Abstract
The threat of a worldwide influenza pandemic has greatly increased over the past decade with the emergence of highly virulent avian influenza strains. The increased frequency of drug-resistant influenza strains against currently available antiviral drugs requires urgent development of new strategies for antiviral therapy, too. The research in the field of therapeutic peptides began to develop extensively in the second half of the 20th century. Since then, the mechanisms of action for several peptides and their antiviral prospect received large attention due to the global threat posed by viruses. Here, we discussed the therapeutic properties of peptides used in influenza treatment. Peptides with antiviral activity against influenza can be divided into three main groups. First, entry blocker peptides such as a Flupep that interact with influenza hemagglutinin, block its binding to host cells and prevent viral fusion. Second, several peptides display virucidal activity, disrupting viral envelopes, e.g., Melittin. Finally, a third set of peptides interacts with the viral polymerase complex and act as viral replication inhibitors such as PB1 derived peptides. Here, we present a review of the current literature describing the antiviral activity, mechanism and future therapeutic potential of these influenza antiviral peptides.
1. Introduction
Generally, some biological active peptides act in compliance with other defense mechanisms of plants or mammals [1,2,3] and can be considered as one of the first forms of “chemical” protection of eukaryotic cells against bacteria, protozoa, fungi, and viruses developed throughout the course of evolution [4,5]. These effects of natural peptides have been studied since 1970s and since then, various therapeutic activities were proposed against Gram-negative and Gram-positive bacteria [6]. The mechanisms of peptide action depend on their structure and can be enhanced by modifications of native peptides or chemically synthesized counterparts. In addition to screening of libraries of native structures, efficient peptides can be selected with commonly used phage display or in silico approaches [7]. Peptides can be designed to mimic or interact with conserved surface proteins and in the case of a variety of pathogens with mutagenic shift the peptide sequence could be modified to preserve therapeutic efficiency. In recent years, researchers have been exploring various methods to improve peptide synthesis technology from solid/liquid phase synthesis up to commercial scale. The economic and biological prospects have been well discussed in the strengths, weaknesses, opportunities and threats (SWOT) analysis by Fosgerau [8]. The good efficacy, safe, selectivity, and predictable metabolism are the strengths of peptide drugs production. On other hand, chemical and physical stability, prone to hydrolysis, and tendency to aggregation are the weaknesses of peptide pharmaceutics.
Influenza is highly contagious, febrile and influenza viruses cause acute respiratory disease. Influenza viruses cause illness with significant morbidity and mortality worldwide and they are considered as potential pandemic agents due to their high mutation rate, which may result in the formation of new subtypes [9,10]. The emerging threat of novel pandemic influenza strains spreading into the human population, as well as increasing resistance against conventional antiviral drug encouraged research efforts to develop new therapies against influenza viruses [11,12,13,14]. In our review, we present a comprehensive overview of peptides with therapeutic potential against specific targets of influenza viruses.
3. Mode of Action of Various Antimicrobial Peptides with Antiviral Activity
The three main mechanisms of antiviral effects of antiviral peptides are: (i) peptides that inhibit attachment of viruses and virus-cell membrane fusion; (ii) peptides that disrupt the viral envelope; and (iii) peptides that inhibit replication of influenza virus by interacting with viral polymerase (Table 1). In this regard, the same mechanism that is described for influenza A, which is the commonly reported type of influenza in publications, have also been reported in the case of influenza B type.
3.1. The Peptides Inhibiting Virus Attachment and Virus-Cell Membrane Fusion
Two mechanisms of inhibition of virus entry by peptides have been proposed. In the first case, the peptides compete with sialic acid (SA) binding by blocking receptor site of HA (Figure 2, Step I.). The second mechanism involves the interference with HA conformation change necessary for viral fusion (Figure 2, Step II.). Thus, the fusion of viral and endosomal membranes is blocked and release of RNA to the host cell is prevented. The viral replication and mechanisms of peptide action are shown in Figure 2.
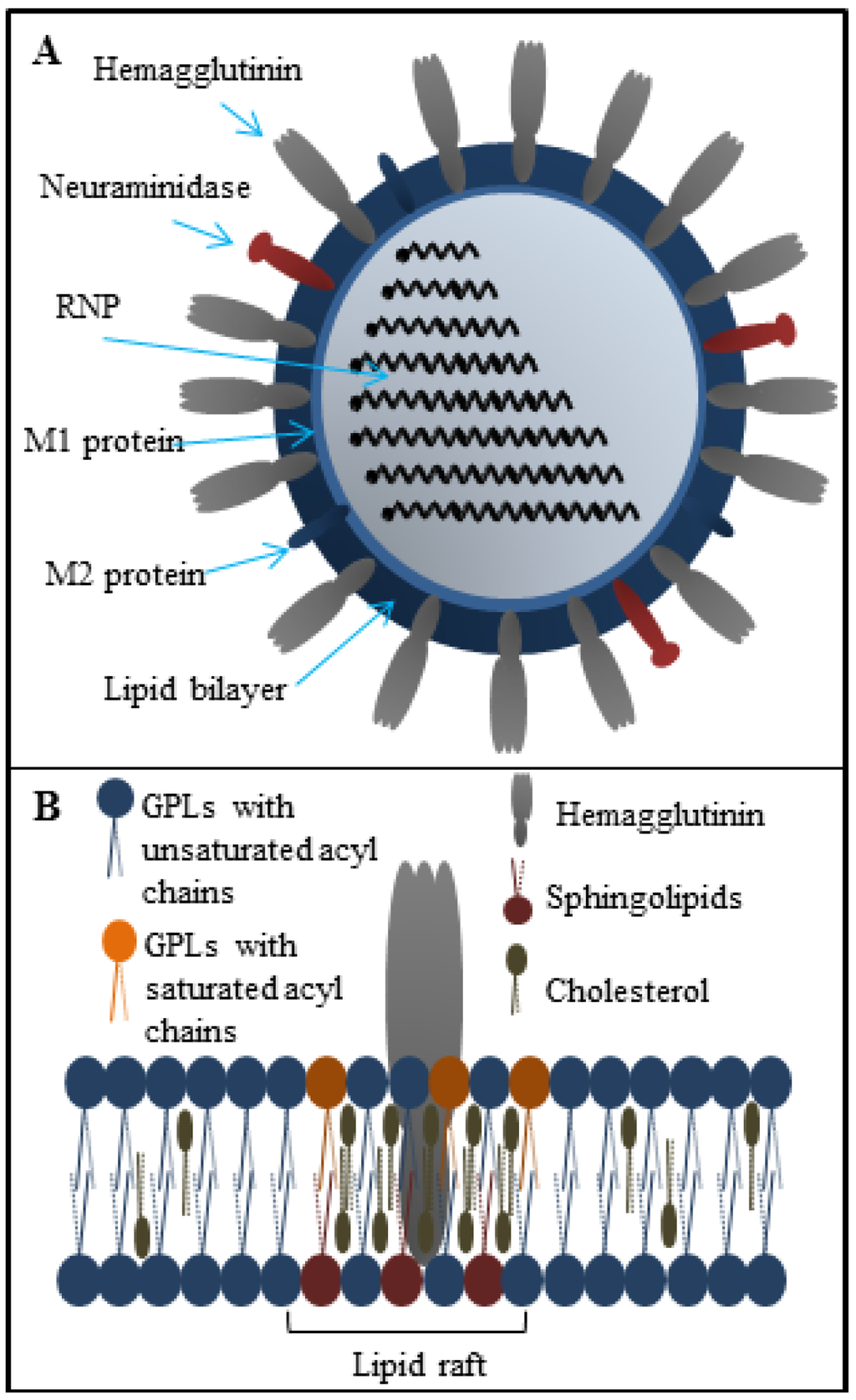
Figure 1.
(A) The structure of influenza virus particle; (B) Structure of the lipid raft localized in the influenza lipid bilayer. The lipid rafts are composed mainly from glycolipids (GPLs), cholesterol and sphingolipids. These microdomains are responsible for the effective viral fusion.
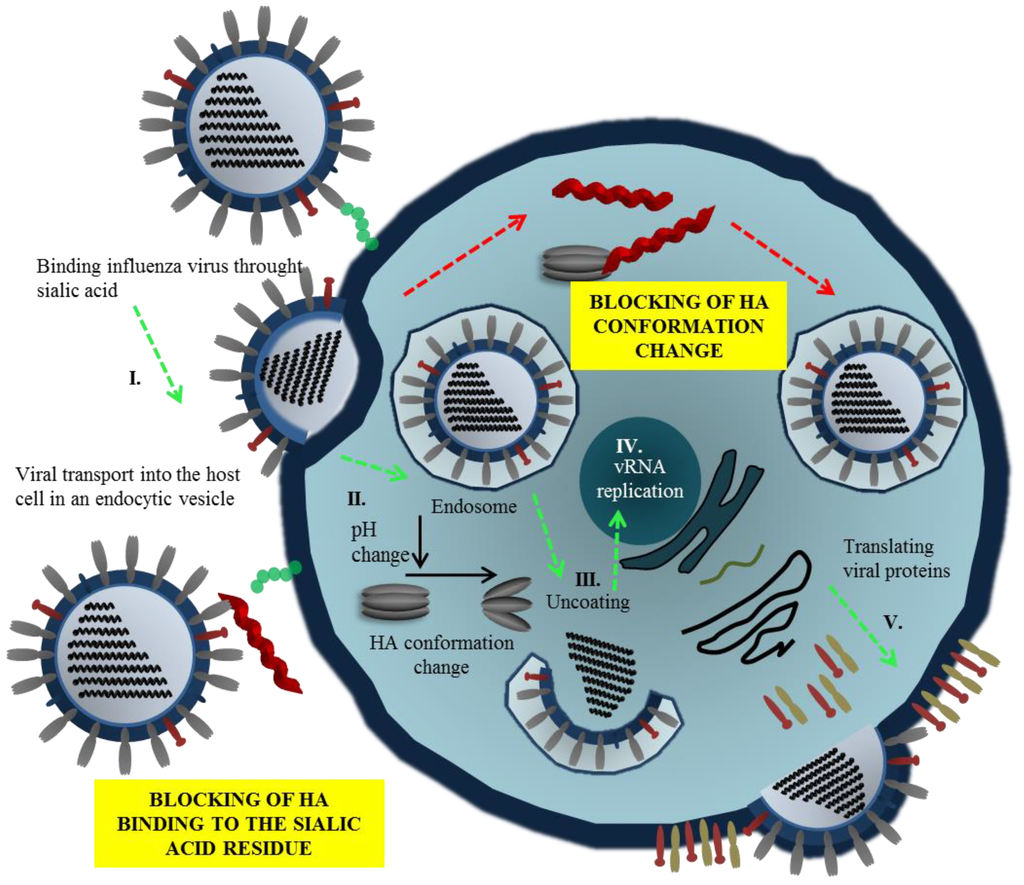
Figure 2.
Mechanisms of inhibition of virus entry by peptides. Viral entry can be blocked via interaction of peptide with hemagglutinin (HA), commonly interacting with residue of sialic acid. This phenomenon results in the alteration of HA functions, and thus influenza virion cannot be attached to the membrane of a host cell. The second antiviral action of peptides may be carried out intracellularly due to blocking of HA conformation change that commonly leads to open of endosome and dissemination of viral genome.

Table 1.
List of antiviral peptides.
| The Peptides Inhibiting Virus Attachment and Virus-Cell Membrane Fusion | |||||||
| Peptide | Influenza Serotype | Sequence | Conformation | Net Charge * | Hydrophobic Residue * | IC50 | Reference |
| EB peptide | Broad spectrum | RRKKAAVALLPAVLLALLAP | linear | 4 | 70 | 3 to 20 µM | [37] |
| Derived EB peptide | Broad spectrum | RRKKLAVLLALLA | linear | 4 | 69 | 3.5 µM | [38] |
| P1 | H9N2 | NDFRSKT | linear | 1 | 14 | 48 µM | [39] |
| P1 cyclic | H9N3 | CNDFRSKTC | cyclic | 1 | 33 | 71 µM | [39] |
| FluPep 1 | H1N1 | WLVFFVIFYFFR | α-helix | 1 | 83 | 0.093 µM | [40] |
| FluPep 2 | H1N1 | WLVFFVIAYFAR | α-helix | 1 | 83 | 0.0009 µM | [40] |
| FluPep 3 | H1N1 | WLVFFVIFYFFRRRKK | α-helix | 5 | 62 | 0.00003 µM | [40] |
| FluPep 4 | H1N1 | RRKKWLVFFVIFYFFR | α-helix | 5 | 62 | 0.00004 µM | [40] |
| FluPep 7 | H1N1 | RRKKIFYFFR | α-helix | 5 | 40 | 0.15 µM | [40] |
| FluPep 8 | H1N1 | WLVFFVRRKK | α-helix | 4 | 60 | 0.63 µM | [40] |
| FluPep 9 | H1N1 | FFVIFYRRKK | α-helix | 4 | 50 | 1.48 µM | [40] |
| C18-s2 | H1N1, H3N2 | C17H35CO-ARLPRTMVHPKPAQP-NH2 | - | 3 | 33 | 11–15 µM | [41] |
| Pal L1 | H5N1 | C16-ARLPRTMVHPKPAQP | micelle | 3 | 33 | - | [42] |
| Pal M1 | H5N1 | C16-ARLPRTMV | micelle | 2 | 50 | - | [42] |
| Pal S1 | H5N1 | C16-ARLPR | micelle | 2 | 40 | - | [42] |
| Flufirvitide | Broad spectrum | - | - | - | - | - | [43] |
| PEP 19-2.5 | H7N7, H3N2, H1N1 | GCKKYRRFRWKFKGKFWFWG | α-helix | 8 | 40 | - | [44] |
| PEP 19-4 | H7N7, H3N2, H1N1 | GKKYRRFRWKFKGKWFWFG | α-helix | 8 | 36 | - | [44] |
| PEP 19-8D | H7N7, H3N2, H1N1 | GFWFKGKWRFKKYRGGRYKKFRWKGKFWFG | α-helix | 12 | 33 | - | [44] |
| PEP 19-CP | H7N7, H3N2, H1N1 | SSNKSTTGSGETTTA | α-helix | 0 | 6 | - | [44] |
| Defensins | H1N1, H3N2 | ACYCRIPACIAGERRYGTCIYQGRLWAFCC | β-sheet | 3 | 53 | - | [45] |
| The Peptides Disrupting Viral Envelope | |||||||
| Peptide | Influenza Serotype | Sequence | Conformation | Net Charge * | Hydrophobic Residue * | IC50 | Reference |
| LF C-lobe peptide 1 | H1H1, H3N2 | SKHSSLDCVLRP | α-helix | 1 | 33 | 4–6 pM | [46] |
| LF C-lobe peptide 2 | H1H1, H3N2 | AGDDQGLDKCVPNSKEK | α-helix | −1 | 23 | 4–7 pM | [46] |
| LF C-lobe peptide 3 | H1H1, H3N2 | NGESSADWAKN | α-helix | −1 | 27 | 22–225 pM | [46] |
| Mucroporin-M1 | H5N1, H1N1 | LFRLIKSLIKRLVSAFK | α-helix | 5 | 58 | 1.03 μM | [47] |
| LL-37 | H1N1, H3N2 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | α-helix | 6 | 35 | - | [48] |
| The Peptides Inhibiting Viral Replication | |||||||
| Peptide | Influenza Serotype | Sequence | Conformation | Net Charge* | Hydrophobic Residue* | IC50 | Reference |
| PB11-25 | Broad spectrum | MDVNPTLLFLKVPAQNAISTTFPYT | α-helix | 0 | 44 | - | [49] |
| PB21-37 | H1N1, H5N1 | MERIKELRDLMSWSRTREILTKTTVDHMAIIKKYTSG | α-helix | 3 | 35 | 375 nM | [50] |
| PB1731–757 | H5N1 | ESGRIKKEEFAEIMKICSTIEELGRQK | α-helix | 0 | 33 | - | [51] |
| PB11–25AT6Y | H1N1, H5N1 | MDVNPYLLFLKVPAQ | α-helix | 0 | 53 | 22–107 nM | [52] |
| Killer peptide | H7N1 | AKVTMTCSAS | α-helix | 1 | 50 | 2.6 µM | [53] |
| HNP-1 | H3N2 | CYCRIPACIAGERRYGTCIYQGRLWAFCC | β-sheet | 3 | 51 | - | [54] |
| Peptid 6 | H1N1, H3N2 | CATCEQIADSQHRSHRQMV | Zn-finger | 0 | 36 | 0.7 nM | [55,56] |
* Calculated by APD2: Antimicrobial Peptide Calculator and Predictor.
The entry blocker peptides are very promising prospective candidates for viral therapy applications. Jones et al. [37] demonstrated the use of a 20 amino acid peptide derived from signal sequence of fibroblast growth factor 4. This study confirmed the broad-spectrum activity of peptides against human, swine, and avian influenza A H1N1, H2N2, H3N2, H5N1, H5N9, and H7N3 strains and influenza B viruses. Pretreatment of mice with peptide shows 100% protection against influenza virus, demonstrated by a decrease in viral titers in the lungs of infected animals. Although postinfection treatment with peptide was not as effective as pretreatment, it should be noted that the peptide was as effective as rimantadine in protecting mice from H5N1 infection. The peptide inhibited chicken red blood cells agglutination with IC50 values ranging from 3 to 20 μM. These results confirmed the ability of peptide inhibit viral attachment. Furthermore, the analysis showed low cytotoxicity of the peptide for the Madin-Darby canine kidney (MDCK) cells ranging in concentrations exceeding 50 μM in medium containing 1% BSA [37]. In a subsequent study, the minimal and optimal sequence, RRKKLAVLLALLA, confers antiviral activity similar to that of EB. In addition, a newly identified peptide, RRKKVALLAVLLALLA, possessing significantly enhanced antiviral and potentially virucidal activity against influenza A was explored. The N-terminus of these peptides with characteristic sequence RRKK influenced their solubility. The results of this study showed that up to four amino acids from C-terminus and up to seven amino acids from N-terminus could be deleted while preserving the antiviral activity [37,38]. Other results indicate that peptide P1 (NDFRSKT) has the ability to interact with HA and exhibits a strong antiviral effects and negligible hemolytic activity.
FluPep is a mix of predominantly hydrophobic α-helical peptides capable of interaction with HA blocking the viral fusion. These peptides are derived from Tkip peptide, which is a mimetic for the suppressor of cytokine signaling protein, known to be active in modulating inflammatory cytokine responses and known as an effective antiviral drug against Poxviruses [57]. A variety of influenza subtypes were inhibited by FluPep in nanomolar concentrations in MDCK cells [40]. Other inhibitory peptides were identified using the Phage display library and the novel alkylated peptide with the sequence C17H35CO-ARLPRTMVHPKPAQP was retrieved. By docking simulation it was proven that the peptide was mimicking sialic acid and was recognized the by receptor-binding site in HA [41]. It seems that RLxRxMxxxK motif is crucial for the inhibitory activity, as it is homologous with highly conserved sequence within HA in many influenza strains. The amino terminal alkyl chain can play an important role in directing peptides into self-assembling micelle, stabilizing the peptide and allowing interaction with multiple binding partners. Huttl et al. described N-modified peptides with palmitic acid (C16-ARLPRTMVHPKPAQP, C16-ARLPRTMV, and C16-ARLPR) [42]. Due to the micelle structure of the peptides, their entropy is reduced [58,59] and affinity to HA is increased in comparison with unmodified linear peptides. Although the mechanism of the binding of the micelle peptide to the HA remains unclear, the concept has the potential of future exploits. Another peptide blocking binding of HA to sialic acid is Flufirvitide, which is currently testing in clinical trials. Besides interfering with the virus entry, it modulates the immune system by activation of production of anti-inflammatory cytokines and chemokines, increasing the activity of neutrophilic cells, and improving phagocytosis of macrophages [43]. A special group of peptides against influenza virus are cyclic delta defensins (retrocyclins), formed by coupling of N- and C-terminal domains. Their occurrence has been described in primates [43,45]. Meanwhile, previous studies have shown their ability to inhibit HIV virus by their ability to bind to HIV surface protein and the similar mechanism is supposed for influenza virus [60,61]. Another class of antiviral peptides are anti-lipopolysaccharide peptides (SALPs). The SALPs are originally based on the LPS-binding domain of Limulus anti-lipopolysaccharide-factor (LALF) and have been discovered by Gutsmann and colleagues as peptides with antimicrobial activity against Gram-positive and Gram-negative bacteria [62]. Recently, SALPs, which show antiviral activity against some enveloped viruses (HIV, HCV and HBV), have been investigated [63]. Hoffman and coworkers reported that SALPs are able to inhibit influenza virus replication of various influenza virus subtypes (H7, H3 and H1) by preventing virus attachment to host cells in vitro and in vivo by binding to N-Acetylneuraminic acids as major components of the influenza virus receptor [44].
3.2. The Peptides Disrupting Viral Envelope
The viral envelope is derived from host cell membranes containing lipid rafts and is rich in sphingolipids and cholesterol [34,35], as shown in Figure 1B. These compounds provide amphipathic character and negative charge [64], which is responsible for electrostatic interactions with the positively charged cationic peptides [65,66]. Generally, peptide-membrane interactions are mediated by electrostatic interactions, while membrane disruption can be accomplished by different means. In the field of antimicrobial peptides, the mechanism of action is quite well understood (the topic is described in detailed in [67]). The viral envelope is adopted from host cell and thus does not exhibit a strong negative charge as bacterial membrane. It may seem that antiviral peptides result in less selectivity, however, several antiviral peptides that can cause the viral envelope disruption have been reported. The mechanism of antiviral action against viruses can either target the viral membrane in general or the lipid rafts rich in cholesterol. The latter case results in destabilization of viral surface proteins that are already enriched in the lipid rafts domains [68]. The common mechanisms of action are summarized in Figure 3A. Cathelicidins are human antiviral peptides that are able to disrupt viral envelope and were shown to elicit a number of host protective mechanisms such as promotion of barrier repairs, chemokine and cytokine production, modulation of dendritic cell differentiation, and T-cell polarization, as well as demonstrate potent anti-sepsis and anti-inflammatory properties [69]. One cathelicidin, namely human LL-37, is produced as a precursor of hCAP-18 that accumulates in neutrophil granules, but it may also be produced in epithelial cells as an acute response to pathogens [70]. Their mechanism of action is related to the interactions between the peptide and viral envelope by the carpet model characterized by formation of continuous layer on lipid bilayer surface resulting in membrane destabilization [71]. The potency of LL-37 against influenza virus seems to be similar to human defensins, involving direct interactions with the virus without affecting viral aggregation or inhibition of binding or uptake of virus by cells. LL-37 may be an important contributor to the initial innate defense against influenza virus [48]. Lactoferrin, widely present in various secretory fluids, is able to interact not only with the viral envelope, but also with receptors on the cell membrane of the host cells by interaction with viral hemagglutinin [72]. Therefore, it is not surprising that bovine lactoferrin was found as a possible agent with an ability to disrupt the virus envelope [73]. Three sequences derived from lactoferrin, SKHSSLDCVLRP, AGDDQGLDKCVPNSKEK, and NGESSADWAKN, inhibit influenza virus (H1H1, H3N2) activity at femtomolar concentrations [46]. To improve stability and circulation time, Balco et al. showed that lactoferrin could be encapsulated in liposomes without loss of activity [74]. Light, ionic strength or pH change stimuli can stimulate cargo release [75]. Thus, the liposomes could be used to preserve peptide drugs through tissues transport. Development of this approach deserves detailed attention in the future. Probably the best-known membrane disrupting peptide is melittin, a 26-amino acid peptide that forms the major component of European honeybee (Apis mellifera) venom [76,77]. Melittin, with a primary structure GIGAVLKVLTTGLPALISWIKRKRQQ, exhibits a variety of effects on lipid bilayer membranes, such as deformation of vesicles, formation of artificial pores, disruption, and lysis [78,79]. Currently, peptide-induced disruption of virion envelopes is vaguely understood. Two of the most discussed models of membrane disruption that are invoked for explaining the release of lipid content of the bilayer are: (i) forming of ruptures [80,81], mostly in the form of toroidal pores characterized by peptide aggregation on the lipid bilayer surface and subsequent perpendicular permeation through lipid bilayer by transmembrane potential change; and/or (ii) lipid bilayer destruction/solubilization [82,83] by the carpet mechanism, mentioned above. Lu et al. used real-time quartz crystal microbalance for tracing the dynamic behavior of lipid bilayers interacting with melittin. These results showed that reaching a threshold peptide concentration (typical for carpet model) followed by mass removal includes the release of lipids, probably as lipid-melittin complex, and the leakage of vesicle components [84], by disrupting the bilayer curvature leading to micellization of released lipids, is crucial [82]. Finally, the virion is destroyed by transient openings in the membrane enabling the passage of low molecular mass molecules prior to complete membrane lysis. Li and co-workers tested mucroporin and its optimized peptide variant mucroporin-M1 LFRLIKSLIKRLVSAFK and employed these peptides for antiviral action against measles, SARS-CoV and influenza H5N1 viruses [85]. Mucoporin M-1 design was based on the protein sequence of mucroporin to enhance the net positive charge of the hydrophilic side by replacing glycine and proline residues with lysine and arginine. It was found that the virucidal activity of mucroporin-M1 was notably increased, whereas the original mucroporin showed no virucidal activity with EC50 of 2.10 μg/mL (1.03 μM) against influenza strain H5N1. The inhibition model could be explained by direct interaction with the virus envelope, thereby decreasing the infectivity of virus. Due to this fact mucroporin-M1 analogues represents a practical tool for developing broad-spectrum antiviral agents, especially against RNA viruses [85].
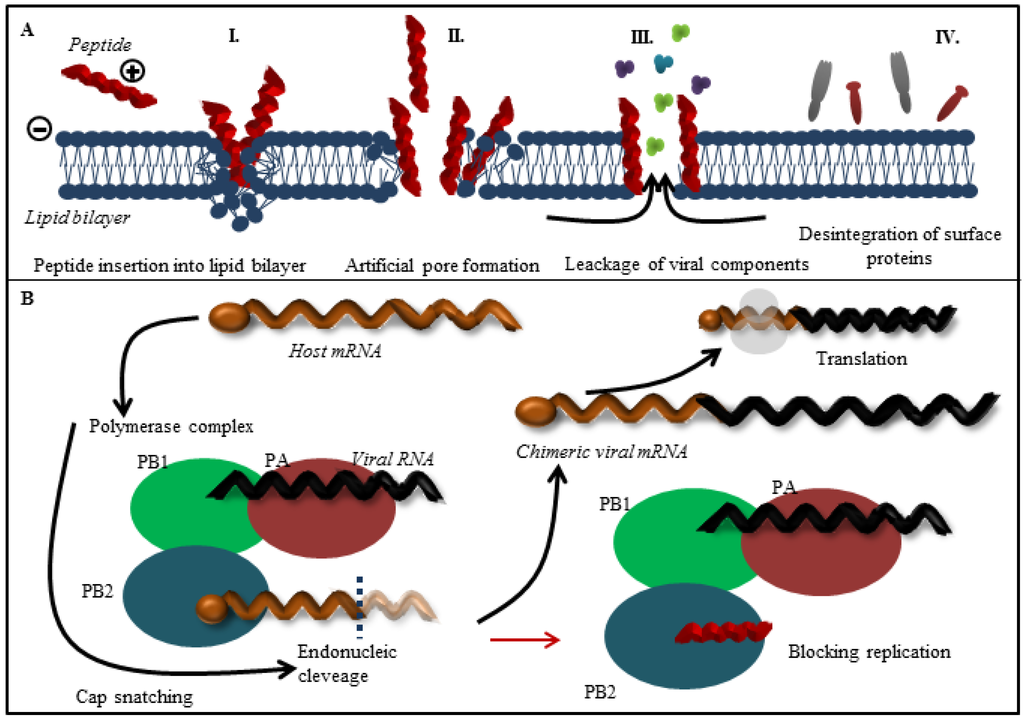
Figure 3.
(A) Overall scheme of the most common interactions between antiviral peptides with an influenza virus lipid bilayer. Due to electrostatic interactions positively charged peptides are attracted by lipid bilayer with negative charge. The peptides insert into lipid bilayer (I.). The critical concentration of peptides triggers the lipid bilayer disruption. These phenomenon results in formation of artificial pores (II.) through which the low mass molecules penetrate into the capsid and contribute to the lipid bilayer destruction and leakage of viral components (III.) as well as disruption of NA and HA functions (IV.); (B) Scheme of function of polymerase assembly in virus replication cycle. Antiviral peptides may bind to PB2 subunit (peptides derived from PB1 subunit) and thus prevent the assembly of influenza polymerase complex via blocking of active binding site of PB2 subunit.
3.3. The Peptides Inhibiting Viral Replication
Viral RNA-dependent RNA polymerase (RdRp), one of the rate-limiting enzymes for influenza virus transcription and replication [86,87], is composed of three polymerase subunits (PB1, PB2 and PA). The PB1 subunit is responsible for polymerization reaction and endonuclease cleavage [88,89], while PB2 is responsible for recognizing and binding the cap structure of host mRNAs [90,91]. The exact role of PA was recently clarified: the N-terminus PA subunit forming the domain with the endonuclease activity and PA endonuclease is responsible for cleavage of host pre-mRNA [92,93]. The RdRp is held together through noncovalent interactions. Disruption of RdRp assembly represents a remarkable opportunity to inhibit the enzyme function and virus replication (Figure 2, Step III.). For this reason, the interaction between PB1 and PA/PB2 is a promising target for design of new anti-influenza drugs (Figure 3B). Numerous authors have used PB1-derived peptides in order to interfere with polymerase function of the enzyme. Ghanem and coworkers tested kinetics of viral polymerase subunit interactions by immunoprecipitation method. PB11-25 and PB1715-740 peptides could bind PA subunit and inhibit influenza replication cycle by interfering with the viral polymerase activity. Preferably, the PB1715-740 peptide binds to conserved site of influenza PA subunit, this approach represents promise tool to block most of influenza A virus strains [49]. Chase et al. described an ELISA-based assay to investigate peptides PB11-25 and PB21-37 capable of impairing polymerase complex formation [50]. The presented system does not include other factors, which could play a role in protein-protein interaction such as other binding domains, binding kinetics, and stabilization through trimer formation. This method enables to test libraries of variant small peptides. In another study, Li and coworkers used PB1731-757 peptide derived from influenza virus strain H5N1. The authors showed that PB1731-757 is capable of inhibiting viral polymerase activity and viral replication [51]. PB1 derived peptide can disrupt the interaction between the C-terminal part of PB1 (corresponding to PB1676-757) and the N-terminal part of PB2 (corresponding to PB21-40) [51,94]. Wunderlich et al. investigated a peptide derived from the PA-binding domain of PB1 and found that the peptide blocked both the polymerase activity and viral spread. This work provides opportunity for developing new antivirals that specifically interfere with the polymerase complex assembly of both influenza A and B viruses [41,52]. Conti and coworkers described anti influenza effect of Killer peptide (KP); a toxin isolated from yeast with proven antimicrobial and anti-human immunodeficiency virus type 1 (HIV-1) activities [53,95]. Treatment with KP demonstrated a significant inhibitory activity on the replication of two influenza A virus strains, as evaluated by hemagglutination, hemadsorption, and plaque assays. In addition, KP demonstrated the complete inhibition of virus particle production and a marked reduction of the synthesis of viral proteins at a KP concentration of 4 µg/mL [53].
3.4. Other Possible Mechanisms of Influenza Virus Inhibition
Numerous studies have shown that a group of antimicrobial peptides called defensins can positively or negatively modulate infection caused by both enveloped and non-enveloped viruses [96,97]. Defensins play direct role in host against microbial infections as innate immune molecules [98] and are able to increase the activity of mucosal epithelia and inhibit the synthesis of viral RNA and proteins [54]. Salvatore and coworkers also showed that human α-defensin-1 (“human neutrophil peptide–1” (HNP-1)) effectively inhibits replication of influenza virus and synthesis of viral proteins when applied soon after infection. Further investigation indicates that viral inhibition could be caused by the modulation of protein kinase C activity in infected cells, suggesting the involvement of the PKC pathway [54]. The proposed strategy involves peptides derived from influenza matrix protein (M1). Peptide 6 was designed corresponding to a zinc finger region of the M1 sequence of influenza virus strain A/PR/8/34 (H1N1), centered around amino acids 148 to 166 [56]. The polymerase inhibitory properties of peptide 6 were evaluated on infections induced in mice by influenza A/PR/8/34 and A/Victoria/3/75 (H3N2) viruses [56]. To avoid the enzymatic breakdown of the peptide, the drug was administrated by intranasal route and was well tolerated up to a dose of 60 mg/kg/day. Based on suggested results, zinc finger peptides may provide a new class of antivirals effective against influenza virus [55].
4. Conclusions
Influenza spreads worldwide in yearly seasonal epidemics, and less frequent pandemics, posing a constant risk; thus, there is a need for new antiviral drugs. The current antiviral therapies have drawbacks such as side effects or selection of resistant strains. Especially, drug resistance of new viral strains compels us to devise new strategies for influenza treatment. Peptides may present the new generation of antiviral drugs with broad-spectrum activity; however, there are potential problems that need to be addressed. Peptide inhibitors of viral polymerase or viral assembly have to target intracellular processes and effective intracellular delivery of peptides still poses a great challenge. Repeated administration of the same peptide has a potential to trigger unwanted immune response. Many membrane disrupting peptides are likely to be cytotoxic through the same mechanism used to disrupt the integrity of viral envelope membrane, which is derived from host cells. Nevertheless, therapeutic applications and testing on animal models have not yet occurred for the majority of the peptides that have been studied. Nonetheless, these are not reasons to ignore the profit of antiviral peptides because careful design could curtail many of the abovementioned problems.
Acknowledgments
Financial support from CEITEC CZ.1.05/1.1.00/02.0068, EXAM CZ.1.05/2.1.00/03.0124, VEGA 2/0146/15 and VEGA 2/0100/13 is highly acknowledged.
Author Contributions
Sylvie Skalickova, Ludmila Krejcova, Zbynek Heger, Vladimir Pekarik and Karel Bastl wrote and discussed the chapter “Design and Characteristics of Antiviral Peptides. Frantisek Kostolansky, Eva Vareckova and Ondrej Zitka prepared and critically reviewed the chapter “Mode of Action of Various Antimicrobial Peptides with Antiviral Activity”. Vojtech Adam conceived of the study, and participated on its design. Rene Kizek participated on design and coordination of the study and drafted manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides the ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.M.; Silva, O.N.; Franco, O.L. Recombinant probiotics with antimicrobial peptides: A dual strategy to improve immune response in immunocompromised patients. Drug Discov. Today 2014, 19, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V.; Ohtake, T.; Lauth, X.; Trowbridge, J.; Rudisill, J.; Dorschner, R.A.; Pestonjamasp, V.; Piraino, J.; Huttner, K.; Gallo, R.L. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 2001, 414, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Tew, G.N.; Clements, D.; Tang, H.Z.; Arnt, L.; Scott, R.W. Antimicrobial activity of an abiotic host defense peptide mimic. Biochim. Biophys. Acta 2006, 1758, 1387–1392. [Google Scholar] [CrossRef]
- Silva, R.R.; Avelino, K.; Ribeiro, K.L.; Franco, O.L.; Oliveira, M.D.L.; Andrade, C.A.S. Optical and dielectric sensors based on an microbial peptides for microorganism diagnosis. Front. Microbiol. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hale, J.D.; Hancock, R.E. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti Infect. Ther. 2007, 5, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Hadley, E.B.; Hancock, R.E.W. Strategies for the discovery and advancement of novel cationic antimicrobial peptides. Curr. Top. Med. Chem. 2010, 10, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Sim, I.S. Potential of new drugs in the prophylaxis and therapy of influenza A virus-infections. Curr. Opin. Infect. Dis. 1989, 2, 411–414. [Google Scholar] [CrossRef]
- Zumla, A.; Memish, Z.A.; Maeurer, M.; Bates, M.; Mwaba, P.; Al-Tawfiq, J.A.; Denning, D.W.; Hayden, F.G.; Hui, D.S. Emerging novel and antimicrobial-resistant respiratory tract infections: New drug development and therapeutic options. Lancet Infect. Dis. 2014, 14, 1136–1149. [Google Scholar] [CrossRef]
- Berkhout, B.; Sanders, R.W. Molecular strategies to design an escape-proof antiviral therapy. Antivir. Res. 2011, 92, 7–14. [Google Scholar] [PubMed]
- Wilson, J.C.; von Itzstein, M. Recent strategies in the search for new anti-influenza therapies. Curr. Drug Targets 2003, 4, 389–408. [Google Scholar] [CrossRef] [PubMed]
- Air, G.M.; Brouillette, W.J. Influenza virus antiviral targets. Antiviral Res. 2009, 187–207. [Google Scholar]
- Krol, E.; Rychowska, M.; Szewczyk, B. Antivirals—Current trends in fighting influenza. Acta Biochim. Pol. 2014, 61, 495–504. [Google Scholar] [PubMed]
- Jitendra, P.K.; Bansal, S.; Banik, A. Noninvasive routes of proteins and peptides drug delivery. Indian J. Pharm. Sci. 2011, 73, 367–375. [Google Scholar] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Liderot, K.; Ahl, M.; Ozenci, V. Secondary bacterial infections in patients with seasonal influenza A and pandemic H1N1. Biomed. Res. Int. 2013, 2013, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic alpha helical antimicrobial peptides—A systematic study of the effects of structural and physical properties on biological activity. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Walker, E.; Egerer, L.; Bunnell, B.A.; Mondal, D.; von Laer, D.; Braun, S.E. The therapeutic potential of secreted antiviral entry inhibitor (SAVE) peptides expressed by transduced MSCs to block HIV infection. Mol. Ther. 2014, 22, S183. [Google Scholar]
- Pessi, A.; Ingallinella, P.; Bianchi, E.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.; Moore, J.; Miller, M. Dramatic increase of antiviral potency of an HIV peptide fusion inhibitor by targeting to lipid rafts via addition of a cholesterol group. Biopolymers 2009, 92, 302. [Google Scholar]
- Abe, K.; Nozaki, A.; Tamura, K.; Ikeda, M.; Naka, K.; Dansako, H.; Hoshino, H.; Tanaka, K.; Kato, N. Tandem repeats of lactoferrin-derived anti-hepatitis C virus peptide enhance antiviral activity in cultured human hepatocytes. Microbiol. Immunol. 2007, 51, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H. Anti herpes simplex virus activity of lactoferrin/lactoferricin—An example of antiviral activity of antimicrobial protein/peptide. Cell. Mol. Life Sci. 2005, 62, 3002–3013. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, D.; Yakoub, A.M.; Bogdanov, A.; Valyi-Nagy, T.; Shukla, D. Characterization of a proteolytically stable d-peptide that suppresses herpes simplex virus 1 infection: Implications for the development of entry-based antiviral therapy. J. Virol. 2015, 89, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Albericio, F.; Kruger, H.G. Therapeutic peptides foreword. Future Med. Chem. 2012, 4, 1527–1531. [Google Scholar] [CrossRef] [PubMed]
- She, R.; Bao, H.; Zhang, Y.; Luo, D. Effect of rabbit sacculus rotundus antimicrobial peptides on serum antibody titers after vaccination with newcastle disease virus vaccine and avian influenza virus vaccine in chickens. Poult. Sci. 2008, 87, 81–86. [Google Scholar]
- Pizzorno, A.; Abed, Y.; Boivin, G. Influenza drug resistance. Semin. Respir. Crit. Care Med. 2011, 32, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Mikulasova, A.; Vareckova, E.; Fodor, E. Transcription and replication of the influenza A virus genome. Acta Virol. 2000, 44, 273–282. [Google Scholar] [PubMed]
- Das, K.; Aramini, J.M.; Ma, L.C.; Krug, R.M.; Arnold, E. Structures of influenza A proteins and insights into antiviral drug targets. Nat. Struct. Mol. Biol. 2010, 17, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Brunotte, L.; Flies, J.; Bolte, H.; Reuther, P.; Vreede, F.; Schwemmle, M. The nuclear export protein of H5N1 influenza A viruses recruits matrix 1 (M1) protein to the viral ribonucleoprotein to mediate nuclear export. J. Biol. Chem. 2014, 289, 20067–20077. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, T.M.; te Velthuis, A.J.W. The RNA-dependent RNA polymerase of the influenza A virus. Future Virol. 2014, 9, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Leser, G.P.; Lamb, R.A. Influenza virus assembly and budding in raft-derived microdomains: A quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology 2005, 342, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Leser, G.P.; Russell, C.J.; Lamb, R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Wang, Y.J.; Wang, J.F. Influenza virus assembly and budding in lipid rafts. Prog. Biochem. Biophys. 2015, 42, 495–500. [Google Scholar]
- Rossman, J.S.; Jing, X.H.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Turpin, E.A.; Bultmann, H.; Brandt, C.R.; Schultz-Cherry, S. Inhibition of influenza virus infection by a novel antiviral peptide that targets viral attachment to cells. J. Virol. 2006, 80, 11960–11967. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Settles, E.W.; Brandt, C.R.; Schultz-Cherry, S. Identification of the minimal active sequence of an anti-influenza virus peptide. Antimicrob. Agents Chemother. 2011, 55, 1810–1813. [Google Scholar] [CrossRef] [PubMed]
- Rajik, M.; Jahanshiri, F.; Omar, A.R.; Ideris, A.; Hassan, S.S.; Yusoff, K. Identification and characterisation of a novel anti-viral peptide against avian influenza virus H9N2. Virol. J. 2009, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nicol, M.Q.; Ligertwood, Y.; Bacon, M.N.; Dutia, B.M.; Nash, A.A. A novel family of peptides with potent activity against influenza A viruses. J. Gen. Virol. 2012, 93, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Onishi, A.; Saito, T.; Shimada, A.; Inoue, H.; Taki, T.; Nagata, K.; Okahata, Y.; Sato, T. Sialic acid-mimic peptides as hemagglutinin inhibitors for anti-influenza therapy. J. Med. Chem. 2010, 53, 4441–4449. [Google Scholar] [CrossRef] [PubMed]
- Huttl, C.; Hettrich, C.; Miller, R.; Paulke, B.R.; Henklein, P.; Rawel, H.; Bier, F.F. Self-assembled peptide amphiphiles function as multivalent binder with increased hemagglutinin affinity. BMC Biotechnol. 2013, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cederlund, A.; Gudmundsson, G.H.; Agerberth, B. Antimicrobial peptides important in innate immunity. FEBS J. 2011, 278, 3942–3951. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Schneider, C.; Heinbockel, L.; Brandenburg, K.; Reimer, R.; Gabriel, G. A new class of synthetic anti-lipopolysaccharide peptides inhibits influenza A virus replication by blocking cellular attachment. Antivir. Res. 2014, 104, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; White, M.R.; Tecle, T.; Gantz, D.; Crouch, E.C.; Jung, G.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Hartshorn, K.L. Interactions of α-, β-, and θ-defensins with influenza A virus and surfactant protein D. J. Immunol. 2009, 182, 7878–7887. [Google Scholar] [CrossRef] [PubMed]
- Ammendolia, M.G.; Agamennone, M.; Pietrantoni, A.; Lannutti, F.; Siciliano, R.A.; de Giulio, B.; Amici, C.; Superti, F. Bovine lactoferrin-derived peptides as novel broad-spectrum inhibitors of influenza virus. Pathog. Glob. Health 2012, 106, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.L.; Zhao, Z.H.; Zhou, D.H.; Chen, Y.Q.; Hong, W.; Cao, L.Y.; Yang, J.Y.; Zhang, Y.; Shi, W.; Cao, Z.J.; et al. Virucidal activity of a scorpion venom peptide variant mucroporin-M1 against measles, SARS-CoV and influenza H5N1 viruses. Peptides 2011, 32, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Tecle, T.; Verma, A.; Crouch, E.; White, M.; Hartshorn, K.L. The human cathelicidin LL-37 inhibits influenza A viruses through a mechanism distinct from that of surfactant protein D or defensins. J. Gen. Virol. 2013, 94, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Mayer, D.; Chase, G.; Tegge, W.; Frank, R.; Kochs, G.; Garcia-Sastre, A.; Schwemmle, M. Peptide-mediated interference with influenza A virus polymerase. J. Virol. 2007, 81, 7801–7804. [Google Scholar] [CrossRef] [PubMed]
- Chase, G.; Wunderlich, K.; Reuther, P.; Schwemmle, M. Identification of influenza virus inhibitors which disrupt of viral polymerase protein-protein interactions. Methods 2011, 55, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, M.M.; Shen, X.T.; Liu, S.W. Influenza A virus entry inhibitors targeting the hemagglutinin. Viruses 2013, 5, 352–373. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, K.; Mayer, D.; Ranadheera, C.; Holler, A.S.; Manz, B.; Martin, A.; Chase, G.; Tegge, W.; Frank, R.; Kessler, U.; et al. Identification of a PA-binding peptide with inhibitory activity against influenza A and B virus replication. PLoS ONE 2009, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Conti, G.; Magliani, W.; Conti, S.; Nencioni, L.; Sgarbanti, R.; Palamara, A.T.; Polonelli, L. Therapeutic activity of an anti-idiotypic antibody-derived killer peptide against influenza A virus experimental infection. Antimicrob. Agents Chemother. 2008, 52, 4331–4337. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, M.; Garcia-Sastre, A.; Ruchala, P.; Lehrer, R.I.; Chang, T.; Klotman, M.E. α-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J. Infect. Dis. 2007, 196, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Judd, A.K.; Sanchez, A.; Bucher, D.J.; Huffman, J.H.; Bailey, K.; Sidwell, R.W. In vivo anti-influenza virus activity of a zinc finger peptide. Antimicrob. Agents Chemother. 1997, 41, 687–692. [Google Scholar] [PubMed]
- Nasser, E.H.; Judd, A.K.; Sanchez, A.; Anastasiou, D.; Bucher, D.J. Antiviral activity of influenza virus M1 zinc finger peptides. J. Virol. 1996, 70, 8639–8644. [Google Scholar] [PubMed]
- Ahmed, C.M.; Dabelic, R.; Waiboci, L.W.; Jager, L.D.; Heron, L.L.; Johnson, H.M. SOCS-1 mimetics protect mice against lethal poxvirus infection: Identification of a novel endogenous antiviral system. J. Virol. 2009, 83, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.P.; Chu, Y.H. Using surface plasmon resonance to directly determine binding affinities of combinatorially selected cyclopeptides and their linear analogs to a streptavidin chip. Anal. Biochem. 2005, 340, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Udugamasooriya, D.G.; Spaller, M.R. Conformational constraint in protein ligand design and the inconsistency of binding entropy. Biopolymers 2008, 89, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cole, A.M.; Hong, T.; Waring, A.J.; Lehrer, R.I. Retrocyclin, an antiretroviral θ-defensin, is a lectin. J. Immunol. 2003, 170, 4708–4716. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; White, M.R.; Tecle, T.; Hartshorn, K.L. Human defensins and LL-37 in mucosal immunity. J. Leukoc. Biol. 2010, 87, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Gutsmann, T.; Razquin-Olazaran, I.; Kowalski, I.; Kaconis, Y.; Howe, J.; Bartels, R.; Hornef, M.; Schurholz, T.; Rossle, M.; Sanchez-Gomez, S.; et al. New antiseptic peptides to protect against endotoxin-mediated shock. Antimicrob. Agents Chemother. 2010, 54, 3817–3824. [Google Scholar] [CrossRef] [PubMed]
- Krepstakies, M.; Lucifora, J.; Nagel, C.H.; Zeisel, M.B.; Holstermann, B.; Hohenberg, H.; Kowalski, I.; Gutsmann, T.; Baumert, T.F.; Brandenburg, K.; et al. A new class of synthetic peptide inhibitors blocks attachment and entry of human pathogenic viruses. J. Infect. Dis. 2012, 205, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Needham, B.D.; Trent, M.S. Fortifying the barrier: The impact of lipid a remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 2013, 11, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Lopez, J.L.; Lopez-Meza, J.E.; Ochoa-Zarzosa, A. Bacterial resistance to cationic antimicrobial peptides. Crit. Rev. Microbiol. 2013, 39, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.K.; O’Neil, D.A. Peptides as the next generation of anti-infectives. Future Med. Chem. 2013, 5, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Marcos, J.F.; Gandia, M. Antimicrobial peptides: To membranes and beyond. Expert Opin. Drug Discov. 2009, 4, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Knoelker, H.-J.; Simons, K. Subcellular targeting strategies for drug design and delivery. Nat. Rev. Drug Discov. 2010, 9, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.-Y.G.; Mookherjee, N. Multiple immune-modulatory functions of cathelicidin host defense peptides. Front. Immun. 2012, 3, 149. [Google Scholar] [CrossRef] [PubMed]
- Barlow, P.G.; Svoboda, P.; Mackellar, A.; Nash, A.A.; York, I.A.; Pohl, J.; Davidson, D.J.; Donis, R.O. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE 2011, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.E.; O’Brien, L.M.; Thwaite, J.E.; Fox, M.A.; Atkins, H.; Ulaeto, D.O. A carpet-based mechanism for direct antimicrobial peptide activity against vaccinia virus membranes. Peptides 2010, 31, 1966–1972. [Google Scholar] [PubMed]
- Pietrantoni, A.; Di Biase, A.M.; Tinari, A.; Marchetti, M.; Valenti, P.; Seganti, L.; Superti, F. Bovine lactoferrin inhibits adenovirus infection by interacting with viral structural polypeptides. Antimicrob. Agents Chemother. 2003, 47, 2688–2691. [Google Scholar] [PubMed]
- Pietrantoni, A.; Dofrelli, E.; Tinari, A.; Ammendolia, M.G.; Puzelli, S.; Fabiani, C.; Donatelli, I.; Superti, F. Bovine lactoferrin inhibits influenza A virus induced programmed cell death in vitro. Biometals 2010, 23, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Balcao, V.M.; Costa, C.I.; Matos, C.M.; Moutinho, C.G.; Amorim, M.; Pintado, M.E.; Gomes, A.P.; Vila, M.M.; Teixeira, J.A. Nanoencapsulation of bovine lactoferrin for food and biopharmaceutical applications. Food Hydrocoll. 2013, 32, 425–431. [Google Scholar] [CrossRef]
- Bibi, S.; Lattmann, E.; Mohammed, A.R.; Perrie, Y. Trigger release liposome systems: Local and remote controlled delivery? J. Microencapsul. 2012, 29, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Hung, W.C.; Chen, F.Y.; Huang, H.W. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 5087–5092. [Google Scholar] [CrossRef] [PubMed]
- Ladokhin, A.S.; White, S.H. ‘Detergent-like’ permeabilization of anionic lipid vesicles by melittin. Biochim. Biophys. Acta 2001, 1514, 253–260. [Google Scholar] [CrossRef]
- Zhang, N.Z.; Qi, J.X.; Feng, S.J.; Gao, F.; Liu, J.; Pan, X.C.; Chen, R.; Li, Q.R.; Chen, Z.S.; Li, X.Y.; et al. Crystal structure of swine major histocompatibility complex class I SLA-1*0401 and identification of 2009 pandemic swine-origin influenza A H1N1 virus cytotoxic t lymphocyte epitope peptides. J. Virol. 2011, 85, 11709–11724. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Grossman, M.; Zimmermann, H.; Wolf, S.G.; Shai, Y.; Goldfarb, D. Investigation of model membrane disruption mechanism by melittin using pulse electron paramagnetic resonance spectroscopy and cryogenic transmission electron microscopy. J. Phys. Chem. B 2012, 116, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Grossman, M.; Gofman, Y.; Zimmermann, H.; Frydman, V.; Shai, Y.; Ben-Tal, N.; Goldfarb, D. A combined pulse EPR and monte carlo simulation study provides molecular insight on peptide-membrane interactions. J. Phys. Chem. B 2009, 113, 15128. [Google Scholar] [CrossRef]
- Oren, Z.; Shai, Y. Mode of action of linear amphipathic α-helical antimicrobial peptides. Biopolymers 1998, 47, 451–463. [Google Scholar] [CrossRef]
- Benachir, T.; Lafleur, M. Study of vesicle leakage induced by melittin. Biochim. Biophys. Acta 1995, 1235, 452–460. [Google Scholar] [CrossRef]
- Lu, N.Y.; Yang, K.; Li, J.L.; Yuan, B.; Ma, Y.Q. Vesicle deposition and subsequent membrane-melittin interactions on different substrates: A QCM-D experiment. Biochim. Biophys. Acta 2013, 1828, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Y.; Yang, X.F.; Jiang, Y.; Wang, B.N.; Yang, Y.; Jiang, Z.H.; Li, M.Y. Inhibition of influenza A virus replication by RNA interference targeted against the PB1 subunit of the RNA polymerase gene. Arch. Virol. 2011, 156, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Huet, S.; Avilov, S.V.; Ferbitz, L.; Daigle, N.; Cusack, S.; Ellenberg, J. Nuclear import and assembly of influenza A virus RNA polymerase studied in live cells by fluorescence cross-correlation spectroscopy. J. Virol. 2010, 84, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Manz, B.; Gotz, V.; Wunderlich, K.; Eisel, J.; Kirchmair, J.; Stech, J.; Stech, O.; Chase, G.; Frank, R.; Schwemmle, M. Disruption of the viral polymerase complex assembly as a novel approach to attenuate influenza A virus. J. Biol. Chem. 2011, 286, 8414–8424. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.H.; Crepin, T.; Hart, D.J.; Cusack, S. Towards an atomic resolution understanding of the influenza virus replication machinery. Curr. Opin. Struct. Biol. 2010, 20, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.C.; Shukla, A.; Puranam, S.; Buss, H.G.; Jreige, N.; Hammond, P.T. Effects of side group functionality and molecular weight on the activity of synthetic antimicrobial polypeptides. Biomacromolecules 2011, 12, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.Q.; Shan, A.S.; Dong, N.; Gu, Y.; Sun, W.Y.; Hu, W.N.; Feng, X.J. Cell selectivity and interaction with model membranes of Val/Arg-rich peptides. J. Pept. Sci. 2011, 17, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Fechter, P.; Mingay, L.; Sharps, J.; Chambers, A.; Fodor, E.; Brownlee, G.G. Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J. Biol. Chem. 2003, 278, 20381–20388. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Bouvier, D.; Crepin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W.H. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.W.; Bartlam, M.; Lou, Z.Y.; Chen, S.D.; Zhou, J.; He, X.J.; Lv, Z.Y.; Ge, R.W.; Li, X.M.; Deng, T.; et al. Crystal structure of an avian influenza polymerase PA(n) reveals an endonuclease active site. Nature 2009, 458, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.Y.; Yu, W.L.; McBride, R.; Li, Y.; Chen, L.M.; Donis, R.O.; Tong, S.X.; Paulson, J.C.; Wilson, I.A. Hemagglutinin homologue from H17N10 bat influenza virus exhibits divergent receptor-binding and pH-dependent fusion activities. Proc. Natl. Acad. Sci. USA 2013, 110, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.; Kazan, K.; Goulter, K.C.; Maclean, D.J.; Manners, J.M. The mode of action of the plant antimicrobial peptide MiAMP1 differs from that of its structural homologue, the yeast killer toxin WmKT. FEMS Microbiol. Lett. 2005, 243, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Chang, T.L. Defensins in viral infection. In Small Wonders: Peptides for Disease Control; American Chemical Society: Washington, DC, USA, 2012; pp. 137–171. [Google Scholar]
- Mohan, T.; Sharma, C.; Bhat, A.A.; Rao, D.N. Modulation of HIV peptide antigen specific cellular immune response by synthetic α- and β-defensin peptides. Vaccine 2013, 31, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).