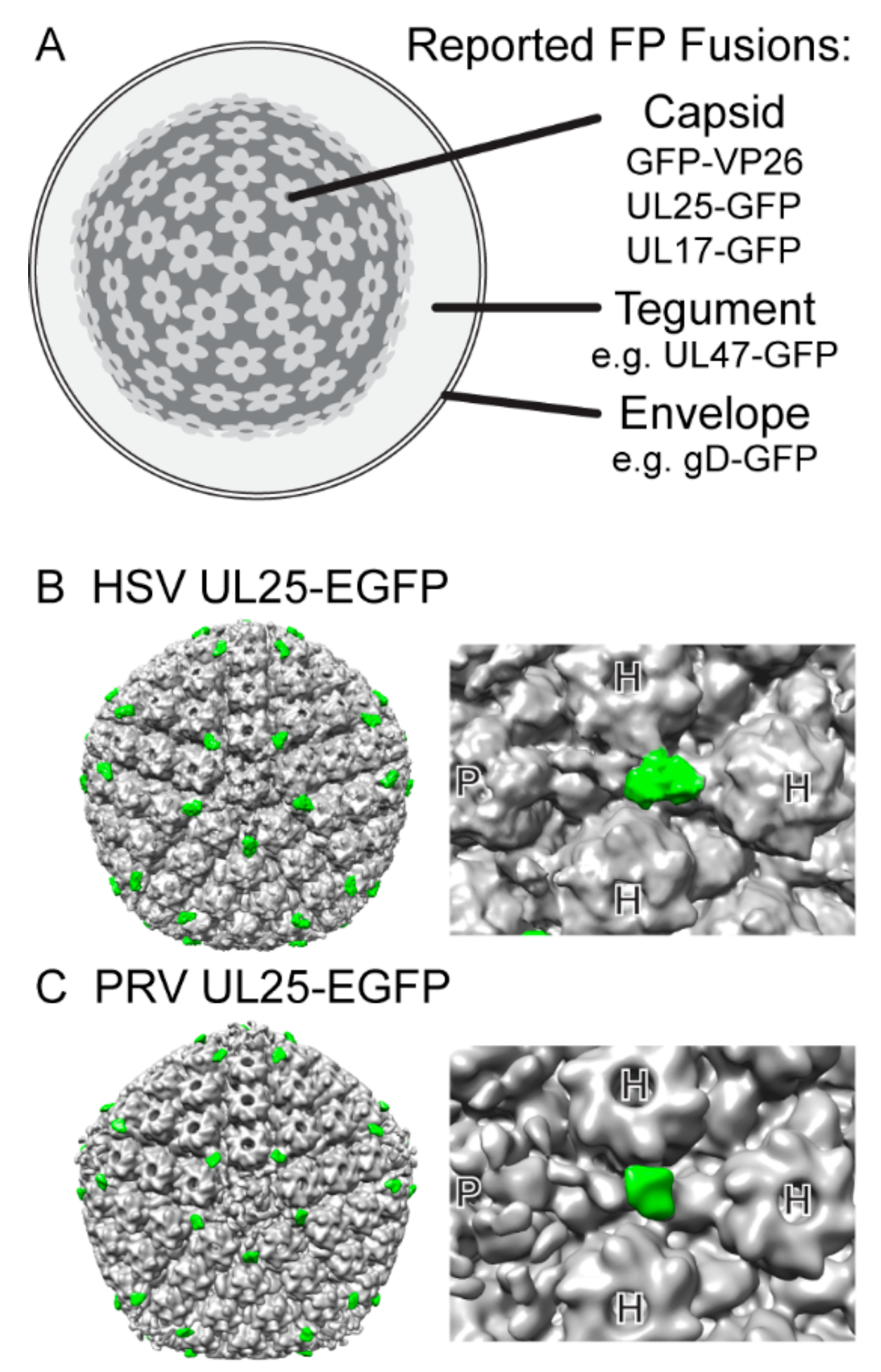
1.3. Fluorescent Protein Fusions to Viral Proteins in HSV-1 and PRV

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Protein Name | Description/Function | Structural Role | Fusion Location | References | |
|---|---|---|---|---|---|
| HSV-1 | PRV | ||||
| Capsid Proteins | |||||
| UL17 | capsid vertex specific component | capsid | C-terminal | [42] | |
| UL25 | capsid vertex specific component | capsid | internal | [43] | [44] |
| UL35 VP26 | small capsid protein | capsid | N-terminal | [41,45] | [46] |
| C-terminal | [47] | ||||
| Tegument Proteins | |||||
| UL13 | protein kinase | tegument | N-terminal | [48] | |
| C-terminal | [48] | ||||
| UL16 | interacts with UL21 | tegument | C-terminal | [49] | [50] |
| UL21 | interacts with UL16 | N-terminal | [51] | ||
| C-terminal | [52]b | ||||
| UL31 | nuclear egress | tegument a | N-terminal | [53,54] | [55] |
| UL36 VP1/2 | large tegument protein | tegument | N-terminal | [56] | |
| C-terminal | [57] | [44] | |||
| UL37 | interacts with UL36 | tegument | N-terminal | [57] | |
| C-terminal | [58] | [56] | |||
| UL41 VHS | RNAase | tegument | N-terminal | [59] | |
| UL46 VP11/12 | most abundant tegument protein | tegument | N-terminal | [57] | |
| C-terminal | [60] | [50] | |||
| UL47 VP13/14 | tegument | N-terminal | [61] | [56] | |
| UL48 VP16 | transactivation of viral gene expression | tegument | N-terminal | [62] | |
| C-terminal | [57,62] | [56] | |||
| UL49 VP22 | secondary envelopment | tegument | N-terminal | [57,63] | [56,64] |
| C-terminal | [40,63] | [64] | |||
| US1 ICP22 | regulation of viral gene expression | tegument | C-terminal | [65] | |
| US3 | protein kinase | tegument | N-terminal | [66] | |
| C-terminal | [67]b | [48,68] | |||
| US10 | unknown | tegument | C-terminal | [69]c | |
| ICP34.5 | tegument | N-terminal | [70] | * | |
| ICP0 (EP0) | transactivation of viral gene expression, E3 ubiquitin ligase | tegument | N-terminal | [71] | |
| C-terminal | [72] | [73] | |||
| internal | [74] | ||||
| ICP4 (IE180) | transactivation of viral gene expression | tegument | N-terminal | [75] | |
| C-terminal | [76] | [77] | |||
| Envelope Proteins | |||||
| UL10 gM | glycoprotein M | envelope | N-terminal, intravirion | [78] | [78] |
| C-terminal, intravirion | [36,79] | ||||
| internal, extravirion | [36] | ||||
| UL11 | lipid-anchored membrane protein | envelope | C-terminal | [49] | [49] |
| UL20 | multipass transmembrane protein | envelope | [80] | ||
| UL22 gH | glycoprotein H, membrane fusion | envelope | N-terminal, extravirion | [81] | |
| UL27 gB | glycoprotein B, receptor binding, membrane fusion | envelope | N-terminal, extravirion | [82,83] | |
| C-terminal, intravirion | [84] | ||||
| UL34 | transmembrane protein, nuclear egress | envelope a | C-terminal | [85]d | |
| UL43 | multipass transmembrane protein | envelope | C-terminal, intravirion | [86]d | |
| UL44 gC | glycoprotein C, receptor binding | envelope | C-terminal, intravirion | [87] | |
| UL49A gN | glycoprotein N | envelope | C-terminal, intravirion | [88] | |
| UL51 | lipid-anchored membrane protein | envelope | C-terminal | [89] | |
| UL53 gK | glycoprotein K | envelope | C-terminal, extravirion | [90]d | |
| US2 | lipid-anchored membrane protein | envelope | N-terminal | [91] | |
| US6 gD | glycoprotein D, receptor binding, membrane fusion | envelope | C-terminal, intravirion | [92] | [93] |
| US7 gI | glycoprotein I, anterograde axonal transport | envelope | N-terminal, extravirion | [94] | |
| US8 gE | glycoprotein E, anterograde axonal transport | envelope | N-terminal, extravirion | [94] | |
| C-terminal, intravirion | [95] | ||||
| US9 | transmembrane protein, anterograde axonal transport | envelope | N-terminal, intravirion | [37] | [79] |
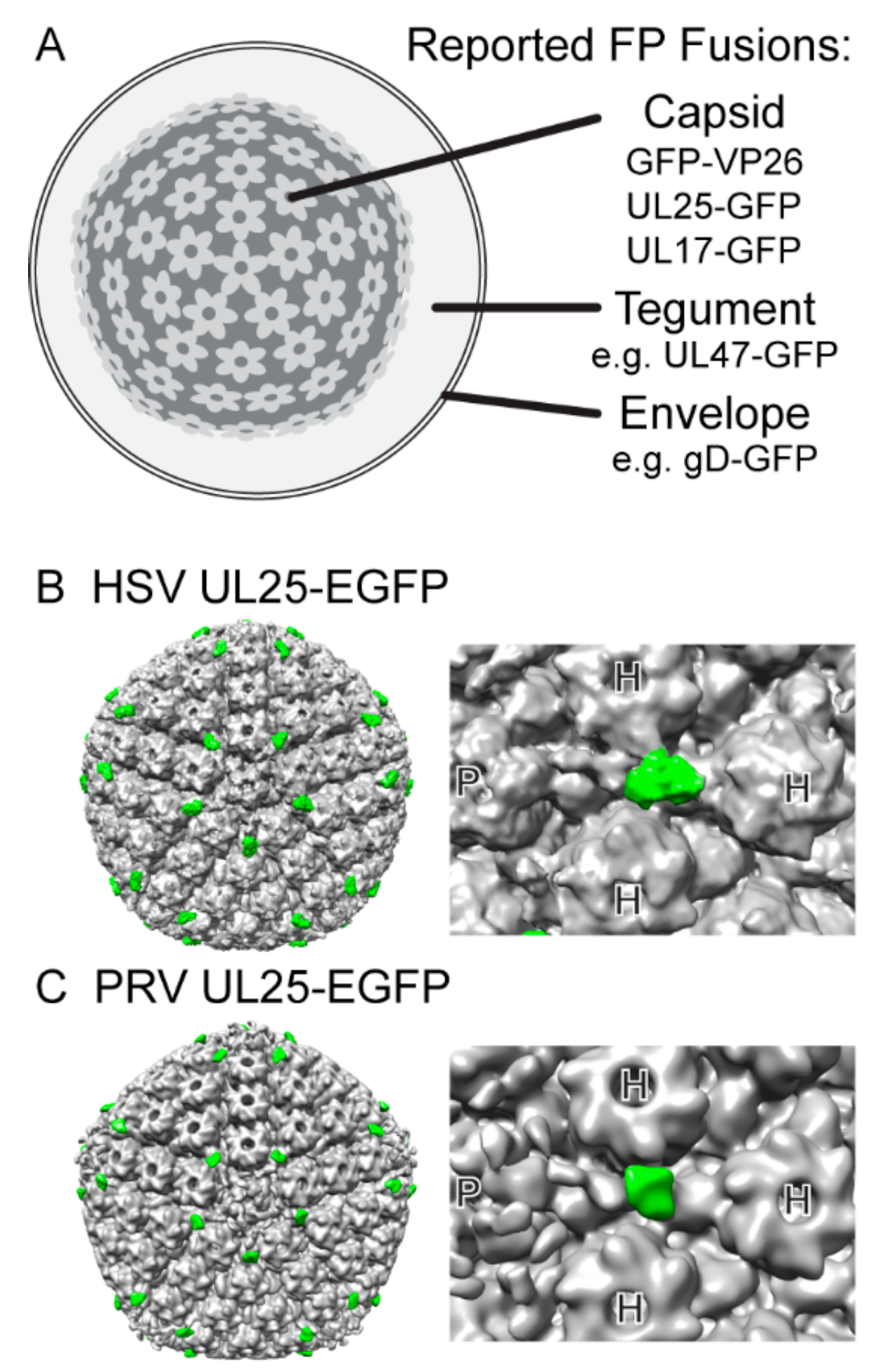
2. Unique Challenges of Fluorescent Protein-based Approaches in Viruses
2.1. Structure of Virion
2.2. Genome Constraints
2.3. Evolvable and Highly Recombinogenic Genomes
3. How-To: General Strategies and Best Practices
3.1. Choice of Fluorescent Protein
3.1.1. Fluorescent Protein Dimerization
3.1.2. Fluorescent Protein Maturation Time
| Fluorescent Protein | t1/2 (min) | References |
|---|---|---|
| Blue | ||
| mTagBFP2 | 12 | [116] |
| Cyan | ||
| SCFP3A | 82 | [117] |
| mCerulean3, mTurquoise2 | a | [118] |
| Green | ||
| wild-type Aequorea GFP | 37–83 b | [119,120] |
| EGFP, mEGFP | 10–65 b | [120,121] |
| Emerald | 8 | [120] |
| mNeonGreen | <10 | [122] |
| TagGFP2 | 18 | [123] |
| Yellow | ||
| Venus, mVenus | 28–72 b | [117,120] |
| SYFP2 | 55 | [117] |
| Orange | ||
| mKO2 | 108 | [124] |
| mOrange2 | 270 | [125] |
| Red | ||
| DsRed | ~600 | [9] |
| mRFP1 | <60 | [9,125] |
| mCherry | 15–40 b | [9,125,126] |
| mStrawberry | 50 | |
| TagRFP-T | 100 | [125] |
| FusionRed | 130 | [127] |
| Far-Red | ||
| mKate2 | <20 | [126] |
| mNeptune | 35 | [128] |
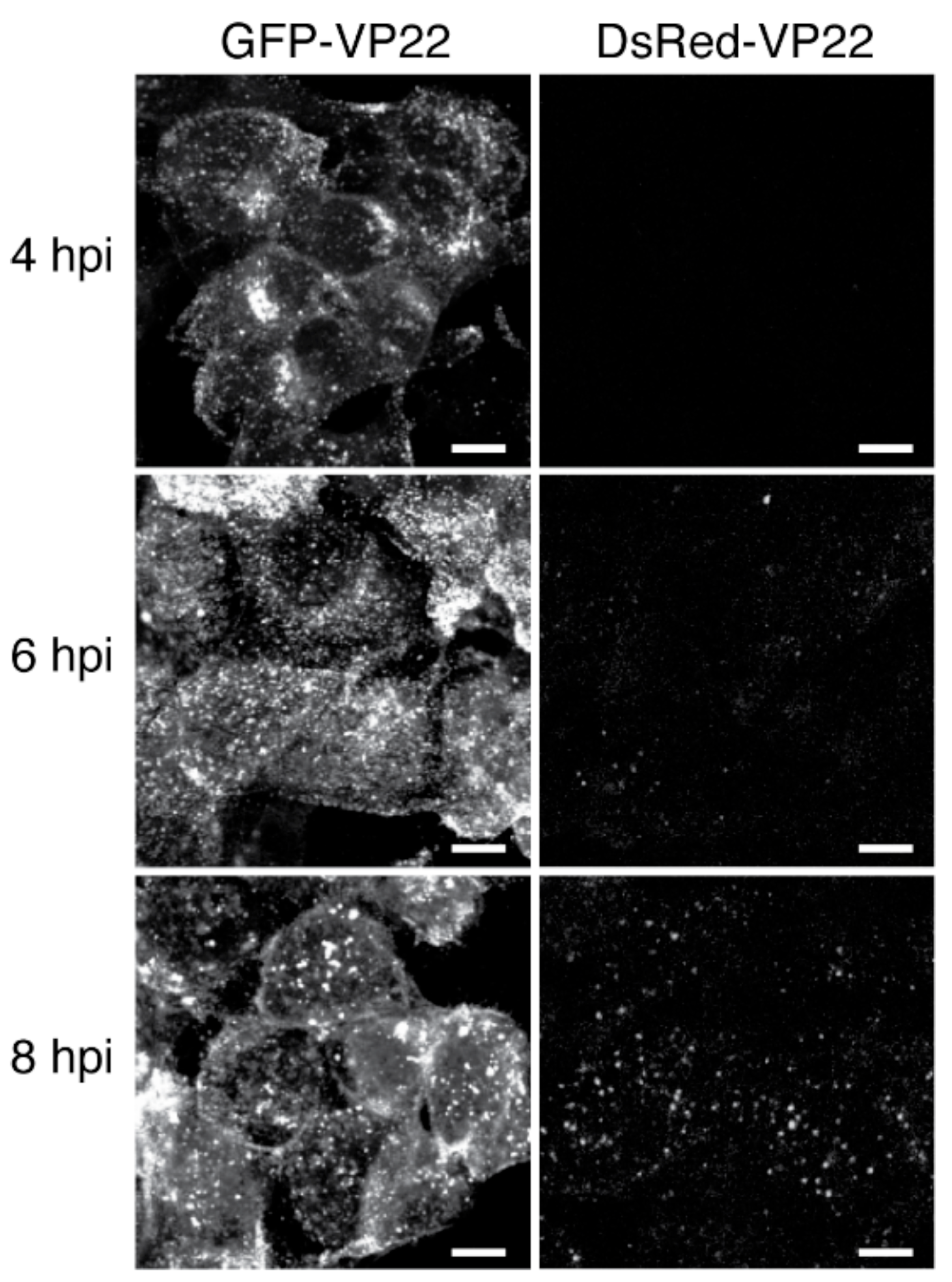
3.1.3. Fluorescent Protein pH Sensitivity
| Compartment | pH | References |
|---|---|---|
| Cytoplasm | 7.2–7.4 | [129,130] |
| Nucleus | 7.4–7.8 | [131,132] |
| Secretory Pathway | ||
| Endoplasmic Reticulum | 7.2 | [129] |
| cis-Golgi | 6.7 | [129] |
| trans-Golgi | 6.0 | [129] |
| Secretory Vesicles | 5.2–5.7 | [129] |
| Endocytic Pathway | ||
| Early and Recycling Endosomes | 6.3–6.5 | [129] |
| Late Endosomes | 6.0 | [129] |
| Lysosomes | 5.5 | [129] |
| Fluorescent Protein | pKa | Notes | Reference |
|---|---|---|---|
| Blue | |||
| mTagBFP2 | 2.7 | [116] | |
| Cyan | as a class, cyan FPs are relatively pH insensitive | ||
| ECFP, mCerulean | 4.7 | [133] | |
| mTFP1 | 4.3 | [134] | |
| mCerulean3 | 3.2 | [135] | |
| mTurquoise2 | 3.1 | [118] | |
| Green | as a class, green FPs are moderately pH sensitive | ||
| mEGFP, mEmerald | 6.0 | [136] | |
| mNeonGreen | 5.7 | [122] | |
| mTagGFP2 | 5.0 | [123] | |
| T-Sapphire | 4.9 | a UV-excited green FP | |
| superecliptic pHluorin | 7.2 | purposely optimized to be very pH sensitive | [137] |
| Yellow | as a class, modern yellow FPs are moderately pH sensitive | ||
| EYFP | 6.9 | very pH sensitive, newer variants are somewhat improved | |
| mVenus, SYFP2 | 6.0 | [117,138] | |
| mCitrine | 5.7 | [136] | |
| Orange | |||
| mKO2 | 5.0–5.5 | [125] | |
| mOrange, mOrange2 | 6.5–7.5 | very pH sensitive | [125] |
| Red | as a class, red FPs are relatively pH insensitive | ||
| mRFP1, mCherry, mStrawberry | ≤4.5 | [9] | |
| TagRFP-T | 4.0 | [125] | |
| FusionRed | 4.6 | [127] | |
| Far-Red | |||
| mKate2 | 5.4 | [126] | |
| mNeptune | 5.8 | [128] |
3.2. Choice of Fluorescent Protein Insertion Site and Linker
3.3. Choice of Excitation Light Sources
4. Case Studies: Discussion of Particular Fluorescent Protein Fusions
4.1. PRV Membrane Protein US9
4.2. HSV-1 and PRV Capsid Protein VP26
4.2.1. Compensatory Mutations that May Affect FP-VP26 Expression
4.2.2. Fluorescent Protein Dimerization
4.2.3. Assemblons
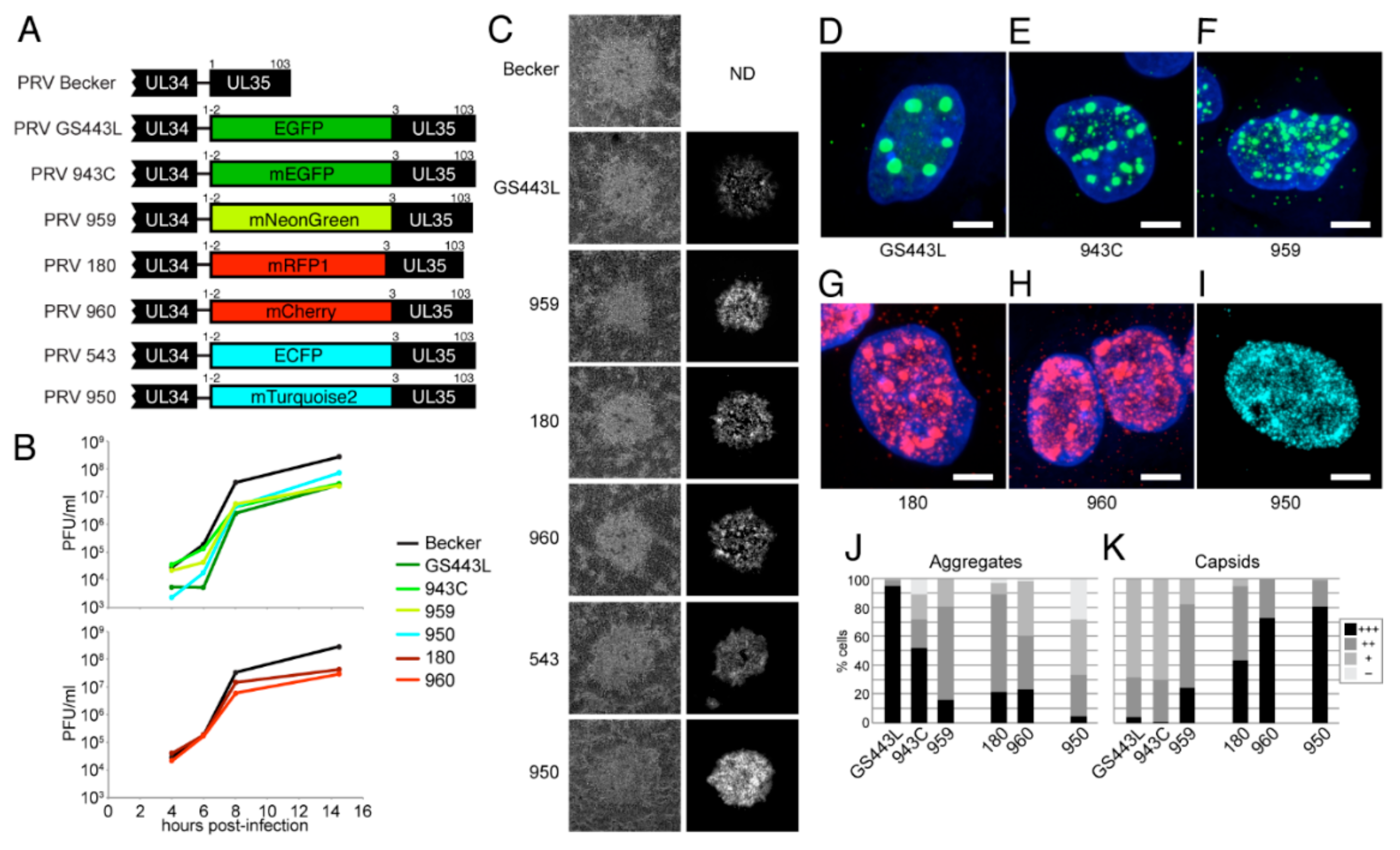
4.2.4. Description and Characterization of New VP26 Fusions in PRV
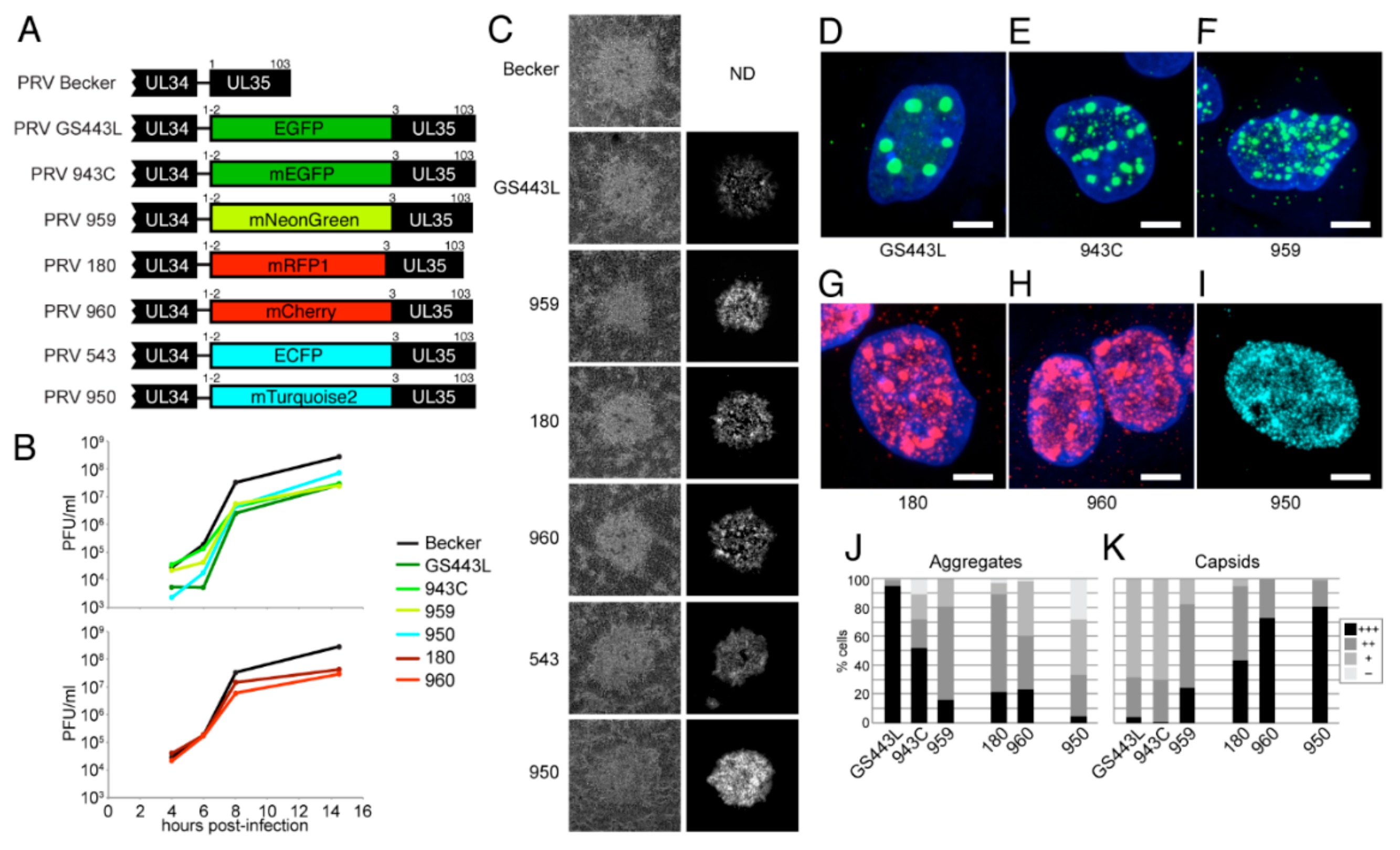
5. Comparison to Immunofluorescence
6. Beyond Traditional Fluorescence Microscopy
6.1. Fluorescent Protein Fusions as Proteomics Probes
6.2. Flow Virometry
6.3. Herpes Past the Diffraction Barrier
Acknowledgments
Conflicts of Interest
References
- Shimomura, O. The discovery of aequorin and green fluorescent protein. J. Microsc. 2005, 217, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Prasher, D.C.; Eckenrode, V.K.; Ward, W.W.; Prendergast, F.G.; Cormier, M.J. Primary structure of the Aequorea victoria green-fluorescent protein. Gene 1992, 111, 229–233. [Google Scholar] [CrossRef]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Inouye, S.; Tsuji, F.I. Aequorea green fluorescent protein. Expression of the gene and fluorescence characteristics of the recombinant protein. FEBS Lett. 1994, 341, 277–280. [Google Scholar] [CrossRef]
- Cubitt, A.B.; Heim, R.; Adams, S.R.; Boyd, A.E.; Gross, L.A.; Tsien, R.Y. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci. 1995, 20, 448–455. [Google Scholar] [CrossRef]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Sci. Signal. 2002, 296, 913. [Google Scholar] [CrossRef] [PubMed]
- Matz, M.V.; Fradkov, A.F.; Labas, Y.A.; Savitsky, A.P.; Zaraisky, A.G.; Markelov, M.L.; Lukyanov, S.A. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 1999, 17, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.G.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Patterson, G.H. Photoactivation and imaging of optical highlighter fluorescent proteins. In Current Protocols Cytometry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Chapter 12, Unit 12.23. [Google Scholar] [CrossRef]
- Nienhaus, K.; Ulrich Nienhaus, G. Fluorescent proteins for live-cell imaging with super-resolution. Chem. Soc. Rev. 2013, 43, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Tantama, M.; Hung, Y.P.; Yellen, G. Optogenetic reporters: Fluorescent protein-based genetically encoded indicators of signaling and metabolism in the brain. Prog. Brain Res. 2012, 196, 235–263. [Google Scholar] [PubMed]
- Bizzarri, R.; Serresi, M.; Luin, S.; Beltram, F. Green fluorescent protein based pH indicators for in vivo use: a review. Anal. Bioanal Chem. 2008, 393, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Adam, V.; Berardozzi, R.; Byrdin, M.; Bourgeois, D. Phototransformable fluorescent proteins: Future challenges. Curr. Opin. Chem. Biol. 2014, 20, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Adam, V. Phototransformable fluorescent proteins: Which one for which application? Histochem. Cell Biol. 2014, 142, 19–41. [Google Scholar] [CrossRef] [PubMed]
- Jöns, A.; Mettenleiter, T.C. Green fluorescent protein expressed by recombinant pseudorabies virus as an in vivo marker for viral replication. J. Virol. Methods 1997, 66, 283–292. [Google Scholar] [CrossRef]
- Foster, T.P.; Rybachuk, G.V.; Kousoulas, K.G. Expression of the enhanced green fluorescent protein by herpes simplex virus type 1 (HSV-1) as an in vitro or in vivo marker for virus entry and replication. J. Virol. Methods 1998, 75, 151–160. [Google Scholar] [CrossRef]
- Zerboni, L.; Sommer, M.; Ware, C.F.; Arvin, A.M. Varicella-Zoster Virus Infection of a Human CD4-Positive T-Cell Line. Virology 2000, 270, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, R.; Wellish, M.; White, T.; Soike, K.; Cohrs, R.; Kleinschmidt-Demasters, B.K.; Gilden, D.H. Infectious simian varicella virus expressing the green fluorescent protein. J. Neurovirol. 1998, 4, 438–444. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hogue, I.B.; Enquist, L.W.; Princeton University, Princeton, NJ, USA. Unpublished work. 2011.
- Klingbeil, K.; Lange, E.; Teifke, J.P.; Mettenleiter, T.C.; Fuchs, W. Immunization of pigs with an attenuated pseudorabies virus recombinant expressing the hemagglutinin of pandemic swine origin H1N1 influenza A virus. J. Gen. Virol. 2014, 95 (Pt 4), 948–959. [Google Scholar] [CrossRef] [PubMed]
- Card, J.P.; Enquist, L.W. Use and Visualization of Neuroanatomical Viral Transneuronal Tracers. In Visualization Techniques; Humana Press: New York, NY, USA, 2012; Volume 70, pp. 225–268. [Google Scholar]
- Wojaczynski, G.J.; Engel, E.A.; Steren, K.E.; Enquist, L.W.; Patrick Card, J. The neuroinvasive profiles of H129 (herpes simplex virus type 1) recombinants with putative anterograde-only transneuronal spread properties. Brain Struct. Funct. 2014, 220, 1395–1420. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.W.; Engel, E.A.; Enquist, L.W. Characterization of a replication-incompetent pseudorabies virus mutant lacking the sole immediate early gene IE180. MBio 2014, 5, e01850. [Google Scholar] [CrossRef] [PubMed]
- DeFalco, J.; Tomishima, M.; Liu, H.; Zhao, C.; Cai, X.; Marth, J.D.; Enquist, L.; Friedman, J.M. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science 2001, 291, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Lo, L.; Anderson, D.J. A Cre-dependent, anterograde transsynaptic viral tracer for mapping output pathways of genetically marked neurons. Neuron 2011, 72, 938–950. [Google Scholar] [CrossRef] [PubMed]
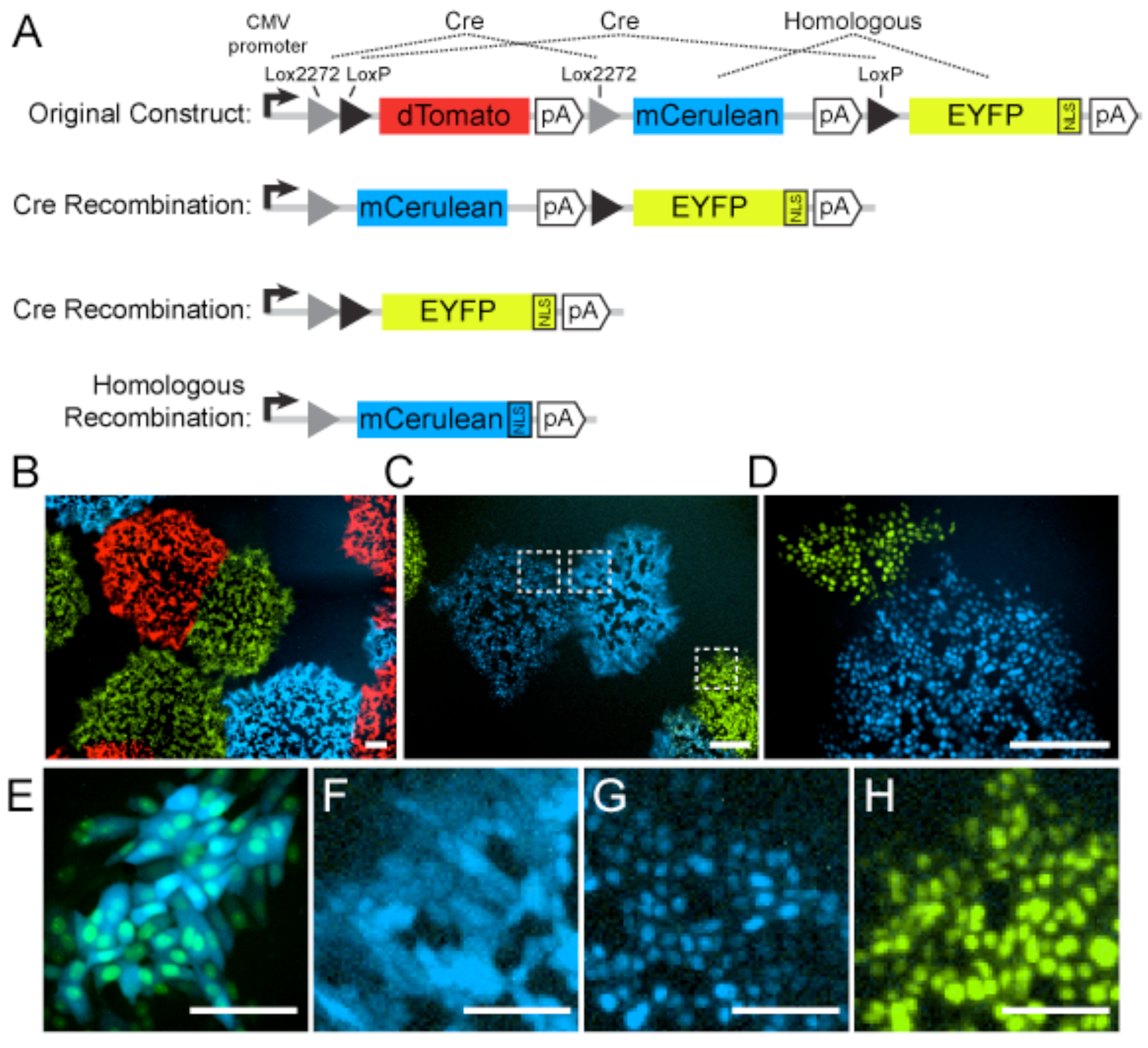
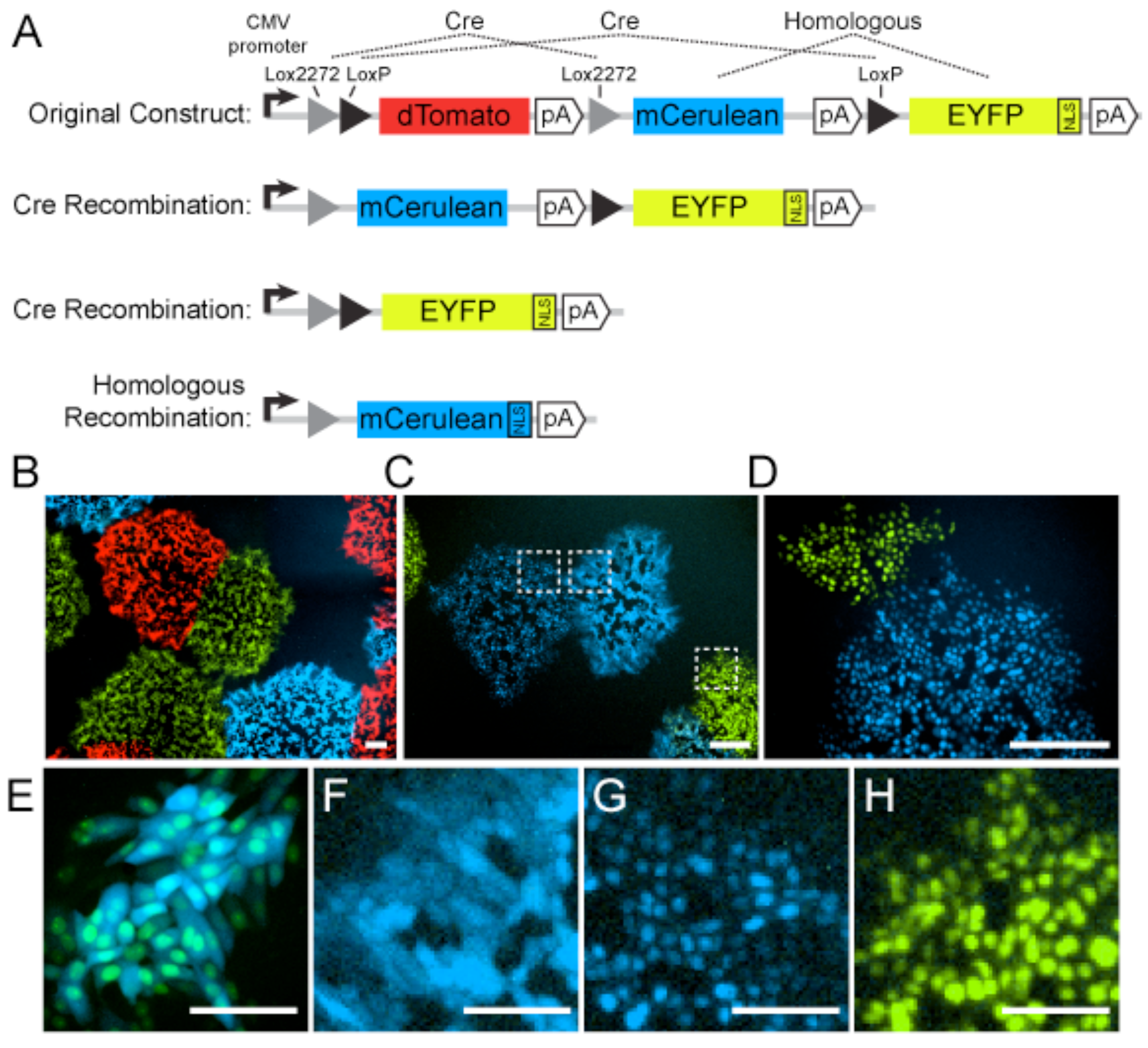
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef] [PubMed]
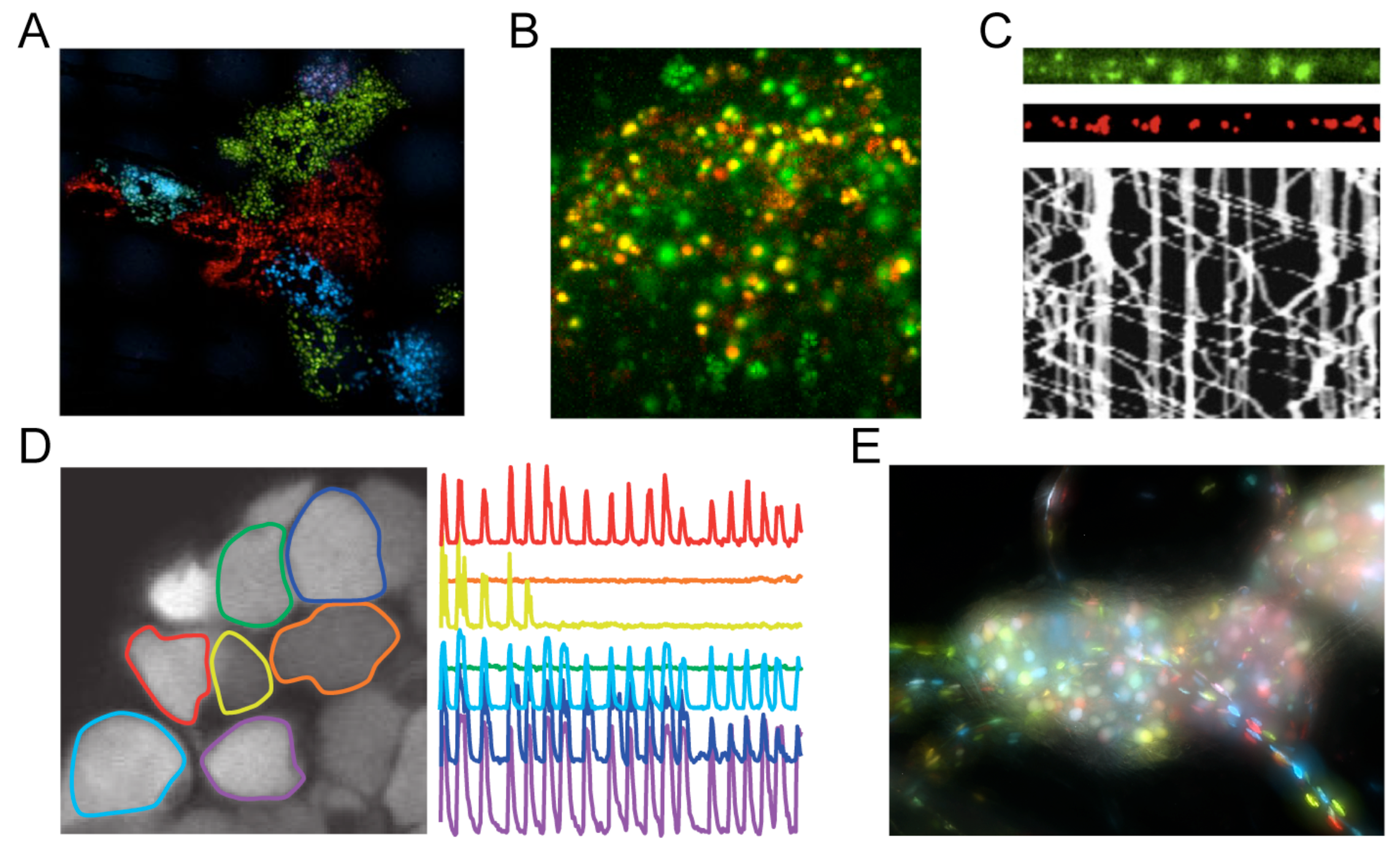
- Card, J.P.; Kobiler, O.; Ludmir, E.B.; Desai, V.; Sved, A.F.; Enquist, L.W. A dual infection pseudorabies virus conditional reporter approach to identify projections to collateralized neurons in complex neural circuits. PLoS ONE 2011, 6, e21141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Card, J.P.; Kobiler, O.; McCambridge, J.; Ebdlahad, S.; Shan, Z.; Raizada, M.K.; Sved, A.F.; Enquist, L.W. Microdissection of neural networks by conditional reporter expression from a Brainbow herpesvirus. Proc. Natl. Acad. Sci. USA 2011, 108, 3377–3382. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.R.; Badura, A.; Pacheco, D.A.; Lynch, L.A.; Schneider, E.R.; Taylor, M.P.; Hogue, I.B.; Enquist, L.W.; Murthy, M.; Wang, S.S.-H. Fast GCaMPs for improved tracking of neuronal activity. Nat. Commun. 2013, 4, 2170. [Google Scholar] [CrossRef] [PubMed]
- Granstedt, A.E.; Bosse, J.B.; Thiberge, S.Y.; Enquist, L.W. In vivo imaging of alphaherpesvirus infection reveals synchronized activity dependent on axonal sorting of viral proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3516–E3525. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.L.; Shieh, M.T.; Sawtell, N.M. Analysis of herpes simplex virus ICP0 promoter function in sensory neurons during acute infection, establishment of latency, and reactivation in vivo. J. Virol. 2003, 77, 12319–12330. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Arii, J.; Akashi, H.; Kawaguchi, Y. Identification of multiple sites suitable for insertion of foreign genes in herpes simplex virus genomes. Microbiol. Immunol. 2009, 53, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Balliet, J.W.; Kushnir, A.S.; Schaffer, P.A. Construction and characterization of a herpes simplex virus type I recombinant expressing green fluorescent protein: Acute phase replication and reactivation in mice. Virology 2007, 361, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Kobiler, O.; Enquist, L.W. Alphaherpesvirus axon-to-cell spread involves limited virion transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 17046–17051. [Google Scholar] [CrossRef] [PubMed]
- Hogue, I.B.; Bosse, J.B.; Hu, J.-R.; Thiberge, S.Y.; Enquist, L.W. Cellular mechanisms of alpha herpesvirus egress: live cell fluorescence microscopy of pseudorabies virus exocytosis. PLoS Pathog. 2014, 10, e1004535. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Greco, T.M.; Taylor, M.P.; Ambrosini, A.E.; Cristea, I.M.; Enquist, L.W. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 2012, 12, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Granstedt, A.E.; Enquist, L.W.; Princeton University, Princeton, NJ, USA. Unpublished work. 2012.
- Smith, G.A. Herpesvirus Transport to the Nervous System and Back Again. Annu. Rev. Microbiol. 2012, 66, 153–176. [Google Scholar] [CrossRef] [PubMed]
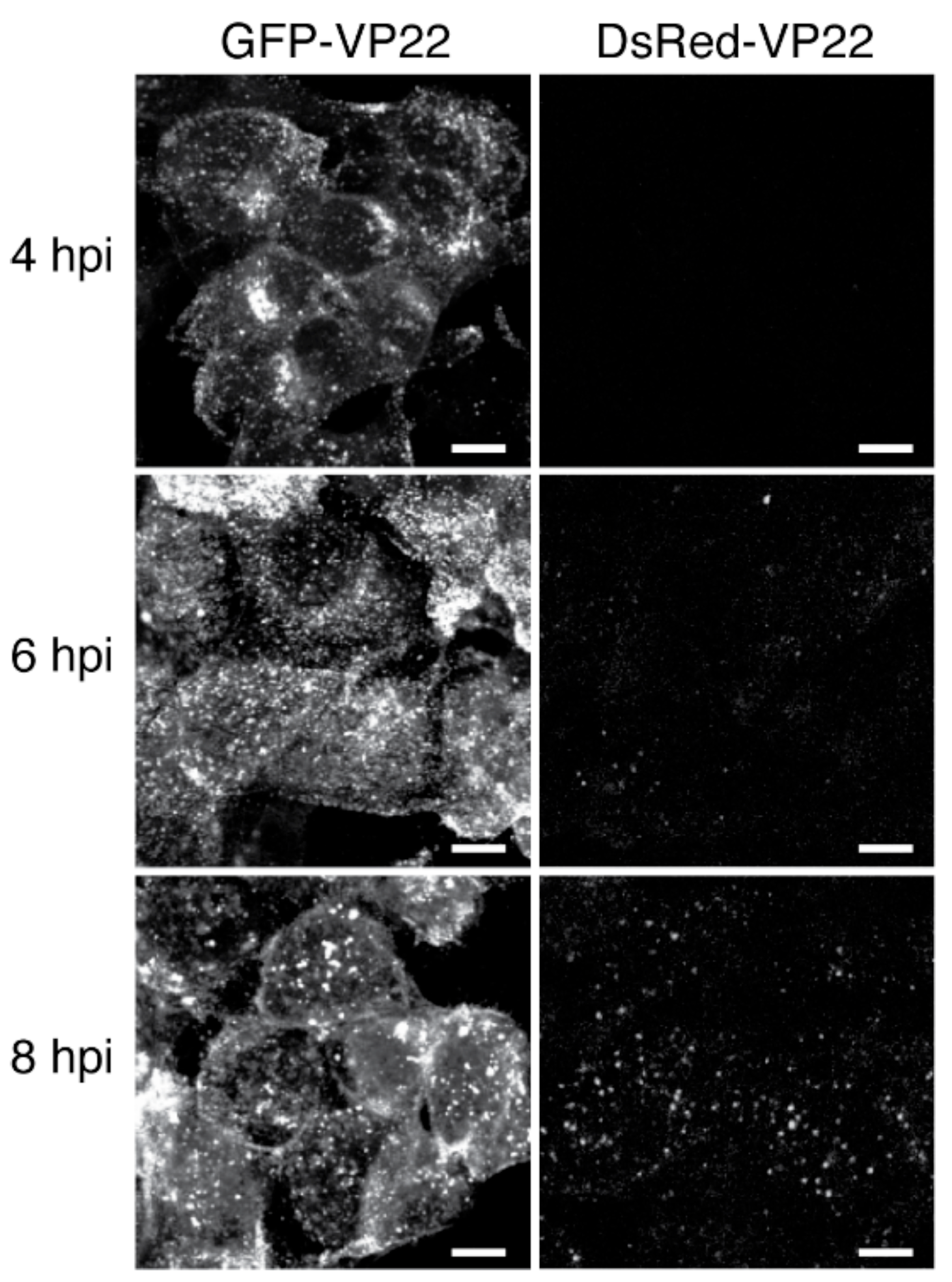
- Elliott, G.; O'Hare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997, 88, 223–233. [Google Scholar] [CrossRef]
- Desai, P.; Person, S. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 1998, 72, 7563–7568. [Google Scholar] [PubMed]
- Toropova, K.; Huffman, J.B.; Homa, F.L.; Conway, J.F. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J. Virol. 2011, 85, 7513–7522. [Google Scholar] [CrossRef] [PubMed]
- Cockrell, S.K.; Sanchez, M.E.; Erazo, A.; Homa, F.L. Role of the UL25 protein in herpes simplex virus DNA encapsidation. J. Virol. 2009, 83, 47–57. [Google Scholar] [CrossRef] [PubMed]
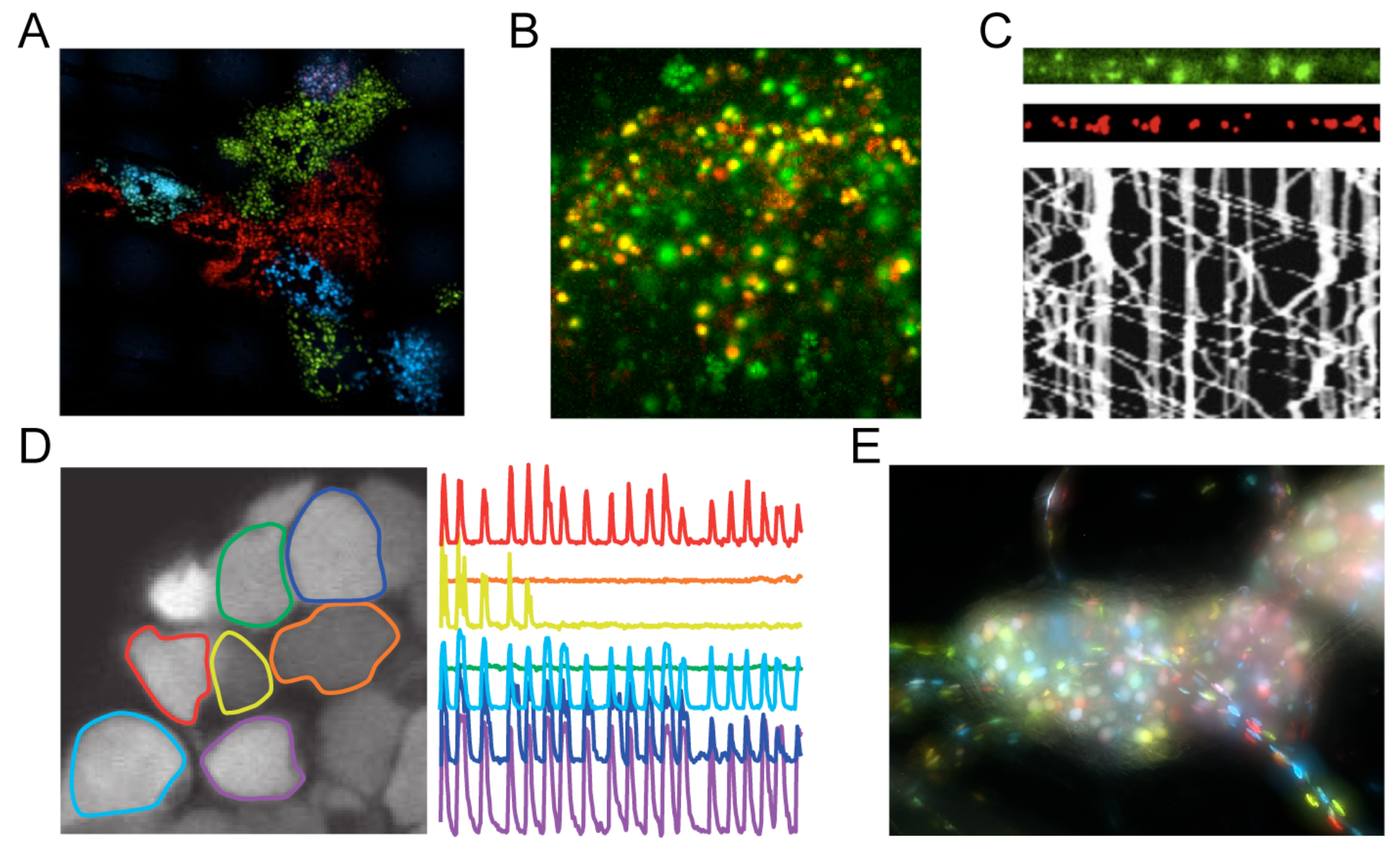
- Bohannon, K.P.; Sollars, P.J.; Pickard, G.E.; Smith, G.A. Fusion of a fluorescent protein to the pUL25 minor capsid protein of pseudorabies virus allows live-cell capsid imaging with negligible impact on infection. J. Gen. Virol. 2011, 93, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Nagel, C.-H.; Döhner, K.; Binz, A.; Bauerfeind, R.; Sodeik, B. Improper Tagging of the Non-Essential Small Capsid Protein VP26 Impairs Nuclear Capsid Egress of Herpes Simplex Virus. PLoS ONE 2012, 7, e44177. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Gross, S.P.; Enquist, L.W. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 3466–3470. [Google Scholar] [CrossRef] [PubMed]
- Hogue, I.B.; Wilson, J.J.; Enquist, L.W.; Princeton University, Princeton, NJ, USA. Unpublished work. 2015.
- Coller, K.E.; Smith, G.A. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 2008, 9, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Loomis, J.S.; Courtney, R.J.; Wills, J.W. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2003, 77, 11417–11424. [Google Scholar] [CrossRef] [PubMed]
- Bohannon, K.P.; Jun, Y.; Gross, S.P.; Smith, G.A. Differential protein partitioning within the herpesvirus tegument and envelope underlies a complex and variable virion architecture. Proc. Natl. Acad. Sci. USA 2013, 110, E1613–E1620. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.L.; Meckes, D.G.; Marsh, J.A.; Ward, M.D.; Yeh, P.-C.; Baird, N.L.; Wilson, C.B.; Semmes, O.J.; Wills, J.W. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J. Virol. 2010, 84, 2963–2971. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, V.; Jung, M.; Alter, J.D.; Wills, E.G.; Johnston, S.M.; Kawaguchi, Y.; Baines, J.D.; Banfield, B.W. The herpes simplex virus 2 UL21 protein is essential for virus propagation. J. Virol. 2013, 87, 5904–5915. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Vollmer, B.; Unsay, J.D.; Klupp, B.G.; García-Sáez, A.J.; Mettenleiter, T.C.; Antonin, W. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J. Biol. Chem. 2015, 290, 6962–6974. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Guo, L.; Yang, E.; Liao, Y.; Liu, L.; Che, Y.; Zhang, Y.; Wang, L.; Wang, J.; Li, Q. HSV-1 nucleocapsid egress mediated by UL31 in association with UL34 is impeded by cellular transmembrane protein 140. Virology 2014, 464–465, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Leelawong, M.; Guo, D.; Smith, G.A. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J. Virol. 2011, 85, 11675–11684. [Google Scholar] [CrossRef] [PubMed]
- Luxton, G.W.G.; Haverlock, S.; Coller, K.E.; Antinone, S.E.; Pincetic, A.; Smith, G.A. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5832–5837. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Smith, G.A. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J. Virol. 2010, 84, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Saksena, M.M.; Wakisaka, H.; Tijono, B.; Boadle, R.A.; Rixon, F.; Takahashi, H.; Cunningham, A.L. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J. Virol. 2006, 80, 3592–3606. [Google Scholar] [CrossRef] [PubMed]
- Zenner, H.L.; Mauricio, R.; Banting, G.; Crump, C.M. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J. Virol. 2013, 87, 13115–13123. [Google Scholar] [CrossRef] [PubMed]
- Willard, M. Rapid directional translocations in virus replication. J. Virol. 2002, 76, 5220–5232. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, M.; Elliott, G. Fluorescent tagging of herpes simplex virus tegument protein VP13/14 in virus infection. J. Virol. 2001, 75, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- La Boissière, S.; Izeta, A.; Malcomber, S.; O'Hare, P. Compartmentalization of VP16 in cells infected with recombinant herpes simplex virus expressing VP16-green fluorescent protein fusion proteins. J. Virol. 2004, 78, 8002–8014. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Huang, J.; Wang, H. The study of the intercellular trafficking of the fusion proteins of herpes simplex virus protein VP22. PLoS ONE 2014, 9, e100840. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, T.; Ch'ng, T.H.; Flood, E.A.; Gross, S.P.; Enquist, L.W. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 2005, 79, 3903–3919. [Google Scholar] [CrossRef] [PubMed]
- Cun, W.; Hong, M.; Liu, L.-D.; Dong, C.-H.; Luo, J.; Li, Q.-H. Structural and functional characterization of herpes simplex virus 1 immediate-early protein infected-cell protein 22. J. Biochem. 2006, 140, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Tanaka, M.; Yamamoto, M.; Asai, R.; Sata, T.; Nishiyama, Y.; Kawaguchi, Y. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 2008, 82, 6172–6189. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Roy, B.B.; Zhang, H.; Banfield, B.W. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 2010, 397, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Calton, C.M.; Randall, J.A.; Adkins, M.W.; Banfield, B.W. The pseudorabies virus serine/threonine kinase Us3 contains mitochondrial, nuclear and membrane localization signals. Virus Genes 2004, 29, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Kim, T.; Cheng, H.H. Visualization of Marek's disease virus in vitro using enhanced green fluorescent protein fused with US10. Virus Genes 2013, 47, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Harland, J.; Dunn, P.; Cameron, E.; Conner, J.; Brown, S.M. The herpes simplex virus (HSV) protein ICP34.5 is a virion component that forms a DNA-binding complex with proliferating cell nuclear antigen and HSV replication proteins. J. Neurovirol. 2003, 9, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, P.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein Vmw110 inhibits progression of cells through mitosis and from G(1) into S phase of the cell cycle. J. Virol. 1999, 73, 9456–9467. [Google Scholar] [PubMed]
- Gu, H.; Poon, A.P.; Roizman, B. During its nuclear phase the multifunctional regulatory protein ICP0 undergoes proteolytic cleavage characteristic of polyproteins. Proc. Natl. Acad. Sci. USA 2009, 106, 19132–19137. [Google Scholar] [CrossRef] [PubMed]
- Paulus, C.; Enquist, L.W.; Princeton University, Princeton, NJ, USA. Unpublished work. 2005.
- Liu, M.; Schmidt, E.E.; Halford, W.P. ICP0 dismantles microtubule networks in herpes simplex virus-infected cells. PLoS ONE 2010, 5, e10975. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Sourvinos, G.; Orr, A. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 2003, 77, 3680–3689. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.A.; Manchak, M.D.; Hager, E.J.; Krummenacher, C.; Whitbeck, J.C.; Levin, M.J.; Freed, C.R.; Wilcox, C.L.; Cohen, G.H.; Eisenberg, R.J.; et al. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J. Neurovirol. 2005, 11, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Dukhovny, A.; Sloutskin, A.; Markus, A.; Yee, M.B.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus infects human embryonic stem cell-derived neurons and neurospheres but not pluripotent embryonic stem cells or early progenitors. J. Virol. 2012, 86, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Crump, C.M.; Bruun, B.; Bell, S.; Pomeranz, L.E.; Minson, T.; Browne, H.M. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J. Gen. Virol. 2004, 85, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Kramer, T.; Lyman, M.G.; Kratchmarov, R.; Enquist, L.W. Visualization of an alphaherpesvirus membrane protein that is essential for anterograde axonal spread of infection in neurons. MBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.-Y.K.; Crump, C.M. HSV-1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 2015, 7, 915–938. [Google Scholar] [CrossRef] [PubMed]
- Lorentzen, E.U.; Eing, B.R.; Hafezi, W.; Manservigi, R.; Kühn, J.E. Replication-competent herpes simplex virus type 1 mutant expressing an autofluorescent glycoprotein H fusion protein. Intervirology 2001, 44, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Uema, M.; Sagara, H.; Tanaka, M.; Sata, T.; Hashimoto, Y.; Kawaguchi, Y. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 2008, 82, 5198–5211. [Google Scholar] [CrossRef] [PubMed]
- Potel, C.; Kaelin, K.; Gautier, I.; Lebon, P.; Coppey, J.; Rozenberg, F. Incorporation of green fluorescent protein into the essential envelope glycoprotein B of herpes simplex virus type 1. J. Virol. Methods 2002, 105, 13–23. [Google Scholar] [CrossRef]
- Potel, C.; Kaelin, K.; Danglot, L.; Triller, A.; Vannier, C.; Rozenberg, F. Herpes simplex virus type 1 glycoprotein B sorting in hippocampal neurons. J. Gen. Virol. 2003, 84, 2613–2624. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.; Rudolph, J.; Brandmüller, C.; Just, F.T.; Osterrieder, N. The equine herpesvirus 1 UL34 gene product is involved in an early step in virus egress and can be efficiently replaced by a UL34-GFP fusion protein. Virology 2002, 300, 189–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, T.; Ma, G.; Osterrieder, N. Equine Herpesvirus 1 Multiply Inserted Transmembrane Protein pUL43 Cooperates with pUL56 in Downregulation of Cell Surface Major Histocompatibility Complex Class I. J. Virol. 2015, 89, 6251–6263. [Google Scholar] [CrossRef] [PubMed]
- Goheen, M. Construction and Characterization of Pseudorabies Virus Mutants Expressing gC-GFP. Master's Thesis, Princeton University, Princeton, NJ, USA, 2007. [Google Scholar]
- El Kasmi, I.; Lippé, R. Herpes simplex virus 1 gN partners with gM to modulate the viral fusion machinery. J. Virol. 2015, 89, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, T.; Kawaguchi, Y.; Nozawa, N.; Mori, I.; Nishiyama, Y. Herpes simplex virus protein UL11 but not UL51 is associated with lipid rafts. Virus Genes 2007, 35, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.; Osterrieder, N. Equine herpesvirus type 1 (EHV-1) glycoprotein K is required for efficient cell-to-cell spread and virus egress. Virology 2004, 329, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, V.; Banfield, B.W. Dysregulation of autophagy in murine fibroblasts resistant to HSV-1 infection. PLoS ONE 2012, 7, e42636. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.S.B.; Nicola, A.V.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 2005, 79, 6655–6663. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Smith, G.A. Two modes of herpesvirus trafficking in neurons: Membrane acquisition directs motion. 2006, 80, 11235–11240. [Google Scholar] [CrossRef] [PubMed]
- Tirabassi, R.S.; Enquist, L.W. Role of the pseudorabies virus gI cytoplasmic domain in neuroinvasion, virulence, and posttranslational N-linked glycosylation. J. Virol. 2000, 74, 3505–3516. [Google Scholar] [CrossRef] [PubMed]
- Kratchmarov, R.; Kramer, T.; Greco, T.M.; Taylor, M.P.; Ch'ng, T.H.; Cristea, I.M.; Enquist, L.W. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J. Virol. 2013, 87, 9431–9440. [Google Scholar] [CrossRef] [PubMed]
- Loret, S.; Guay, G.; Lippé, R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 2008, 82, 8605–8618. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Greco, T.M.; Enquist, L.W.; Cristea, I.M. Proteomic characterization of pseudorabies virus extracellular virions. J. Virol. 2011, 85, 6427–6441. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Gong, D.; Wu, T.-T.; Sun, R.; Zhou, Z.H. Organization of capsid-associated tegument components in Kaposi's sarcoma-associated herpesvirus. J. Virol. 2014, 88, 12694–12702. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.F.; Cockrell, S.K.; Copeland, A.M.; Newcomb, W.W.; Brown, J.C.; Homa, F.L. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J. Mol. Biol. 2010, 397, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Homa, F.L.; Huffman, J.B.; Toropova, K.; Lopez, H.R.; Makhov, A.M.; Conway, J.F. Structure of the pseudorabies virus capsid: Comparison with herpes simplex virus type 1 and differential binding of essential minor proteins. J. Mol. Biol. 2013, 425, 3415–3428. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.B.; Enquist, L.W.; Princeton University, Princeton, NJ, USA. Unpublished work. 2013.
- Krautwald, M.; Maresch, C.; Klupp, B.G.; Fuchs, W.; Mettenleiter, T.C. Deletion or green fluorescent protein tagging of the pUL35 capsid component of pseudorabies virus impairs virus replication in cell culture and neuroinvasion in mice. J. Gen. Virol. 2008, 89, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Northwestern University, Chicago, IL. Personal communication, 2015.
- Demmin, G.L.; Clase, A.C.; Randall, J.A.; Enquist, L.W.; Banfield, B.W. Insertions in the gG gene of pseudorabies virus reduce expression of the upstream Us3 protein and inhibit cell-to-cell spread of virus infection. J. Virol. 2001, 75, 10856–10869. [Google Scholar] [CrossRef] [PubMed]
- Haseloff, J.; Siemering, K.R.; Prasher, D.C.; Hodge, S. Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc. Natl. Acad. Sci. USA 1997, 94, 2122–2127. [Google Scholar] [CrossRef] [PubMed]
- Gurskaya, N.G.; Staroverov, D.B.; Fradkov, A.F.; Lukyanov, K.A. The coding region of far-red fluorescent protein Katushka contains a strong donor splice site. Russ. J. Bioorg. Chem. 2011, 37, 380–382. [Google Scholar] [CrossRef]
- Gee, P.; Ando, Y.; Kitayama, H.; Yamamoto, S.P.; Kanemura, Y.; Ebina, H.; Kawaguchi, Y.; Koyanagi, Y. APOBEC1-mediated editing and attenuation of herpes simplex virus 1 DNA indicate that neurons have an antiviral role during herpes simplex encephalitis. J. Virol. 2011, 85, 9726–9736. [Google Scholar] [CrossRef] [PubMed]
- Suspène, R.; Aynaud, M.-M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.-P.; Meyerhans, A.; Wain-Hobson, S. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef] [PubMed]
- Elde, N.C.; Child, S.J.; Eickbush, M.T.; Kitzman, J.O.; Rogers, K.S.; Shendure, J.; Geballe, A.P.; Malik, H.S. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 2012, 150, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Bestvater, F.; Knoch, T.A.; Langowski, J.; Spiess, E. Construct conversions caused by simultaneous co-transfection: "GFP-walking". BioTechniques 2002, 32, 844–854. [Google Scholar] [PubMed]
- Smith, G.A.; Banfield, B.W. The development and use of alpha-herpesviruses-expressing fluorescent proteins. In Alpha Herpesviruses: Molecular and Cellular Biology; Sandri-Goldin, R.M., Ed.; Caister Academic Press: Wymondham, UK, 2006; pp. 205–218. [Google Scholar]
- Brown, S.M.; Ritchie, D.A.; Subak-Sharpe, J.H. Genetic studies with herpes simplex virus type 1. The isolation of temperature-sensitive mutants, their arrangement into complementation groups and recombination analysis leading to a linkage map. J. Gen. Virol. 1973, 18, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Cohen, K.B.; Luo, T.; Lichtman, J.W.; Sanes, J.R. Improved tools for the Brainbow toolbox. Nat. Methods 2013, 10, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, P.E.; Lehman, I.R. Herpes simplex virus DNA replication. Annu. Rev. Biochem. 1997, 66, 347–384. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.B.; Hogue, I.B.; Feric, M.; Thiberge, S.Y.; Sodeik, B.; Brangwynne, C.P.; Enquist, L.W. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc. Natl. Acad. Sci. USA 2015, 112, E5725–E5733. [Google Scholar] [CrossRef] [PubMed]
- Subach, O.M.; Cranfill, P.J.; Davidson, M.W.; Verkhusha, V.V. An Enhanced Monomeric Blue Fluorescent Protein with the High Chemical Stability of the Chromophore. PLoS ONE 2011, 6, e28674. [Google Scholar] [CrossRef] [PubMed]
- Kremers, G.-J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W.J. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Förster radius. Biochemistry 2006, 45, 6570–6580. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W.J.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Heim, R.; Cubitt, A.B.; Tsien, R.Y. Improved green fluorescence. Nature 1995, 373, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, R.; Yamagishi-Shirasaki, M.; Funatsu, T. Kinetic study of de novo chromophore maturation of fluorescent proteins. Anal. Biochem. 2011, 414, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Evdokimov, A.G.; Pokross, M.E.; Egorov, N.S.; Zaraisky, A.G.; Yampolsky, I.V.; Merzlyak, E.M.; Shkoporov, A.N.; Sander, I.; Lukyanov, K.A.; Chudakov, D.M. Structural basis for the fast maturation of Arthropoda green fluorescent protein. EMBO Rep. 2006, 7, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Subach, O.M.; Gundorov, I.S.; Yoshimura, M.; Subach, F.V.; Zhang, J.; Grüenwald, D.; Souslova, E.A.; Chudakov, D.M.; Verkhusha, V.V. Conversion of Red Fluorescent Protein into a Bright Blue Probe. Chem. Biol. 2008, 15, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Strack, R.L.; Keenan, R.J.; Glick, B.S. Noncytotoxic DsRed Derivatives for Whole-Cell Labeling. In Methods in Molecular Biology; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 699, pp. 355–370. [Google Scholar]
- Shaner, N.C.; Lin, M.Z.; McKeown, M.R.; Steinbach, P.A.; Hazelwood, K.L.; Davidson, M.W.; Tsien, R.Y. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat. Methods 2008, 5, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Shcherbo, D.; Murphy, C.S.; Ermakova, G.V.; Solovieva, E.A.; Chepurnykh, T.V.; Shcheglov, A.S.; Verkhusha, V.V.; Pletnev, V.Z.; Hazelwood, K.L.; Roche, P.M.; et al. Far-red fluorescent tags for protein imaging in living tissues. Biochem. J. 2009, 418, 567. [Google Scholar] [CrossRef] [PubMed]
- Shemiakina, I.I.; Ermakova, G.V.; Cranfill, P.J.; Baird, M.A.; Evans, R.A.; Souslova, E.A.; Staroverov, D.B.; Gorokhovatsky, A.Y.; Putintseva, E.V.; Gorodnicheva, T.V.; et al. A monomeric red fluorescent protein with low cytotoxicity. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.Z.; McKeown, M.R.; Ng, H.-L.; Aguilera, T.A.; Shaner, N.C.; Campbell, R.E.; Adams, S.R.; Gross, L.A.; Ma, W.; Alber, T.; et al. Autofluorescent Proteins with Excitation in the Optical Window for Intravital Imaging in Mammals. Chem. Biol. 2009, 16, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Paroutis, P.; Touret, N.; Grinstein, S. The pH of the secretory pathway: Measurement, determinants, and regulation. Physiology (Bethesda) 2004, 19, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Llopis, J.; McCaffery, J.M.; Miyawaki, A.; Farquhar, M.G.; Tsien, R.Y. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 6803–6808. [Google Scholar] [CrossRef] [PubMed]
- Seksek, O.; Bolard, J. Nuclear pH gradient in mammalian cells revealed by laser microspectrofluorimetry. J. Cell Sci. 1996, 109 (Pt 1), 257–262. [Google Scholar] [PubMed]
- Benink, H.; McDougall, M.; Klaubert, D.; Los, G. Direct pH measurements by using subcellular targeting of 5(and 6-) carboxyseminaphthorhodafluor in mammalian cells. BioTechniques 2009, 47, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.A.; Springer, G.H.; Granada, B.; Piston, D.W. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 2004, 22, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.-W.; Henderson, J.N.; Remington, S.J.; Campbell, R.E. Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: Structural characterization and applications in fluorescence imaging. Biochem. J. 2006, 400, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, M.L.; Kremers, G.-J.; Kraft, C.A.; Ray, K.; Cranfill, P.J.C.; Wilson, K.A.; Day, R.N.; Wachter, R.M.; Davidson, M.W.; Rizzo, M.A. An Improved Cerulean Fluorescent Protein with Enhanced Brightness and Reduced Reversible Photoswitching. PLoS ONE 2011, 6, e17896. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, S.; de Angelis, D.; Rothman, J.E.; Ryan, T.A. The Use of pHluorins for Optical Measurements of Presynaptic Activity. Biophys. J. 2000, 79, 2199–2208. [Google Scholar] [CrossRef]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Schwarzländer, M.; Logan, D.C.; Fricker, M.D.; Sweetlove, L.J. The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide “flashes”. Biochem. J. 2011, 437, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Melikyan, G.B.; Barnard, R.J.O.; Abrahamyan, L.G.; Mothes, W.; Young, J.A.T. Imaging individual retroviral fusion events: from hemifusion to pore formation and growth. Proc. Natl. Acad. Sci. USA 2005, 102, 8728–8733. [Google Scholar] [CrossRef] [PubMed]
- Miesenböck, G.; de Angelis, D.A.; Rothman, J.E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 1998, 394, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Ruzsics, Z.; Koszinowski, U.H. Herpesvirus genetics has come of age. Trends Microbiol. 2002, 10, 318–324. [Google Scholar] [CrossRef]
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus infections in the nervous system. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.G.; Feierbach, B.; Curanovic, D.; Bisher, M.; Enquist, L.W. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J. Virol. 2007, 81, 11363–11371. [Google Scholar] [CrossRef] [PubMed]
- Tomishima, M.J.; Enquist, L.W. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J. Cell Biol. 2001, 154, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Kratchmarov, R.; Taylor, M.P.; Enquist, L.W. Role of Us9 phosphorylation in axonal sorting and anterograde transport of pseudorabies virus. PLoS ONE 2013, 8, e58776. [Google Scholar] [CrossRef] [PubMed]
- Brideau, A.D.; del Rio, T.; Wolffe, E.J.; Enquist, L.W. Intracellular trafficking and localization of the pseudorabies virus Us9 type II envelope protein to host and viral membranes. J. Virol. 1999, 73, 4372–4384. [Google Scholar] [PubMed]
- Brideau, A.D.; Banfield, B.W.; Enquist, L.W. The Us9 gene product of pseudorabies virus, an alphaherpesvirus, is a phosphorylated, tail-anchored type II membrane protein. J. Virol. 1998, 72, 4560–4570. [Google Scholar] [PubMed]
- Lyman, M.G.; Curanovic, D.; Brideau, A.D.; Enquist, L.W. Fusion of enhanced green fluorescent protein to the pseudorabies virus axonal sorting protein Us9 blocks anterograde spread of infection in mammalian neurons. J. Virol. 2008, 82, 10308–10311. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Bosse, J.B.; Enquist, L.W.; Princeton University, Princeton, NJ, USA. Unpublished work. 2012.
- Szpara, M.L.; Tafuri, Y.R.; Parsons, L.; Shamim, S.R.; Verstrepen, K.J.; Legendre, M.; Enquist, L.W. A Wide Extent of Inter-Strain Diversity in Virulent and Vaccine Strains of Alphaherpesviruses. PLoS Pathog. 2011, 7, e1002282. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.L.; Ogle, W.O.; Roizman, B. Assemblons: nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 1996, 70, 4623–4631. [Google Scholar] [PubMed]
- Lamberti, C.; Weller, S.K. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 1998, 72, 2463–2473. [Google Scholar] [PubMed]
- De Bruyn Kops, A.; Uprichard, S.L.; Chen, M.; Knipe, D.M. Comparison of the Intranuclear Distributions of Herpes Simplex Virus Proteins Involved in Various Viral Functions. Virology 1998, 252, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Forest, T.; Barnard, S.; Baines, J.D. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 2005, 7, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Rupp, B.; Ruzsics, Z.; Sacher, T.; Koszinowski, U.H. Conditional cytomegalovirus replication in vitro and in vivo. J. Virol. 2005, 79, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Song, R.; Greco, T.M.; Cristea, I.M.; Enquist, L.W. The number of alphaherpesvirus particles infecting axons and the axonal protein repertoire determines the outcome of neuronal infection. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Brodersen, P.; Taylor, M.P.; Ludmir, E.B.; Enquist, L.W. Herpesvirus replication compartments originate with single incoming viral genomes. MBio 2011, 2, e00278-11. [Google Scholar] [CrossRef] [PubMed]
- Coons, A.H.; Creech, H.J.; Jones, R.N.; Berliner, E. The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J. Immunol. 1942, 45, 3159–3170. [Google Scholar]
- Weller, T.H.; Coons, A.H. Fluorescent antibody studies with agents of varicella and herpes zoster propagated in vitro. Proc. Soc. Exp. Biol. Med. 1954, 86, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, J. Cellular localization of Herpes simplex virus by means of fluorescent antibody. Virology 1956, 2, 496–510. [Google Scholar] [CrossRef]
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N.G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Zaichick, S.V.; Smith, G.A. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 2010, 84, 13019–13030. [Google Scholar] [CrossRef] [PubMed]
- Wisner, T.W.; Sugimoto, K.; Howard, P.W.; Kawaguchi, Y.; Johnson, D.C. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J. Virol. 2011, 85, 5919–5928. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, W.W.; Brown, J.C. Time-dependent transformation of the herpesvirus tegument. J. Virol. 2009, 83, 8082–8089. [Google Scholar] [CrossRef] [PubMed]
- Nagel, C.-H.; Döhner, K.; Fathollahy, M.; Strive, T.; Borst, E.M.; Messerle, M.; Sodeik, B. Nuclear egress and envelopment of herpes simplex virus capsids analyzed with dual-color fluorescence HSV1(17+). J. Virol. 2008, 82, 3109–3124. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2008, 18, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Kratchmarov, R.; Taylor, M.P.; Enquist, L.W. Making the case: Married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 2012, 22, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.; Miranda-Saksena, M.; Diefenbach, R.; Johnson, D. Letter in response to: Making the case: Married versus Separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 2013, 23, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Einhauer, A.; Jungbauer, A. The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins. J. Biochem. Biophys. Methods 2001, 49, 455–465. [Google Scholar] [CrossRef]
- Engel, E.A.; Song, R.; Koyuncu, O.O.; Enquist, L.W. Investigating the biology of alpha herpesviruses with MS-based proteomics. Proteomics 2015, 15, 1943–1956. [Google Scholar] [CrossRef] [PubMed]
- Cristea, I.M.; Williams, R.; Chait, B.T.; Rout, M.P. Fluorescent proteins as proteomic probes. Mol. Cell. Proteom. 2005, 4, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Fulwyler, M.J. Electronic separation of biological cells by volume. Science 1965, 150, 910–911. [Google Scholar] [CrossRef] [PubMed]
- Hulett, H.R.; Bonner, W.A.; Barrett, J.; Herzenberg, L.A. Cell sorting: automated separation of mammalian cells as a function of intracellular fluorescence. Science 1969, 166, 747–749. [Google Scholar] [CrossRef]
- Bailey, J.E.; Fazel-Makjlessi, J.; McQuitty, D.N.; Lee, Y.N.; Allred, J.C.; Oro, J.A. Characterization of bacterial growth by means of flow microfluorometry. Science 1977, 198, 1175–1176. [Google Scholar] [CrossRef] [PubMed]
- Cavelier, L.; Johannisson, A.; Gyllensten, U. Analysis of mtDNA copy number and composition of single mitochondrial particles using flow cytometry and PCR. Exp. Cell Res. 2000, 259, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Rajotte, D.; Stearns, C.D.; Kabcenell, A.K. Isolation of mast cell secretory lysosomes using flow cytometry. Cytom. A 2003, 55, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.F. Analysis and isolation of endocytic vesicles by flow cytometry and sorting: demonstration of three kinetically distinct compartments involved in fluid-phase endocytosis. Proc. Natl. Acad. Sci. USA 1985, 82, 8523–8526. [Google Scholar] [CrossRef] [PubMed]
- Nolte-'t Hoen, E.N.M.; van der Vlist, E.J.; Aalberts, M.; Mertens, H.C.H.; Bosch, B.J.; Bartelink, W.; Mastrobattista, E.; van Gaal, E.V.B.; Stoorvogel, W.; Arkesteijn, G.J.A.; et al. Quantitative and qualitative flow cytometric analysis of nanosized cell-derived membrane vesicles. Nanomedicine 2012, 8, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.B. Flow cytometer for measurement of the light scattering of viral and other submicroscopic particles. Cytometry A 2004, 57, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Hercher, M.; Mueller, W.; Shapiro, H.M. Detection and discrimination of individual viruses by flow cytometry. J. Histochem. Cytochem. 1979, 27, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.; Marie, D.; Bratbak, G. Flow cytometric detection of viruses. J. Virol. Methods 2000, 85, 175–182. [Google Scholar] [CrossRef]
- Stepp, P.C.; Ranno, K.A.; Ferris, M.M. New Method for Rapid Virus Quantification. Genet. Eng. Biotechnol. News 2010, 30, 24. [Google Scholar]
- Arakelyan, A.; Fitzgerald, W.; Margolis, L.; Grivel, J.-C. Nanoparticle-based flow virometry for the analysis of individual virions. J. Clin. Invest. 2013, 123, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, D.; Yeh, H.-Y.; Mulchandani, A.; Yates, M.V.; Chen, W. Use of flow cytometry for rapid, quantitative detection of poliovirus-infected cells via TAT peptide-delivered molecular beacons. Appl. Environ. Microbiol. 2013, 79, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.L.; Wu, M.C.; Chiu, Y.H.; Lin, J.L.; Wu, Y.C.; Yueh, Y.Y.; Chen, L.K.; Shaio, M.F.; King, C.C. Flow cytometry compared with indirect immunofluorescence for rapid detection of dengue virus type 1 after amplification in tissue culture. J. Clin. Microbiol. 2001, 39, 3672–3677. [Google Scholar] [CrossRef] [PubMed]
- Loret, S.; El Bilali, N.; Lippé, R. Analysis of herpes simplex virus type I nuclear particles by flow cytometry. Cytom. A 2012, 81, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Lippé, R.; University of Montreal, Montreal, QC, Canada. Personal communication, 2015.
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Archiv für mikroskopische Anatomie 1873, 9, 413–418. [Google Scholar] [CrossRef]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Heilemann, M. Shedding new light on viruses: Super-resolution microscopy for studying human immunodeficiency virus. Trends Microbiol. 2013, 21, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Grove, J. Super-resolution microscopy: A virus' eye view of the cell. Viruses 2014, 6, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Colberg-Poley, A.M.; Patterson, G.H.; Salka, K.; Bhuvanendran, S.; Yang, D.; Jaiswal, J.K. Superresolution imaging of viral protein trafficking. Med. Microbiol. Immunol. 2015, 204, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Laine, R.F.; Albecka, A.; van de Linde, S.; Rees, E.J.; Crump, C.M.; Kaminski, C.F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat. Commun. 2015, 6, 5980. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.B.; Grange, M.; Enquist, L.W.; Schermelleh, L.; Grünewald, K.; Oxford University, Oxford, UK. Unpublished work. 2013.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hogue, I.B.; Bosse, J.B.; Engel, E.A.; Scherer, J.; Hu, J.-R.; Del Rio, T.; Enquist, L.W. Fluorescent Protein Approaches in Alpha Herpesvirus Research. Viruses 2015, 7, 5933-5961. https://doi.org/10.3390/v7112915
Hogue IB, Bosse JB, Engel EA, Scherer J, Hu J-R, Del Rio T, Enquist LW. Fluorescent Protein Approaches in Alpha Herpesvirus Research. Viruses. 2015; 7(11):5933-5961. https://doi.org/10.3390/v7112915
Chicago/Turabian StyleHogue, Ian B., Jens B. Bosse, Esteban A. Engel, Julian Scherer, Jiun-Ruey Hu, Tony Del Rio, and Lynn W. Enquist. 2015. "Fluorescent Protein Approaches in Alpha Herpesvirus Research" Viruses 7, no. 11: 5933-5961. https://doi.org/10.3390/v7112915
APA StyleHogue, I. B., Bosse, J. B., Engel, E. A., Scherer, J., Hu, J.-R., Del Rio, T., & Enquist, L. W. (2015). Fluorescent Protein Approaches in Alpha Herpesvirus Research. Viruses, 7(11), 5933-5961. https://doi.org/10.3390/v7112915