
Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance


Abstract
:1. Introduction
2. The RNA-Dependent RNA Polymerase (RdRp)
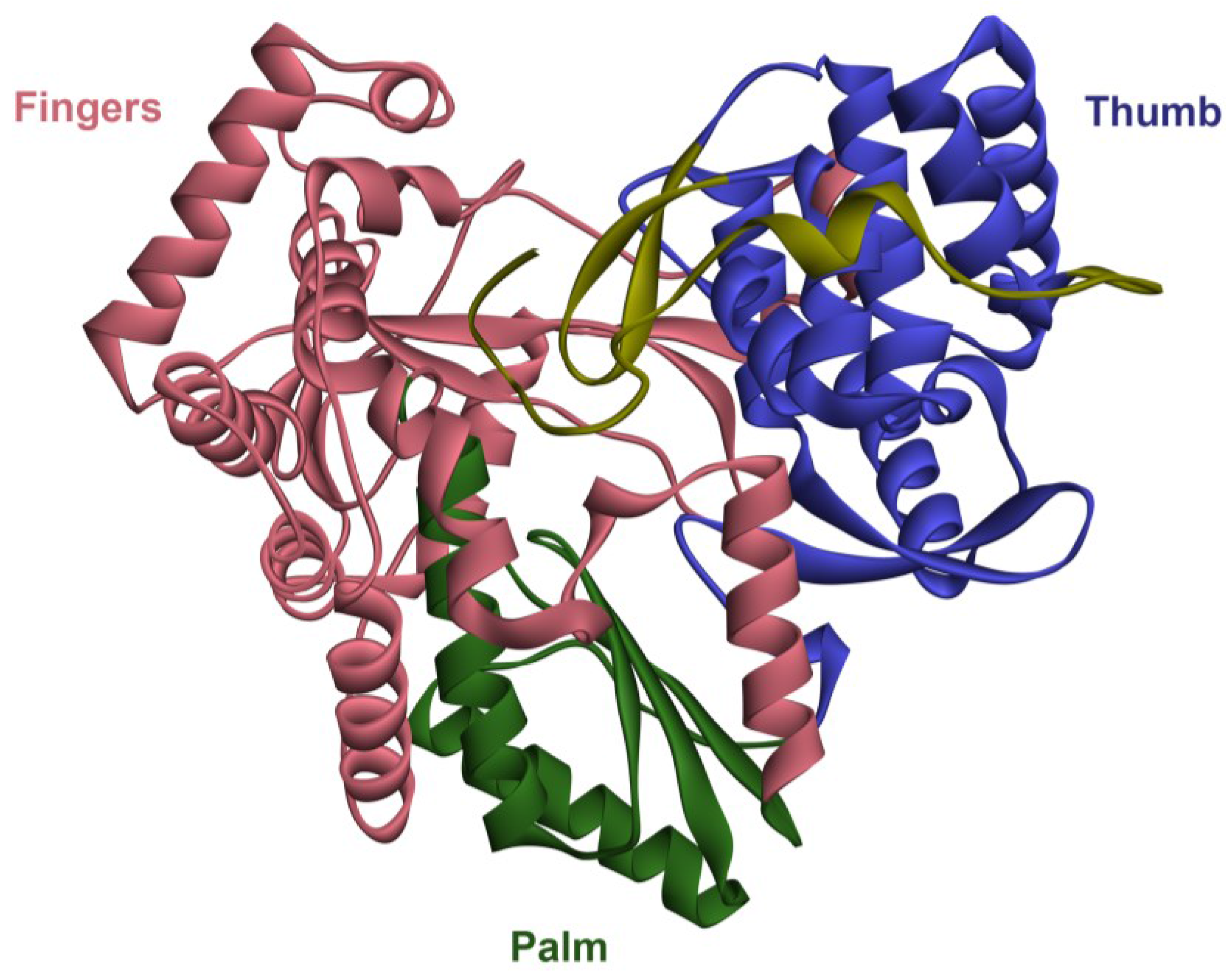
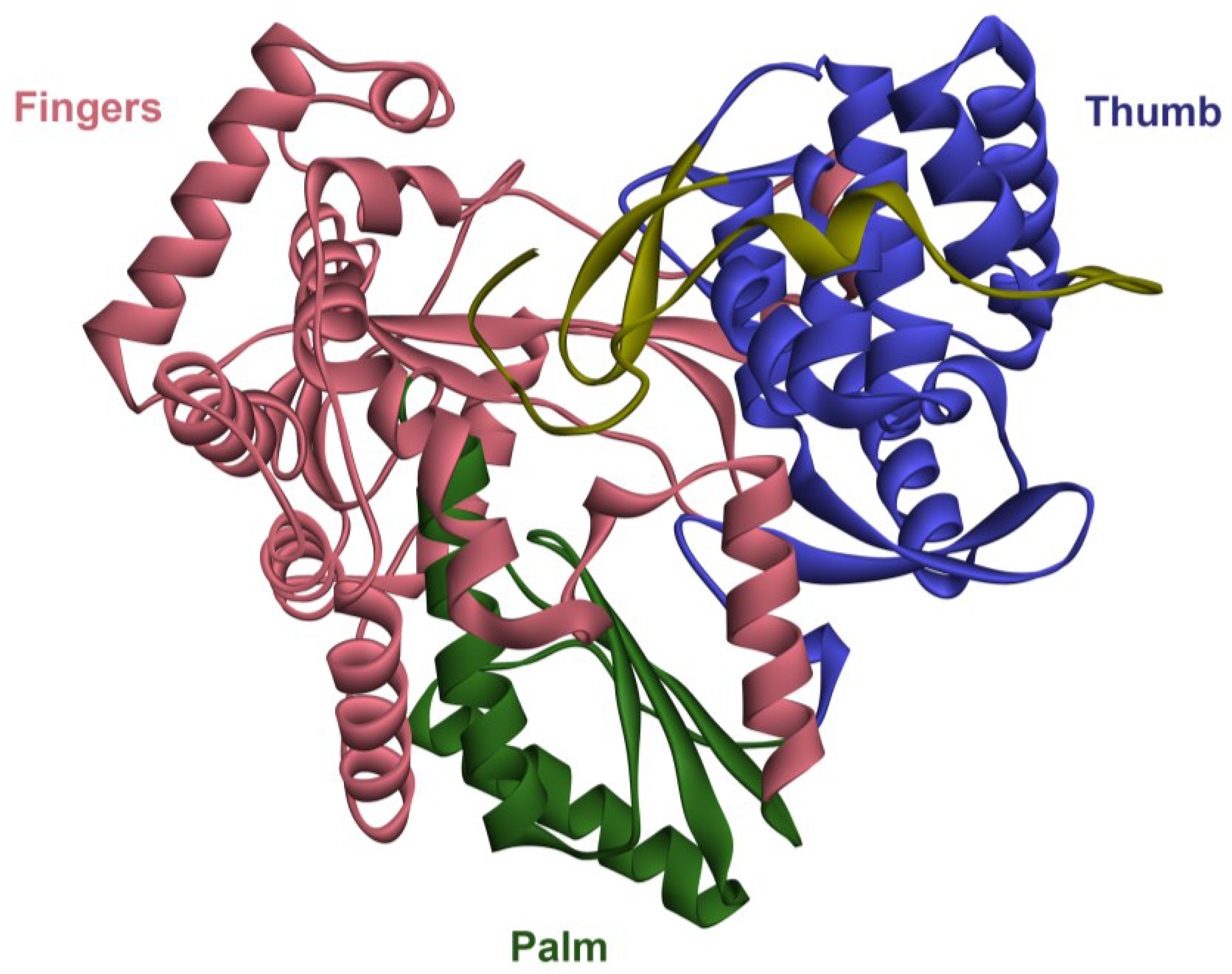
3. Therapies for HCV in the Past
4. HCV RdRp as a Target for DAAs

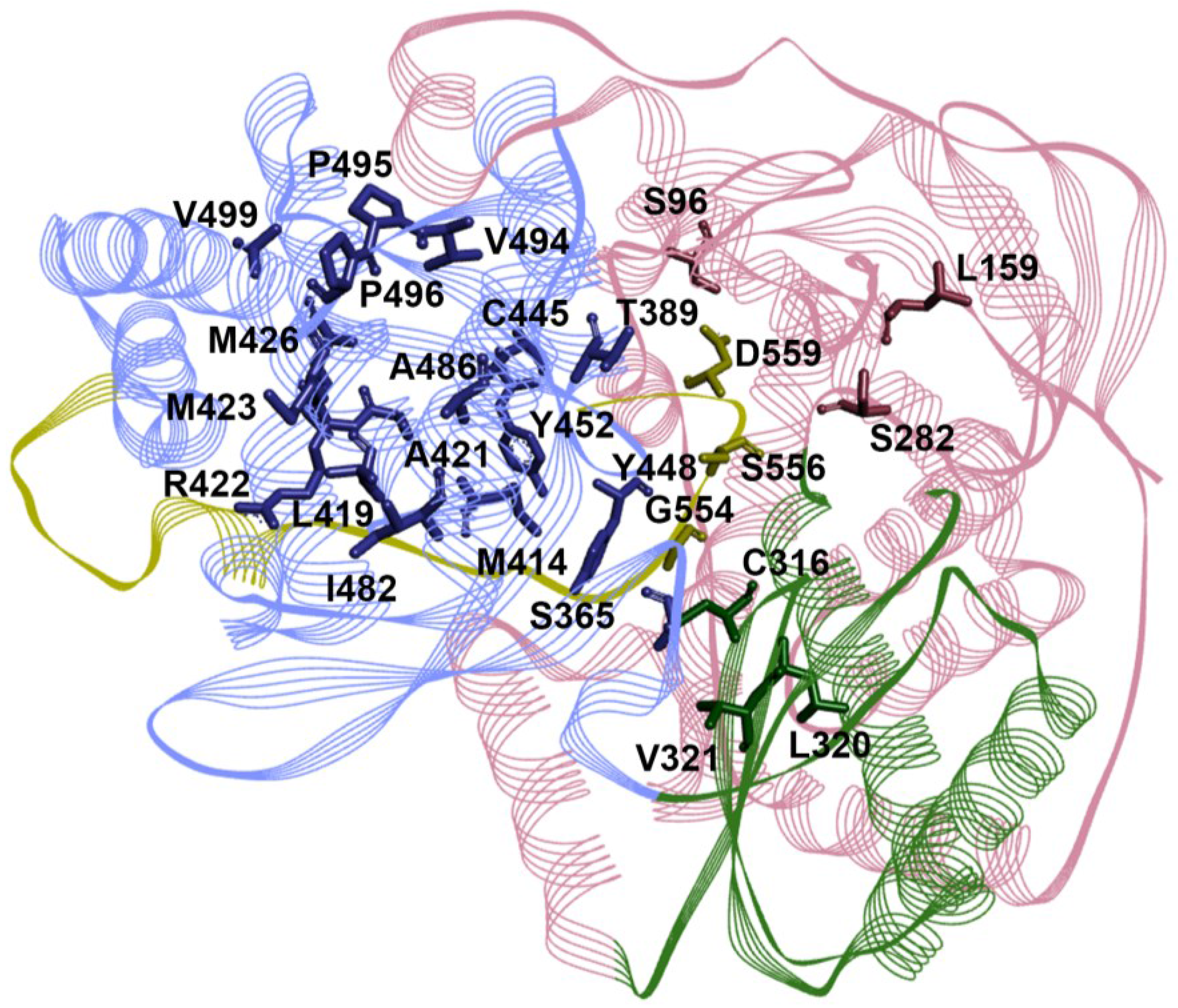
5. Mechanism of Action for RdRp NNIs
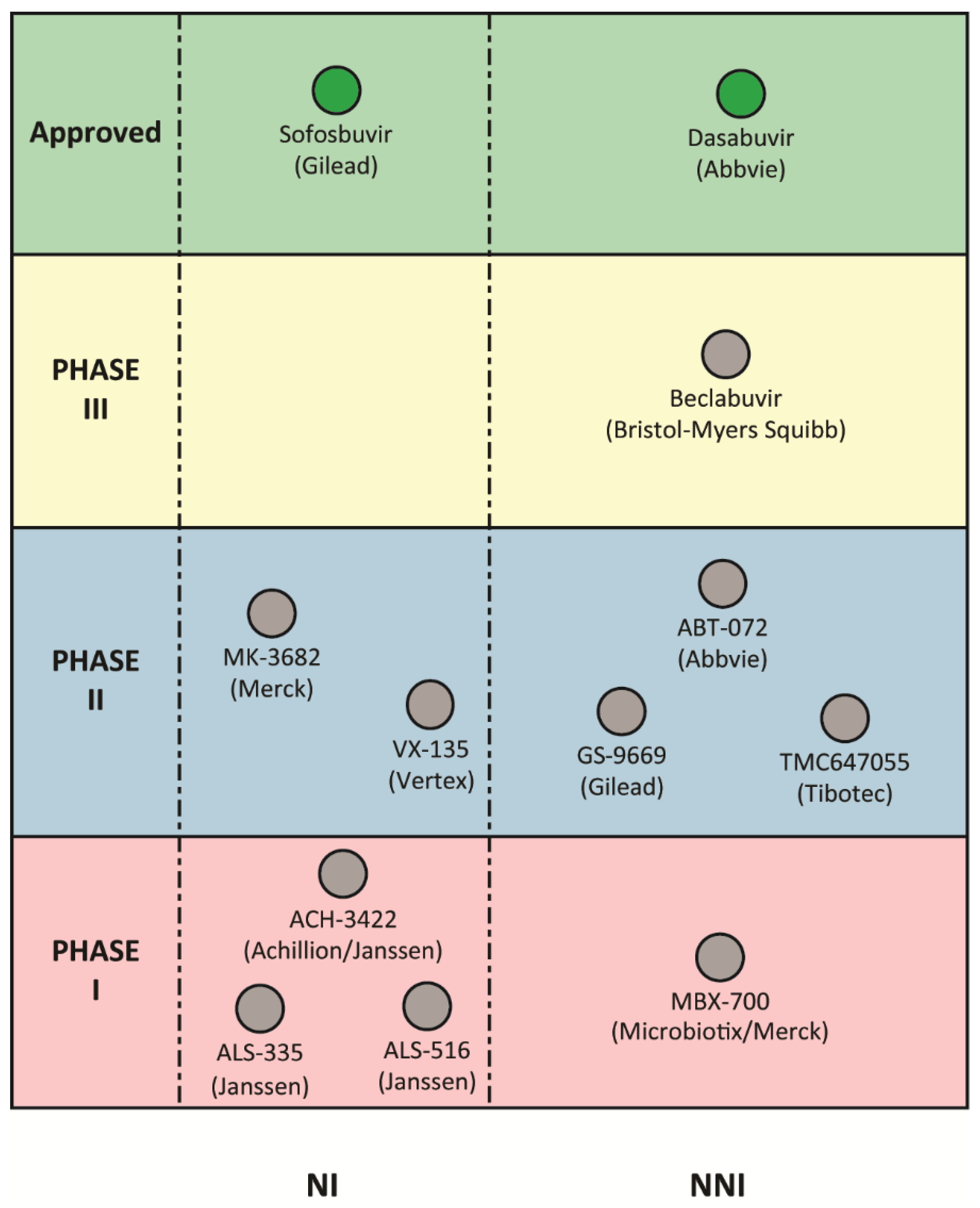
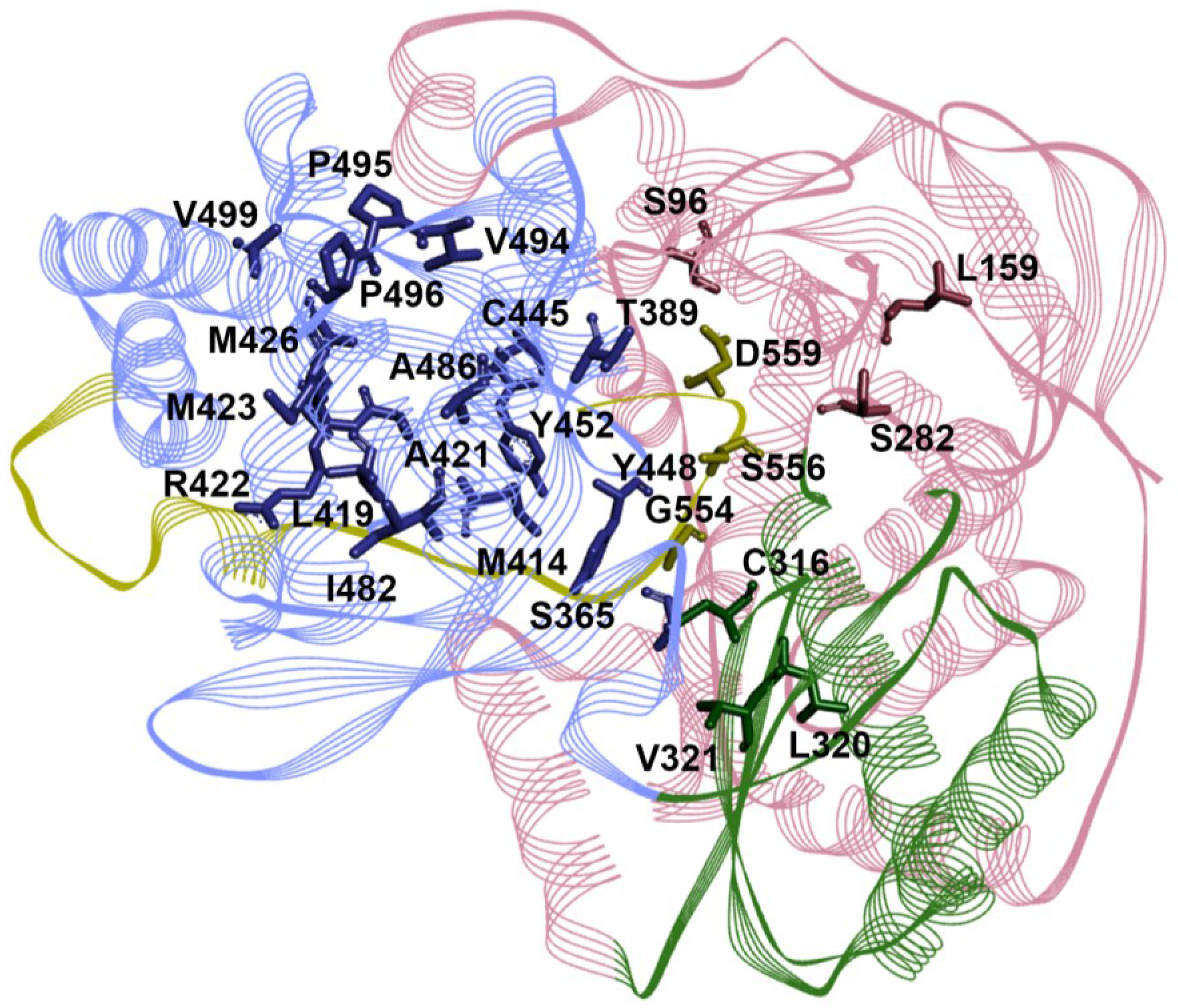
6. Genotype Coverage of RdRp Inhibitors

{kind=link}
{kind=link}
{kind=link}
| RdRp Site | Resistance Substitutions | Prevalence of Resistant Variants (%) | Refs. | |
|---|---|---|---|---|
| GT1a | GT1b | |||
| NI | S96T | 0 | 0 | [126,127,128,129,130] |
| L159F | ND | ND | ||
| S282T | 0–0.6 | 0–1.1 | ||
| L320I/F | 0 | 0 | ||
| V321I | 0.19–3.1 | 2.51–3.3 | ||
| NNI-T1 | T389S/A | ND | ND | |
| L392I | ND | ND | ||
| A421V | 0–17.84 | 0–6.28 | ||
| P495A/L/S | 0 | 0 | ||
| P496A/S/T | 0 | 0–0.84 | ||
| V499A # | 94.4–100 | 5–14.23 | ||
| NNI-T2 | L419M/V/I | 0 | 0–0.9 | |
| R422K | 0–0.56 | 0 | ||
| M423T/I/V | 0–2.8 | 0–0.42 | ||
| M426T/V | 0–3.1 | 0–6.6 | ||
| I482L/V/T | 0–0.2 | 0–0.3 | ||
| A486V | 0 | 0 | ||
| V494I/A | 0 | 0–0.8 | ||
| NNI-P1 | C316Y/N * | 0–0.19 | 10.88–36.6 | |
| M414T/L * | 0–0.38 | 0–1.68 | ||
| G554D | 0 | 0 | ||
| S556G | 0–0.4 | 0–8.2 | ||
| D559G/N | 0–0.57 | 0 | ||
| NNI-P2 | S365T/N | 0 | 0 | |
| NNI-P-β | C445F | 0 | 0–0.42 | |
| Y448C/H | 0–3.1 | 0–1.26 | ||
| Y452H | 0–3.1 | 0.42–3.3 | ||
7. Prevalence of RAVs in Treatment Naïve Patients
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mohd Hanafiah, K.; Groeger, J.; Flaxman, A.D.; Wiersma, S.T. Global epidemiology of hepatitis C virus infection: New estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013, 57, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Tsukiyama-Kohara, K.; Iizuka, N.; Kohara, M.; Nomoto, A. Internal ribosome entry site within hepatitis C virus RNA. J. Virol. 1992, 66, 1476–1483. [Google Scholar] [PubMed]
- Wang, C.; Sarnow, P.; Siddiqui, A. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J. Virol. 1993, 67, 3338–3344. [Google Scholar] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Gonzalez-Candelas, F.; Moya, A.; Sanjuan, R. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J. Virol. 2009, 83, 5760–5764. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.U.; Lam, N.P.; Dahari, H.; Gretch, D.R.; Wiley, T.E.; Layden, T.J.; Perelson, A.S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-α therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Martell, M.; Esteban, J.; Quer, J.; Genesca, J.; Weiner, A.; Esteban, R.; Guardia, J.; Gomez, J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: Quasispecies nature of HCV genome distribution. J. Virol. 1992, 66, 3225–3229. [Google Scholar] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2005, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Genetic diversity and evolution of hepatitis C virus—15 Years on. J. Gen. Virol. 2004, 85, 3173–3188. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.H.; Purcell, R.H. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc. Nat. Acad. Sci. 1990, 87, 2057–2061. [Google Scholar] [CrossRef] [PubMed]
- Behrens, S.-E.; Tomei, L.; de Francesco, R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996, 15, 12–22. [Google Scholar] [PubMed]
- Hwang, S.B.; Park, K.-J.; Kim, Y.-S.; Sung, Y.C.; Lai, M. Hepatitis C virus NS5B protein is a membrane-associated phosphoprotein with a predominantly perinuclear localization. Virology 1997, 227, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Wright-Minogue, J.; Fang, J.W.; Baroudy, B.M.; Lau, J.Y.; Hong, Z. Characterization of soluble hepatitis C virus RNA-dependent RNA polymerase expressed in Escherichia coli. J. Virol. 1999, 73, 1649–1654. [Google Scholar] [PubMed]
- Yamashita, T.; Kaneko, S.; Shirota, Y.; Qin, W.; Nomura, T.; Kobayashi, K.; Murakami, S. RNA-dependent RNA polymerase activity of the soluble recombinant hepatitis C virus NS5B protein truncated at the C-terminal region. J. Biol. Chem. 1998, 273, 15479–15486. [Google Scholar] [CrossRef] [PubMed]
- Eltahla, A.A.; Lackovic, K.; Marquis, C.; Eden, J.S.; White, P.A. A fluorescence-based high-throughput screen to identify small compound inhibitors of the genotype 3a hepatitis C virus RNA polymerase. J. Biomol. Screen 2013, 18, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Bressanelli, S.; Tomei, L.; Roussel, A.; Incitti, I.; Vitale, R.L.; Mathieu, M.; De Francesco, R.; Rey, F.A. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Nat. Acad. Sci. 1999, 96, 13034–13039. [Google Scholar] [CrossRef] [PubMed]
- Lesburg, C.A.; Cable, M.B.; Ferrari, E.; Hong, Z.; Mannarino, A.F.; Weber, P.C. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 1999, 6, 937–943. [Google Scholar] [PubMed]
- Ago, H.; Adachi, T.; Yoshida, A.; Yamamoto, M.; Habuka, N.; Yatsunami, K.; Miyano, M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 1999, 7, 1417–1426. [Google Scholar] [CrossRef]
- Butcher, S.J.; Grimes, J.M.; Makeyev, E.V.; Bamford, D.H.; Stuart, D.I. A mechanism for initiating RNA-dependent RNA polymerization. Nature 2001, 410, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Mosley, J.W.; Operskalski, E.A.; Tobler, L.H.; Andrews, W.W.; Phelps, B.; Dockter, J.; Giachetti, C.; Busch, M.P. Viral and host factors in early hepatitis C virus infection. Hepatology 2005, 42, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Bressanelli, S.; Tomei, L.; Rey, F.A.; de Francesco, R. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 2002, 76, 3482–3492. [Google Scholar] [CrossRef] [PubMed]
- Dutartre, H.; Boretto, J.; Guillemot, J.C.; Canard, B. A relaxed discrimination of 2'-O-methyl-GTP relative to GTP between de novo and elongative RNA synthesis by the hepatitis C RNA-dependent RNA polymerase NS5B. J. Biol. Chem. 2005, 280, 6359–6368. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Körner, F.; Herian, U.; Bartenschlager, R. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 1997, 71, 8416–8428. [Google Scholar] [PubMed]
- Luo, G.; Hamatake, R.K.; Mathis, D.M.; Racela, J.; Rigat, K.L.; Lemm, J.; Colonno, R.J. De novo initiation of RNA synthesis by the RNA-dependent RNA polymerase (NS5B) of hepatitis C virus. J. Virol. 2000, 74, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Uss, A.S.; Ferrari, E.; Lau, J.Y.; Hong, Z. De novo initiation of RNA synthesis by hepatitis C virus nonstructural protein 5B polymerase. J. Virol. 2000, 74, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.C.; Singh, P.; Ecker, D.J. De novo initiation of viral RNA-dependent RNA synthesis. Virology 2001, 287, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Harrus, D.; Ahmed-El-Sayed, N.; Simister, P.C.; Miller, S.; Triconnet, M.; Hagedorn, C.H.; Mahias, K.; Rey, F.A.; Astier-Gin, T.; Bressanelli, S. Further insights into the roles of GTP and the C terminus of the hepatitis C virus polymerase in the initiation of RNA synthesis. J. Biol. Chem. 2010, 285, 32906–32918. [Google Scholar] [CrossRef] [PubMed]
- Scrima, N.; Caillet-Saguy, C.; Ventura, M.; Harrus, D.; Astier-Gin, T.; Bressanelli, S. Two crucial early steps in RNA synthesis by the hepatitis C virus polymerase involve a dual role of residue 405. J. Virol. 2012, 86, 7107–7117. [Google Scholar] [CrossRef] [PubMed]
- Ghany, M.G.; Strader, D.B.; Thomas, D.L.; Seeff, L.B. Diagnosis, management, and treatment of hepatitis C: An update. Hepatology 2009, 49, 1335–1374. [Google Scholar] [CrossRef] [PubMed]
- Craxì, A. EASL Clinical Practice Guidelines: Management of hepatitis C virus infection. J. Hepatol. 2011, 55, 245–264. [Google Scholar]
- Manns, M.P.; Wedemeyer, H.; Cornberg, M. Treating viral hepatitis C: Efficacy, side effects, and complications. Gut 2006, 55, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Dusheiko, G.M.; Salupere, R.; Mangia, A.; Flisiak, R.; Hyland, R.H.; Illeperuma, A.; Svarovskaia, E.; Brainard, D.M.; Symonds, W.T. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N. Engl. J. Med. 2014, 370, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Mangia, A.; Wyles, D.; Rodriguez-Torres, M.; Hassanein, T.; Gordon, S.C.; Schultz, M.; Davis, M.N.; Kayali, Z.; Reddy, K.R. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 2013, 368, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Lalezari, J.P.; Hassanein, T.; Kowdley, K.V.; Poordad, F.F.; Sheikh, A.M.; Afdhal, N.H.; Bernstein, D.E.; DeJesus, E.; Freilich, B. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: A randomised, double-blind, phase 2 trial. Lancet Inf. Dis. 2013, 13, 401–408. [Google Scholar] [CrossRef]
- Jacobson, I.M.; Gordon, S.C.; Kowdley, K.V.; Yoshida, E.M.; Rodriguez-Torres, M.; Sulkowski, M.S.; Shiffman, M.L.; Lawitz, E.; Everson, G.; Bennett, M. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N. Engl. J. Med. 2013, 368, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Stedman, C.A.; Hyland, R.H.; Ding, X.; Svarovskaia, E.; Symonds, W.T.; Hindes, R.G.; Berrey, M.M. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N. Engl. J. Med. 2013, 368, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Vogt, P.K. Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Springer: Berlin, German, 2013. [Google Scholar]
- De Clercq, E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 2007, 6, 1001–1018. [Google Scholar] [CrossRef] [PubMed]
- Delang, L.; Neyts, J.; Vliegen, I.; Abrignani, S.; Neddermann, P.; de Francesco, R. Hepatitis C virus-specific directly acting antiviral drugs. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Springer: Berlin, German, 2013; pp. 289–320. [Google Scholar]
- Herlihy, K.J.; Graham, J.P.; Kumpf, R.; Patick, A.K.; Duggal, R.; Shi, S.T. Development of intergenotypic chimeric replicons to determine the broad-spectrum antiviral activities of hepatitis C virus polymerase inhibitors. Antimicrob. Agents Chemother. 2008, 52, 3523–3531. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M.; Najera, I.; Jacobson, I. Resistance to mericitabine, a nucleoside analogue inhibitor of HCV RNA-dependent RNA polymerase. Antivir. Ther. 2012, 17, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Nguyen, T.; Younes, Z.; Santoro, J.; Gitlin, N.; McEniry, D.; Chasen, R.; Goff, J.; Knox, S.; Kleber, K. Valopicitabine (NM283) plus PEG-interferon in treatment-naive hepatitis C patients with HCV genotype-1 infection: HCV RNA clearance during 24 weeks of treatment. Hepatology 2006, 44, 223A–223A. [Google Scholar]
- Pockros, P.; Jensen, D.; Tsai, N.; Taylor, R.; Ramji, A.; Cooper, C.; Dickson, R.; Tice, A.; Stande, S.; Ipe, D. First SVR data with the nucleoside analogue polymerase inhibitor Mericitabine (RG7128) combined with Peginterferon/Ribavirin in treatment-naive HCV g1/4 patients: interim analysis from the JUMP-C trial. J. Hepatol. 2011, 54. [Google Scholar] [CrossRef]
- Gane, E.; Pockros, P.; Zeuzem, S.; Marcellin, P.; Shikhman, A.; Bernaards, C.; Yetzer, E.; Shulman, N.; Tong, X.; Najera, I. Interferon-free treatment with a combination of Mericitabine and Danoprevir with or without Ribavirin in treatment-naive HCV genotype 1-infected patients. J. Hepatol. 2012, 56, S555–S556. [Google Scholar] [CrossRef]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.R. Discovery of a β-d-2ʹ-deoxy-2ʹ-α-fluoro-2ʹ-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Gordon, S.C.; Reddy, K.R.; Rossaro, L.; Bernstein, D.E.; Lawitz, E.; Shiffman, M.L.; Schiff, E.; Ghalib, R.; Ryan, M. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N. Engl. J. Med. 2014, 370, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Sulkowski, M.S.; Ghalib, R.; Rodriguez-Torres, M.; Younossi, Z.M.; Corregidor, A.; DeJesus, E.; Pearlman, B.; Rabinovitz, M.; Gitlin, N. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: The COSMOS randomised study. Lancet 2014, 384, 1756–1765. [Google Scholar] [CrossRef]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Steuer, H.M.M.; Niu, C.; Zennou, V.; Keilman, M.; Zhu, Y.; Lan, S.; Otto, M.J. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Migliaccio, G.; Tomassini, J.E.; Carroll, S.S.; Tomei, L.; Altamura, S.; Bhat, B.; Bartholomew, L.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 2003, 278, 49164–49170. [Google Scholar] [CrossRef] [PubMed]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D.; Wetmore, D.R.; McGrath, M.E.; Ray, A.S. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 2015, 347, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Harrington, P.R.; O'Rear, J.J.; Naeger, L.K. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology 2015, 61, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Svarovskaia, E.S.; Dvory-Sobol, H.; Parkin, N.; Hebner, C.; Gontcharova, V.; Martin, R.; Ouyang, W.; Han, B.; Xu, S.; Ku, K. Infrequent development of resistance in genotype 1–6 hepatitis C virus-infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin. Infect. Dis. 2014, 59, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Pockros, P.J.; Jensen, D.; Tsai, N.; Taylor, R.; Ramji, A.; Cooper, C.; Dickson, R.; Tice, A.; Kulkarni, R.; Vierling, J.M. JUMP-C: A randomized trial of mericitabine plus pegylated interferon α-2a/ribavirin for 24 weeks in treatment-naïve HCV genotype 1/4 patients. Hepatology 2013, 58, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Jensen, D.; Herring, R.; Ferenci, P.; Ma, M.M.; Zeuzem, S.; Rodriguez-Torres, M.; Bzowej, N.; Pockros, P.; Vierling, J. PROPEL: A randomized trial of mericitabine plus peginterferon α-2a/ribavirin therapy in treatment-naïve HCV genotype 1/4 patients. Hepatology 2013, 58, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; le Pogam, S.; Li, L.; Haines, K.; Piso, K.; Baronas, V.; Yan, J.M.; So, S.S.; Klumpp, K.; Nájera, I. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low-level resistance to the HCV polymerase inhibitors mericitabine and sofosbuvir. J. Infect. Dis. 2013, 209, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Pockros, P.J.; Zeuzem, S.; Marcellin, P.; Shikhman, A.; Bernaards, C.; Zhou, J.; Yetzer, E.S.; Ballester, R.; Dwyer, C. Mericitabine and ritonavir-boosted danoprevir with or without ribavirin in treatment-naive HCV genotype 1 patients: INFORM-SVR study. Liver Int. 2015, 35, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.; Stedman, C.; Garg, V.; George, S.; Kieffer, T.; Krop, J.; Lawal, A.; Lan, L.; Rubin, R. An interferon-and ribavirin-free 12-week regimen of once-daily VX-135 and daclatasvir in treatment-naïve patients with genotype 1 HCV infection. J. Hepatol. 2014, 60, S528–S529. [Google Scholar] [CrossRef]
- Sarrazin, C.; Hezode, C.; Zeuzem, S.; Pawlotsky, J.M. Antiviral strategies in hepatitis C virus infection. J. Hepatol. 2012, 56, S88–S100. [Google Scholar] [CrossRef]
- Arnold, J.J.; Sharma, S.D.; Feng, J.Y.; Ray, A.S.; Smidansky, E.D.; Kireeva, M.L.; Cho, A.; Perry, J.; Vela, J.E.; Park, Y. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. 2012. [Google Scholar] [CrossRef]
- Caillet-Saguy, C.; Simister, P.C.; Bressanelli, S. An objective assessment of conformational variability in complexes of hepatitis C virus polymerase with non-nucleoside inhibitors. J. Mol. Biol. 2011, 414, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Eltahla, A.A.; Lim, K.L.; Eden, J.S.; Kelly, A.G.; Mackenzie, J.M.; White, P.A. Nonnucleoside inhibitors of norovirus RNA polymerase: Scaffolds for rational drug design. Antimicrob. Agents Chemother. 2014, 58, 3115–3123. [Google Scholar] [CrossRef] [PubMed]
- Vliegen, I.; Paeshuyse, J.; de Burghgraeve, T.; Lehman, L.S.; Paulson, M.; Shih, I.H.; Mabery, E.; Boddeker, N.; de Clercq, E.; Reiser, H.; et al. Substituted imidazopyridines as potent inhibitors of HCV replication. J. Hepatol. 2009, 50, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.H.; Vliegen, I.; Peng, B.; Yang, H.; Hebner, C.; Paeshuyse, J.; Purstinger, G.; Fenaux, M.; Tian, Y.; Mabery, E.; et al. Mechanistic characterization of GS-9190 (Tegobuvir), a novel nonnucleoside inhibitor of hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 2011, 55, 4196–4203. [Google Scholar] [CrossRef] [PubMed]
- Kneteman, N.M.; Howe, A.Y.; Gao, T.; Lewis, J.; Pevear, D.; Lund, G.; Douglas, D.; Mercer, D.F.; Tyrrell, D.L.J.; Immermann, F. HCV796: A selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 2009, 49, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Vermehren, J.; Sarrazin, C. New HCV therapies on the horizon. Clin. Microbiol. Infect. 2011, 17, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Aghemo, A.; de Francesco, R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013, 58, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Asselah, T.; Angus, P.; Zarski, J.P.; Larrey, D.; Müllhaupt, B.; Gane, E.; Schuchmann, M.; Lohse, A.; Pol, S. Efficacy of the protease inhibitor BI 201335, polymerase inhibitor BI 207127, and ribavirin in patients with chronic HCV infection. Gastroenterology 2011, 141, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, P.; Anderson, P.; Brochu, C.; Bos, M.; Cordingley, M.; Duan, J.; Garneau, M.; Lagrace, L.; Marquis, M.; McKercher, G. Preclinical characterization of the hepatitis C virus NS5B polymerase non-nucleoside inhibitor BI 207127. J. Hepatol. 2012, 56. [Google Scholar] [CrossRef]
- Zeuzem, S.; Soriano, V.; Asselah, T.; Bronowicki, J.-P.; Lohse, A.W.; Müllhaupt, B.; Schuchmann, M.; Bourlière, M.; Buti, M.; Roberts, S.K. Faldaprevir and deleobuvir for HCV genotype 1 infection. N. Engl. J. Med. 2013, 369, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Devogelaere, B.; Berke, J.M.; Vijgen, L.; Dehertogh, P.; Fransen, E.; Cleiren, E.; van der Helm, L.; Nyanguile, O.; Tahri, A.; Amssoms, K. TMC647055, a potent nonnucleoside hepatitis C virus NS5B polymerase inhibitor with cross-genotypic coverage. Antimicrob. Agents Chemother. 2012, 56, 4676–4684. [Google Scholar] [CrossRef] [PubMed]
- Lemm, J.A.; Liu, M.; Gentles, R.G.; Ding, M.; Voss, S.; Pelosi, L.A.; Wang, Y.-K.; Rigat, K.L.; Mosure, K.W.; Bender, J.A. Preclinical characterization of BMS-791325, an allosteric inhibitor of hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 2014, 58, 3485–3495. [Google Scholar] [CrossRef] [PubMed]
- Rigat, K.L.; Lu, H.; Wang, Y.-K.; Argyrou, A.; Fanslau, C.; Beno, B.; Wang, Y.; Marcinkeviciene, J.; Ding, M.; Gentles, R.G. Mechanism of inhibition for BMS-791325, a novel non-nucleoside inhibitor of hepatitis C virus NS5B polymerase. J. Biol. Chem. 2014, 289, 33456–33289. [Google Scholar] [CrossRef] [PubMed]
- McPhee, D.; Falk, P.; Fracasso, P.; Lemm, J.; Liu, M.; Kirk, M.; Hernandez, D.; Cooney, E.; Hughes, E.; Gao, M. Characterization of viral escape in HCV genotype 1-infected patients treated with BMS-791325 and pegylated interferon-alfa and ribavirin. J. Hepatol. 2012, 56. [Google Scholar] [CrossRef]
- Poordad, F.; Sievert, W.; Mollison, L.; Bräu, N.; Levin, J.; Sepe, T.; Lee, S.; Boyer, N.; Bronowicki, J. All-Oral, Fixed-Dose Combination Therapy with Daclatasvir/Asunaprevir/Beclabuvir for Non-Cirrhotic Patients with Chronic HCV Genotype 1 Infection: Unity-1 Phase 3 SVR12 Results. In Proceedings of 65th Annual Meeting of the American Association for the Study of Liver Diseases, Boston, MA, USA, 2014.
- Chan, L.; Das, S.K.; Reddy, T.J.; Poisson, C.; Proulx, M.; Pereira, O.; Courchesne, M.; Roy, C.; Wang, W.; Siddiqui, A. Discovery of thiophene-2-carboxylic acids as potent inhibitors of HCV NS5B polymerase and HCV subgenomic RNA replication. Part 1: Sulfonamides. Bioorg. Med. Chem. Lett. 2004, 14, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, M.; Eng, S.; Leavitt, S.A.; Lee, Y.-J.; Mabery, E.M.; Tian, Y.; Byun, D.; Canales, E.; Clarke, M.O.; Doerffler, E. Preclinical characterization of GS-9669, a thumb site II inhibitor of the hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 2013, 57, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Larouche, R.; Bourgault, B.; Chauret, N.; Proulx, L. Safety, tolerability and pharmacokinetics of the HCV polymerase inhibitor VCH-222 following single dose administration in healthy volunteers and antiviral activity in HCV-infected individuals. J. Hepatol. 2009, 50, S342. [Google Scholar] [CrossRef]
- Gane, E.J.; Stedman, C.A.; Hyland, R.H.; Ding, X.; Svarovskaia, E.; Subramanian, G.M.; Symonds, W.T.; McHutchison, J.G.; Pang, P.S. Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology 2014, 146, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tatlock, J.; Linton, A.; Gonzalez, J.; Jewell, T.; Patel, L.; Ludlum, S.; Drowns, M.; Rahavendran, S.V.; Skor, H. Discovery of (R)-6-cyclopentyl-6-(2-(2, 6-diethylpyridin-4-yl) ethyl)-3-((5, 7-dimethyl-[1,2,4] triazolo [1, 5-a] pyrimidin-2-yl) methyl)-4-hydroxy-5, 6-dihydropyran-2-one (PF-00868554) as a potent and orally available hepatitis C virus polymerase inhibitor. J. Med. Chem. 2009, 52, 1255–1258. [Google Scholar] [PubMed]
- Shi, S.T.; Herlihy, K.J.; Graham, J.P.; Nonomiya, J.; Rahavendran, S.V.; Skor, H.; Irvine, R.; Binford, S.; Tatlock, J.; Li, H. Preclinical characterization of PF-00868554, a potent nonnucleoside inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 2009, 53, 2544–2552. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, I.; Pockros, P.; Lalezari, J.; Lawitz, E.; Rodriguez-Torres, M.; DeJesus, E.; Haas, F.; Martorell, C. Antiviral activity of filibuvir in combination with pegylated interferon alfa-2a and ribavirin for 28 days in treatment naive patients chronically infected with HCV genotype 1. J. Hepatol. 2009, 50, S382–S383. [Google Scholar] [CrossRef]
- Yi, G.; Deval, J.; Fan, B.; Cai, H.; Soulard, C.; Ranjith-Kumar, C.T.; Smith, D.B.; Blatt, L.; Beigelman, L.; Kao, C.C. Biochemical study of the comparative inhibition of hepatitis C virus RNA polymerase by VX-222 and filibuvir. Antimicrob. Agents Chemother. 2012, 56, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Le Pogam, S.; Kang, H.; Harris, S.F.; Leveque, V.; Giannetti, A.M.; Ali, S.; Jiang, W.R.; Rajyaguru, S.; Tavares, G.; Oshiro, C.; et al. Selection and characterization of replicon variants dually resistant to thumb- and palm-binding nonnucleoside polymerase inhibitors of the hepatitis C virus. J. Virol. 2006, 80, 6146–6154. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, E.Z.; Ardzinski, A.; Tigges, A.; Davis, A.; Sullivan, J.C.; Nelson, M.; Spanks, J.; Dorrian, J.; Nicolas, O. Genotypic and phenotypic analyses of hepatitis C virus variants observed in clinical studies of VX-222, a nonnucleoside NS5B polymerase inhibitor. Antimicrob. Agents Chemother. 2014, 58, 5456–5465. [Google Scholar] [CrossRef] [PubMed]
- Di Bisceglie, A.M.; Sulkowski, M.; Gane, E.; Jacobson, I.M.; Nelson, D.; DeSouza, C.; Alves, K.; George, S.; Kieffer, T.; Zhang, E.Z. VX-222, a non-nucleoside NS5B polymerase inhibitor, in telaprevir-based regimens for genotype 1 hepatitis C virus infection. Eur. J. Gastroenterol. Hepatol. 2014, 26, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.; Thompson, R.; Kantaridis, C.; Simpson, P.; Troke, P.J.; Jagannatha, S.; Neelakantan, S.; Purohit, V.S.; Hammond, J.L. Antiviral activity of the hepatitis C virus polymerase inhibitor filibuvir in genotype 1-infected patients. Hepatology 2011, 54, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Dhanak, D.; Duffy, K.J.; Johnston, V.K.; Lin-Goerke, J.; Darcy, M.; Shaw, A.N.; Gu, B.; Silverman, C.; Gates, A.T.; Nonnemacher, M.R.; et al. Identification and biological characterization of heterocyclic inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 2002, 277, 38322–38327. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Kowdley, K.V.; Coakley, E.; Sigal, S.; Nelson, D.R.; Crawford, D.; Weiland, O.; Aguilar, H.; Xiong, J.; Pilot-Matias, T. Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N. Engl. J. Med. 2014, 370, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Poordad, F.; Lawitz, E.; Kowdley, K.; Everson, G.; Freilich, B.; Cohen, D.; Siggelkow, S.; Heckaman, M.; Menon, R.; Pilot-Matias, T. 12-week interferon-free regimen of ABT-450/R+ ABT-333+ ribavirin achieved SVR 12 in more than 90% of treatment-naive HCV genotype-1-infected subjects and 47% of previous non-responders. J. Hepatol. 2012, 56, S549–S550. [Google Scholar] [CrossRef]
- Lawitz, E.; Rodriquez-Torres, M.; Rustgi, V.K.; Hassanein, T.; Rahimy, M.H.; Crowley, C.A.; Freddo, J.L.; Muir, A.; McHutchison, J. Safety and antiviral activity of ANA598 in combination with pegylated interferon α2a plus ribavirin in treatment-naive genotype-1 chronic HCV patients. J. Hepatol. 2010, 52. [Google Scholar] [CrossRef]
- Jensen, D.; Brunda, M.; Elston, R. Interferon-free regimen containing setrobuvir in combination with ritonavir-boosted danoprevir and ribavirin with or without mericitabine in HCV genotype 1 treatment-naive patients: Interim results from the ANNAPURNA study. Hepatology 2013, 58 (Suppl. 4), 849A. [Google Scholar]
- Thompson, P.A.; Patel, R.; Showalter, R.E.; Li, C.; Applemon, J.R.; Steffy, K. In vitro studies demonstrate that combinations of antiviral agents that include HCV polymerase inhibitor ANA598 have the potential to overcome viral resistance. Hepatology 2008, 48, 1164A. [Google Scholar]
- Le Pogam, S.; Seshaadri, A.; Kosaka, A.; Chiu, S.; Kang, H.; Hu, S.; Rajyaguru, S.; Symons, J.; Cammack, N.; Nájera, I. Existence of hepatitis C virus NS5B variants naturally resistant to non-nucleoside, but not to nucleoside, polymerase inhibitors among untreated patients. J. Antimicrob. Chemother. 2008, 61, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, A.; Chopra, R.; Lim, K.; Ciszewski, G.; Shi, M.; Curran, K.J.; Sukits, S.F.; Svenson, K.; Bard, J.; Ellingboe, J.W. Discovery of proline sulfonamides as potent and selective hepatitis C virus NS5b polymerase inhibitors. Evidence for a new NS5b polymerase binding site. J. Med. Chem. 2006, 49, 3052–3055. [Google Scholar] [CrossRef] [PubMed]
- Nyanguile, O.; Pauwels, F.; van den Broeck, W.; Boutton, C.W.; Quirynen, L.; Ivens, T.; van der Helm, L.; Vandercruyssen, G.; Mostmans, W.; Delouvroy, F.; et al. 1,5-benzodiazepines, a novel class of hepatitis C virus polymerase nonnucleoside inhibitors. Antimicrob. Agents Chemother. 2008, 52, 4420–4431. [Google Scholar] [CrossRef] [PubMed]
- Haudecoeur, R.; Peuchmaur, M.; Ahmed-Belkacem, A.; Pawlotsky, J.M.; Boumendjel, A. Structure-activity relationships in the development of allosteric hepatitis C virus RNA-dependent RNA polymerase inhibitors: Ten years of research. Med. Res. Rev. 2013, 33, 934–984. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.Y.; Cheng, H.; Johann, S.; Mullen, S.; Chunduru, S.K.; Young, D.C.; Bard, J.; Chopra, R.; Krishnamurthy, G.; Mansour, T. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 2008, 52, 3327–3338. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Jacobson, I.; Godofsky, E.; Foster, G.R.; Filisiak, R. A Phase 2b Trial comparing 24 to 48 weeks treatment with tegobuvir (GS-9190)/PEG/RBV to 48 weeks treatment with PEG/RBV for chronic genotype 1 HCV infection. J. Hepatol. 2011, 54. [Google Scholar] [CrossRef]
- Zeuzem, S.; Buggisch, P.; Agarwal, K.; Marcellin, P.; Sereni, D.; Klinker, H.; Moreno, C.; Zarski, J.P.; Horsmans, Y.; Mo, H.; et al. The protease inhibitor, GS-9256, and non-nucleoside polymerase inhibitor tegobuvir alone, with ribavirin, or pegylated interferon plus ribavirin in hepatitis C. Hepatology 2012, 55, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Vliegen, I.; Paeshuyse, J.; Zhong, W.; Neyts, J. In vitro combinations containing Tegobuvir are highly efficient in curing cells from HCV replicon and in delaying/preventing the development of drug resistance. Antivir. Res. 2015, 120, 112–121. [Google Scholar] [CrossRef]
- Tomei, L.; Altamura, S.; Bartholomew, L.; Biroccio, A.; Ceccacci, A.; Pacini, L.; Narjes, F.; Gennari, N.; Bisbocci, M.; Incitti, I.; et al. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2003, 77, 13225–13231. [Google Scholar] [CrossRef] [PubMed]
- McKercher, G.; Beaulieu, P.L.; Lamarre, D.; LaPlante, S.; Lefebvre, S.; Pellerin, C.; Thauvette, L.; Kukolj, G. Specific inhibitors of HCV polymerase identified using an NS5B with lower affinity for template/primer substrate. Nucleic Acids Res. 2004, 32, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, S.; Volpari, C.; Tomei, L.; Altamura, S.; Harper, S.; Narjes, F.; Koch, U.; Rowley, M.; De Francesco, R.; Migliaccio, G. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J. Biol. Chem. 2005, 280, 29765–29770. [Google Scholar] [CrossRef]
- Winquist, J.; Abdurakhmanov, E.; Baraznenok, V.; Henderson, I.; Vrang, L.; Danielson, U.H. Resolution of the interaction mechanisms and characteristics of non-nucleoside inhibitors of hepatitis C virus polymerase. Antivir. Res. 2013, 97, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Biswal, B.K.; Cherney, M.M.; Wang, M.; Chan, L.; Yannopoulos, C.G.; Bilimoria, D.; Nicolas, O.; Bedard, J.; James, M.N.G. Crystal structures of the RNA-dependent RNA polymerase genotype 2a of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J. Biol. Chem. 2005, 280, 18202–18210. [Google Scholar] [CrossRef] [PubMed]
- Biswal, B.K.; Wang, M.; Cherney, M.M.; Chan, L.; Yannopoulos, C.G.; Bilimoria, D.; Bedard, J.; James, M.N. Non-nucleoside inhibitors binding to hepatitis C virus NS5B polymerase reveal a novel mechanism of inhibition. J. Mol. Biol. 2006, 361, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Eltahla, A.A.; Tay, E.; Douglas, M.W.; White, P.A. Cross-genotypic examination of hepatitis C virus polymerase inhibitors reveals a novel mechanism of action for thumb binders. Antimicrob. Agents Chemother. 2014, 58, 7215–7224. [Google Scholar] [CrossRef] [PubMed]
- Hebner, C.M.; Han, B.; Brendza, K.M.; Nash, M.; Sulfab, M.; Tian, Y.; Hung, M.; Fung, W.; Vivian, R.W.; Trenkle, J.; et al. The HCV non-nucleoside inhibitor Tegobuvir utilizes a novel mechanism of action to inhibit NS5B polymerase function. PLoS ONE 2012, 7, e39163. [Google Scholar] [CrossRef] [PubMed]
- Mosley, R.T.; Edwards, T.E.; Murakami, E.; Lam, A.M.; Grice, R.L.; Du, J.; Sofia, M.J.; Furman, P.A.; Otto, M.J. Structure of hepatitis C virus polymerase in complex with primer-template RNA. J. Virol. 2012, 86, 6503–6511. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.P.; Piper, D.E.; Li, Y.; Mayorga, V.; Anzola, J.; Chen, J.M.; Jaen, J.C.; Lee, G.; Liu, J.; Peterson, M.G. SAR and mode of action of novel non-nucleoside inhibitors of hepatitis C NS5b RNA polymerase. J. Med. Chem. 2006, 49, 1034–1046. [Google Scholar] [CrossRef]
- Hang, J.Q.; Yang, Y.; Harris, S.F.; Leveque, V.; Whittington, H.J.; Rajyaguru, S.; Ao-Ieong, G.; McCown, M.F.; Wong, A.; Giannetti, A.M. Slow binding inhibition and mechanism of resistance of non-nucleoside polymerase inhibitors of hepatitis C virus. J. Biol. Chem. 2009, 284, 15517–15529. [Google Scholar] [CrossRef] [PubMed]
- Tomei, L.; Altamura, S.; Bartholomew, L.; Bisbocci, M.; Bailey, C.; Bosserman, M.; Cellucci, A.; Forte, E.; Incitti, I.; Orsatti, L. Characterization of the inhibition of hepatitis C virus RNA replication by nonnucleosides. J. Virol. 2004, 78, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Johnston, V.K.; Gutshall, L.L.; Nguyen, T.T.; Gontarek, R.R.; Darcy, M.G.; Tedesco, R.; Dhanak, D.; Duffy, K.J.; Kao, C.C. Arresting initiation of hepatitis C virus RNA synthesis using heterocyclic derivatives. J. Biol. Chem. 2003, 278, 16602–16607. [Google Scholar] [CrossRef] [PubMed]
- Zein, N.N. Clinical significance of hepatitis C virus genotypes. Clin. Microbiol. Rev. 2000, 13, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Bartenschlager, R. On the History of Hepatitis C Virus Cell Culture Systems: Miniperspective. J. Med. Chem. 2013, 57, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Corsa, A.C.; Xu, S.; Peng, B.; Gong, R.; Lee, Y.J.; Chan, K.; Mo, H.; Delaney, W., IV; Cheng, G. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antivir. Res. 2013, 100, 439–445. [Google Scholar] [CrossRef] [PubMed]
- May, M.M.; Lorengel, H.; Kreuter, J.; Zimmermann, H.; Ruebsamen-Schaeff, H.; Urban, A. RNA-dependent RNA polymerases from different hepatitis C virus genotypes reveal distinct biochemical properties and drug susceptibilities. Biochim. Biophys. Acta 2011, 1814, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.A.; Xu, S.; Martin, R.; Miller, M.D.; Mo, H. Tegobuvir (GS-9190) potency against HCV chimeric replicons derived from consensus NS5B sequences from genotypes 2b, 3a, 4a, 5a, and 6a. Virology 2012, 429, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, F.; Mostmans, W.; Quirynen, L.M.; van der Helm, L.; Boutton, C.W.; Rueff, A.S.; Cleiren, E.; Raboisson, P.; Surleraux, D.; Nyanguile, O.; et al. Binding-site identification and genotypic profiling of hepatitis C virus polymerase inhibitors. J. Virol. 2007, 81, 6909–6919. [Google Scholar] [CrossRef] [PubMed]
- Maring, C.; Wagner, R.; Hutchinson, D.; Flentge, C.; Kati, W.; Koev, G.; Liu, Y.; Beno, D.; Shen, J.; Lau, Y. Preclinical potency, pharmacokinetic and AMDE characterization of ABT-333, a novel non-nucleoside HCV polymerase inhibitor. J. Hepatol. 2009, 50, S347. [Google Scholar] [CrossRef]
- Paolucci, S.; Fiorina, L.; Mariani, B.; Gulminetti, R.; Novati, S.; Barbarini, G.; Bruno, R.; Baldanti, F. Naturally occurring resistance mutations to inhibitors of HCV NS5A region and NS5B polymerase in DAA treatment-naïve patients. Virol J. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Gaudieri, S.; Rauch, A.; Pfafferott, K.; Barnes, E.; Cheng, W.; McCaughan, G.; Shackel, N.; Jeffrey, G.P.; Mollison, L.; Baker, R. Hepatitis C virus drug resistance and immune-driven adaptations: Relevance to new antiviral therapy. Hepatology 2009, 49, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Queiroz, A.T.L.; Pessoa, M.G.; da Silva, E.; Mazo, D.F.C.; Carrilho, F.J.; Carvalho-Filho, R.; Carvalho, I.M.V.G.D. The presence of resistance mutations to protease and polymerase inhibitors in Hepatitis C virus sequences from the Los Alamos databank. J. Viral. Hepat. 2013, 20, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Kuntzen, T.; Timm, J.; Berical, A.; Lennon, N.; Berlin, A.M.; Young, S.K.; Lee, B.; Heckerman, D.; Carlson, J.; Reyor, L.L. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naïve patients. Hepatology 2008, 48, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Bartels, D.J.; Sullivan, J.C.; Zhang, E.Z.; Tigges, A.M.; Dorrian, J.L.; de Meyer, S.; Takemoto, D.; Dondero, E.; Kwong, A.D.; Picchio, G. HCV variants with decreased sensitivity to direct acting antivirals were rarely observed in DAA-naïve patients prior to treatment. J. Virol. 2012, 87, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Margeridon-Thermet, S.; Le Pogam, S.; Li, L.; Liu, T.F.; Shulman, N.; Shafer, R.W.; Najera, I. Similar prevalence of low-abundance drug-resistant variants in treatment-naive patients with genotype 1a and 1b hepatitis C virus infections as determined by ultradeep pyrosequencing. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Applegate, T.L.; Gaudieri, S.; Plauzolles, A.; Chopra, A.; Grebely, J.; Lucas, M.; Hellard, M.; Luciani, F.; Dore, G.J.; Matthews, G.V. Naturally occurring dominant drug resistance mutations occur infrequently in the setting of recently acquired hepatitis C. Antivir. Ther. 2014, 20, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Lauck, M.; Alvarado-Mora, M.V.; Becker, E.A.; Bhattacharya, D.; Striker, R.; Hughes, A.L.; Carrilho, F.J.; O’Connor, D.H.; Pinho, J.R.R. Analysis of hepatitis C virus intrahost diversity across the coding region by ultradeep pyrosequencing. J. Virol. 2012, 86, 3952–3960. [Google Scholar] [CrossRef] [PubMed]
- Nasu, A.; Marusawa, H.; Ueda, Y.; Nishijima, N.; Takahashi, K.; Osaki, Y.; Yamashita, Y.; Inokuma, T.; Tamada, T.; Fujiwara, T. Genetic heterogeneity of hepatitis C virus in association with antiviral therapy determined by ultra-deep sequencing. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Powdrill, M.H.; Tchesnokov, E.P.; Kozak, R.A.; Russell, R.S.; Martin, R.; Svarovskaia, E.S.; Mo, H.; Kouyos, R.D.; Götte, M. Contribution of a mutational bias in hepatitis C virus replication to the genetic barrier in the development of drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 20509–20513. [Google Scholar] [CrossRef] [PubMed]
- Kukolj, G.; McGibbon, G.A.; McKercher, G.; Marquis, M.; Lefèbvre, S.; Thauvette, L.; Gauthier, J.; Goulet, S.; Poupart, M.A.; Beaulieu, P.L. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 2005, 280, 39260–39267. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.D.; Sarrazin, C. Antiviral therapy of hepatitis C in 2014: Do we need resistance testing? Antivir. Res. 2014, 105, 64–71. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eltahla, A.A.; Luciani, F.; White, P.A.; Lloyd, A.R.; Bull, R.A. Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance. Viruses 2015, 7, 5206-5224. https://doi.org/10.3390/v7102868
Eltahla AA, Luciani F, White PA, Lloyd AR, Bull RA. Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance. Viruses. 2015; 7(10):5206-5224. https://doi.org/10.3390/v7102868
Chicago/Turabian StyleEltahla, Auda A., Fabio Luciani, Peter A. White, Andrew R. Lloyd, and Rowena A. Bull. 2015. "Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance" Viruses 7, no. 10: 5206-5224. https://doi.org/10.3390/v7102868
APA StyleEltahla, A. A., Luciani, F., White, P. A., Lloyd, A. R., & Bull, R. A. (2015). Inhibitors of the Hepatitis C Virus Polymerase; Mode of Action and Resistance. Viruses, 7(10), 5206-5224. https://doi.org/10.3390/v7102868