The Coronavirus Nucleocapsid Is a Multifunctional Protein



Abstract
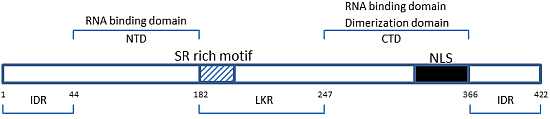
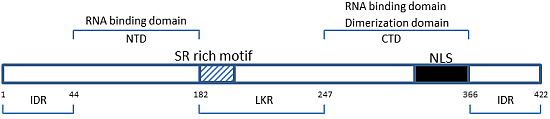
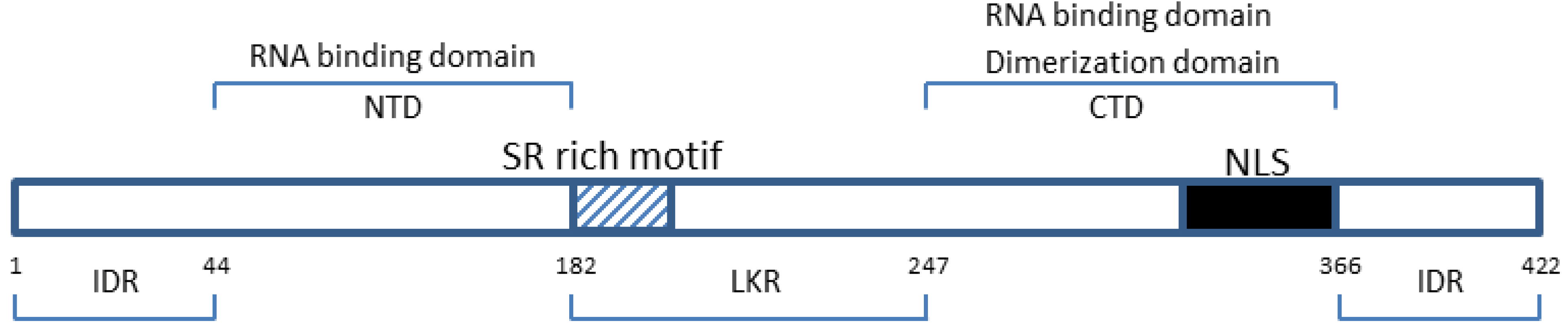
:
1. Introduction
2. Topology of CoV N and RNA Binding

3. Intracellular Localization of the Nucleocapsid Protein
4. Functions of the Nucleocapsid
4.1. Virus Life Cycle
4.1.1. CoV N and Viral Core Formation
4.1.2. CoV N and Viral Assembly
4.1.3. CoV N and Virus Budding/Envelope Formation
4.1.4. Genomic mRNA Replication/Genomic RNA Synthesis
4.2. Cellular Response
4.2.1. Chaperone Activity
4.2.2. Cell Cycle Regulation
4.2.3. Cell Stress Responses—Host Translational Shutoff
4.2.4. Viral Pathogenesis—Immune System Interference
4.2.5. Signal Transduction

{kind=link}
{kind=link}
| 1. Virus Life Cycle | Function |
|---|---|
| 1.1 Viral Core Formation |
|
| 1.2 Viral Assembly | |
| 1.3 Virus Budding/envelope formation | |
| 1.4 Genomic mRNA replication/genomic RNA synthesis | |
| 2. Cellular Response | |
| 2.1. Chaperone activity | |
| 2.2. Cell cycle regulation |
|
| 2.3. Cell stress responses—host translational shutoff |
|
| 2.4. Viral pathogenesis—Immune system interference | |
| 2.5. Signal transduction |
|
5. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef]
- Fielding, B.C. Human coronavirus NL63: A clinically important virus? Future Microbiol. 2011, 6, 153–159. [Google Scholar] [CrossRef]
- The International Committee for Taxonomy of Viruses (ICTV). Available online: htp://talk.ictvonline.org/files/ictv_documents/m/msl/4090.aspx (accessed on 27 June 2014).
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. Adv. Virus Res. 1997, 48, 1–100. [Google Scholar]
- Pyrc, K.; Berkhout, B.; van der Hoek, L. The novel human coronaviruses NL63 and HKU1. J. Virol. 2007, 81, 3051–3057. [Google Scholar] [CrossRef]
- Fang, S.G.; Shen, H.; Wang, J.; Tay, F.P.; Liu, D.X. Proteolytic processing of polyproteins 1a and 1ab between non-structural proteins 10 and 11/12 of Coronavirus infectious bronchitis virus is dispensable for viral replication in cultured cells. Virology 2008, 379, 175–180. [Google Scholar] [CrossRef]
- Brierley, I.; Boursnell, M.E.; Binns, M.M.; Bilimoria, B.; Blok, V.C.; Brown, T.D.; Inglis, S.C. An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J. 1987, 6, 3779–3785. [Google Scholar]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.; Guan, Y.; Rozanov, M.; Spaan, W.J.; Gorbalenya, A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weissbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84 Pt 9, 2305–2315. [Google Scholar]
- Laude, H.; Masters, P. The coronavirus nucleocapsid protein. In Coronaviruses and Arteriviruses; Plenum Press: New York, NY, USA, 1995; pp. 141–163. [Google Scholar]
- Lai, M.; Cavanagh, D. The molecular biology of coronaviruses. Adv. Virus Res. 1997, 48, 1–100. [Google Scholar]
- Masters, P. Localization of an RNA-binding domain in the nucleocapsid protein of the coronavirus mouse hepatitis virus. Arch. Virol. 1992, 125, 141–160. [Google Scholar] [CrossRef]
- Parker, M.M.; Masters, P.S. Sequence comparison of the N genes of five strains of the coronavirus mouse hepatitis virus suggests a three domain structure for the nucleocapsid protein. Virology 1990, 179, 463–468. [Google Scholar] [CrossRef]
- Huang, Q.; Yu, L.; Petros, A.M.; Gunasekera, A.; Liu, Z.; Xu, N.; Hajduk, P.; Mack, J.; Fesik, S.W.; Olejniczak, E.T. Structure of the N-terminal RNA-binding domain of the SARS CoV nucleocapsid protein. Biochemistry 2004, 43, 6059–6063. [Google Scholar] [CrossRef]
- Chang, C.K.; Sue, S.C.; Yu, T.H.; Hsieh, C.M.; Tsai, C.K.; Chiang, Y.C.; Lee, S.J.; Hsiao, H.H.; Wu, W.J.; Chang, C.F.; et al. The dimer interface of the SARS coronavirus nucleocapsid protein adapts a porcine respiratory and reproductive syndrome virus-like structure. FEBS Lett. 2005, 579, 5663–5668. [Google Scholar] [CrossRef]
- Keane, S.C.; Liu, P.; Leibowitz, J.L.; Giedroc, D.P. Functional transcriptional regulatory sequence (TRS) RNA binding and helix destabilizing determinants of murine hepatitis virus (MHV) nucleocapsid (N) protein. J. Biol. Chem. 2012, 287, 7063–7073. [Google Scholar]
- Chang, C.K.; Hou, M.H.; Chang, C.F.; Hsiao, C.D.; Huang, T.H. The SARS coronavirus nucleocapsid protein—Forms and functions. Antivir. Res. 2014, 103C, 39–50. [Google Scholar]
- Jayaram, H.; Fan, H.; Bowman, B.R.; Ooi, A.; Jayaram, J.; Collisson, E.W.; Lescar, J.; Prasad, B.V. X-ray structures of the N- and C-terminal domains of a coronavirus nucleocapsid protein: Implications for nucleocapsid formation. J. Virol. 2006, 80, 6612–6620. [Google Scholar] [CrossRef]
- Fan, H.; Ooi, A.; Tan, Y.W.; Wang, S.; Fang, S.; Liu, D.X.; Lescar, J. The nucleocapsid protein of coronavirus infectious bronchitis virus: Crystal structure of its N-terminal domain and multimerization properties. Structure 2005, 13, 1859–1868. [Google Scholar] [CrossRef]
- Spencer, K.A.; Hiscox, J.A. Characterisation of the RNA binding properties of the coronavirus infectious bronchitis virus nucleocapsid protein amino-terminal region. FEBS Lett. 2006, 580, 5993–5998. [Google Scholar] [CrossRef]
- Tan, Y.W.; Fang, S.; Fan, H.; Lescar, J.; Liu, D.X. Amino acid residues critical for RNA-binding in the N-terminal domain of the nucleocapsid protein are essential determinants for the infectivity of coronavirus in cultured cells. Nucleic Acids Res. 2006, 34, 4816–4825. [Google Scholar] [CrossRef]
- Oubridge, C.; Ito, N.; Evans, P.R.; Teo, C.H.; Nagai, K. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature 1994, 372, 432–438. [Google Scholar] [CrossRef]
- Valegard, K.; Murray, J.B.; Stockley, P.G.; Stonehouse, N.J.; Liljas, L. Crystal structure of an RNA bacteriophage coat protein-operator complex. Nature 1994, 371, 623–626. [Google Scholar] [CrossRef]
- Grossoehme, N.E.; Li, L.; Keane, S.C.; Liu, P.; Dann, C.E., 3rd; Leibowitz, J.L.; Giedroc, D.P. Coronavirus N protein N-terminal domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS-cTRS RNA duplexes. J. Mol. Biol. 2009, 394, 544–557. [Google Scholar] [CrossRef]
- Ma, Y.; Tong, X.; Xu, X.; Li, X.; Lou, Z.; Rao, Z. Structures of the N- and C-terminal domains of MHV-A59 nucleocapsid protein corroborate a conserved RNA-protein binding mechanism in coronavirus. Protein Cell 2010, 1, 688–697. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L. A new model for coronavirus transcription. Adv. Exp. Med. Biol. 1998, 440, 215–219. [Google Scholar] [CrossRef]
- Zuniga, S.; Sola, I.; Alonso, S.; Enjuanes, L. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 2004, 78, 980–994. [Google Scholar] [CrossRef]
- Zuniga, S.; Sola, I.; Moreno, J.L.; Sabella, P.; Plana-Duran, J.; Enjuanes, L. Coronavirus nucleocapsid protein is an RNA chaperone. Virology 2007, 357, 215–227. [Google Scholar] [CrossRef]
- Zuniga, S.; Cruz, J.L.; Sola, I.; Mateos-Gomez, P.A.; Palacio, L.; Enjuanes, L. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 2010, 84, 2169–2175. [Google Scholar] [CrossRef]
- Urbaneja, M.A.; Wu, M.; Casas-Finet, J.R.; Karpel, R.L. HIV-1 nucleocapsid protein as a nucleic acid chaperone: Spectroscopic study of its helix-destabilizing properties, structural binding specificity, and annealing activity. J. Mol. Biol. 2002, 318, 749–764. [Google Scholar] [CrossRef]
- Monaghan, A.; Webster, A.; Hay, R.T. Adenovirus DNA binding protein: Helix destabilising properties. Nucleic Acids Res. 1994, 22, 742–748. [Google Scholar] [CrossRef]
- Hurst, K.R.; Koetzner, C.A.; Masters, P.S. Identification of in vivo-interacting domains of the murine coronavirus nucleocapsid protein. J. Virol. 2009, 83, 7221–7234. [Google Scholar] [CrossRef]
- You, J.; Dove, B.K.; Enjuanes, L.; DeDiego, M.L.; Alvarez, E.; Howell, G.; Heinen, P.; Zambon, M.; Hiscox, J.A. Subcellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein. J. Gen. Virol. 2005, 86 Pt 12, 3303–3310. [Google Scholar]
- Stohlman, S.A.; Baric, R.S.; Nelson, G.N.; Soe, L.H.; Welter, L.M.; Deans, R.J. Specific interaction between coronavirus leader RNA and nucleocapsid protein. J. Virol. 1988, 62, 4288–4295. [Google Scholar]
- Chang, C.K.; Hsu, Y.L.; Chang, Y.H.; Chao, F.A.; Wu, M.C.; Huang, Y.S.; Hu, C.K.; Huang, T.H. Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: Implications for ribonucleocapsid protein packaging. J. Virol. 2009, 83, 2255–2264. [Google Scholar] [CrossRef]
- Peng, T.Y.; Lee, K.R.; Tarn, W.Y. Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J. 2008, 275, 4152–4163. [Google Scholar] [CrossRef]
- Surjit, M.; Kumar, R.; Mishra, R.N.; Reddy, M.K.; Chow, V.T.; Lal, S.K. The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J. Virol. 2005, 79, 11476–11486. [Google Scholar] [CrossRef]
- Wu, C.H.; Yeh, S.H.; Tsay, Y.G.; Shieh, Y.H.; Kao, C.L.; Chen, Y.S.; Wang, S.H.; Kuo, T.J.; Chen, D.S.; Chen, P.J. Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J. Biol. Chem. 2009, 284, 5229–5239. [Google Scholar] [CrossRef]
- Luo, H.; Chen, Q.; Chen, J.; Chen, K.; Shen, X.; Jiang, H. The nucleocapsid protein of SARS coronavirus has a high binding affinity to the human cellular heterogeneous nuclear ribonucleoprotein A1. FEBS Lett. 2005, 579, 2623–2628. [Google Scholar] [CrossRef]
- Chang, C.K.; Sue, S.C.; Yu, T.H.; Hsieh, C.M.; Tsai, C.K.; Chiang, Y.C.; Lee, S.J.; Hsiao, H.H.; Wu, W.J.; Chang, W.L.; et al. Modular organization of SARS coronavirus nucleocapsid protein. J. Biomed. Sci. 2006, 13, 59–72. [Google Scholar] [CrossRef]
- Nelson, G.W.; Stohlman, S.A.; Tahara, S.M. High affinity interaction between nucleocapsid protein and leader/intergenic sequence of mouse hepatitis virus RNA. J. Gen. Virol. 2000, 81, 181–188. [Google Scholar]
- He, R.; Dobie, F.; Ballantine, M.; Leeson, A.; Li, Y.; Bastien, N.; Cutts, T.; Andonov, A.; Cao, J.; Booth, T.F.; et al. Analysis of multimerization of the SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 2004, 316, 476–483. [Google Scholar] [CrossRef]
- Chang, C.K.; Chen, C.M.; Chiang, M.H.; Hsu, Y.L.; Huang, T.H. Transient oligomerization of the SARS-COV N protein—Implication for virus ribonucleoprotein packaging. PLoS One 2013, 8, e65045. [Google Scholar]
- Luo, H.; Ye, F.; Chen, K.; Shen, X.; Jiang, H. SR-rich motif plays a pivotal role in recombinant SARS coronavirus nucleocapsid protein multimerization. Biochemistry 2005, 44, 15351–15358. [Google Scholar] [CrossRef]
- Yu, I.M.; Oldham, M.L.; Zhang, J.; Chen, J. Crystal structure of the severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein dimerization domain reveals evolutionary linkage between corona- and arteriviridae. J. Biol. Chem. 2006, 281, 17134–17139. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Verheije, M.H.; Hagemeijer, M.C.; Ulasli, M.; Reggiori, F.; Rottier, P.J.; Masters, P.S.; de Haan, C.A. The coronavirus nucleocapsid protein is dynamically associated with the replication-transcription complexes. J. Virol. 2010, 84, 11575–11579. [Google Scholar] [CrossRef]
- Keane, S.C.; Giedroc, D.P. Solution structure of mouse hepatitis virus (MHV) nsp3a and determinants of the interaction with MHV nucleocapsid (N) protein. J. Virol. 2013, 87, 3502–3515. [Google Scholar] [CrossRef]
- Hurst, K.R.; Ye, R.; Goebel, S.J.; Jayaraman, P.; Masters, P.S. An interaction between the nucleocapsid protein and a component of the replicase-transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J. Virol 2010, 84, 10276–10288. [Google Scholar] [CrossRef]
- Lo, Y.S.; Lin, S.Y.; Wang, S.M.; Wang, C.T.; Chiu, Y.L.; Huang, T.H.; Hou, M.H. Oligomerization of the carboxyl terminal domain of the human coronavirus 229E nucleocapsid protein. FEBS Lett. 2013, 587, 120–127. [Google Scholar] [CrossRef]
- Yu, I.M.; Gustafson, C.L.; Diao, J.; Burgner, J.W., 2nd; Li, Z.; Zhang, J.; Chen, J. Recombinant severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein forms a dimer through its C-terminal domain. J. Biol. Chem. 2005, 280, 23280–23286. [Google Scholar]
- Chen, C.Y.; Chang, C.K.; Chang, Y.W.; Sue, S.C.; Bai, H.I.; Riang, L.; Hsiao, C.D.; Huang, T.H. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 2007, 368, 1075–1086. [Google Scholar] [CrossRef]
- Surjit, M.; Liu, B.; Kumar, P.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of the SARS coronavirus is capable of self-association through a C-terminal 209 amino acid interaction domain. Biochem. Biophys. Res. Commun. 2004, 317, 1030–1036. [Google Scholar] [CrossRef]
- Takeda, M.; Chang, C.K.; Ikeya, T.; Guntert, P.; Chang, Y.H.; Hsu, Y.L.; Huang, T.H.; Kainosho, M. Solution structure of the c-terminal dimerization domain of SARS coronavirus nucleocapsid protein solved by the SAIL-NMR method. J. Mol. Biol. 2008, 380, 608–622. [Google Scholar] [CrossRef]
- Zlotnick, A. Theoretical aspects of virus capsid assembly. J. Mol. Recognit. 2005, 18, 479–490. [Google Scholar] [CrossRef]
- Hsieh, P.K.; Chang, S.C.; Huang, C.C.; Lee, T.T.; Hsiao, C.W.; Kou, Y.H.; Chen, I.Y.; Chang, C.K.; Huang, T.H.; Chang, M.F. Assembly of severe acute respiratory syndrome coronavirus RNA packaging signal into virus-like particles is nucleocapsid dependent. J. Virol. 2005, 79, 13848–13855. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, Z.Y.; Kong, W.P.; Nabel, G.J. Generation of synthetic severe acute respiratory syndrome coronavirus pseudoparticles: Implications for assembly and vaccine production. J. Virol. 2004, 78, 12557–12565. [Google Scholar] [CrossRef]
- Hurst, K.R.; Kuo, L.; Koetzner, C.A.; Ye, R.; Hsue, B.; Masters, P.S. A major determinant for membrane protein interaction localizes to the carboxy-terminal domain of the mouse coronavirus nucleocapsid protein. J. Virol. 2005, 79, 13285–13297. [Google Scholar] [CrossRef]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 473–484. [Google Scholar]
- Garner, E.; Romero, P.; Dunker, A.K.; Brown, C.; Obradovic, Z. Predicting Binding Regions within Disordered Proteins. Genome Inform. Workshop Genome Inform. 1999, 10, 41–50. [Google Scholar]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Luo, H.; Chen, J.; Chen, K.; Shen, X.; Jiang, H. Carboxyl terminus of severe acute respiratory syndrome coronavirus nucleocapsid protein: Self-association analysis and nucleic acid binding characterization. Biochemistry 2006, 45, 11827–11835. [Google Scholar] [CrossRef]
- He, R.; Leeson, A.; Ballantine, M.; Andonov, A.; Baker, L.; Dobie, F.; Li, Y.; Bastien, N.; Feldmann, H.; Strocher, U.; et al. Characterization of protein-protein interactions between the nucleocapsid protein and membrane protein of the SARS coronavirus. Virus Res. 2004, 105, 121–125. [Google Scholar] [CrossRef]
- Hiscox, J.A.; Wurm, T.; Wilson, L.; Britton, P.; Cavanagh, D.; Brooks, G. The coronavirus infectious bronchitis virus nucleoprotein localizes to the nucleolus. J. Virol. 2001, 75, 506–512. [Google Scholar]
- Reed, M.L.; Dove, B.K.; Jackson, R.M.; Collins, R.; Brooks, G.; Hiscox, J.A. Delineation and modelling of a nucleolar retention signal in the coronavirus nucleocapsid protein. Traffic 2006, 7, 833–848. [Google Scholar] [CrossRef]
- Cawood, R.; Harrison, S.M.; Dove, B.K.; Reed, M.L.; Hiscox, J.A. Cell cycle dependent nucleolar localization of the coronavirus nucleocapsid protein. Cell Cycle 2007, 6, 863–867. [Google Scholar] [CrossRef]
- Wurm, T.; Chen, H.; Hodgson, T.; Britton, P.; Brooks, G.; Hiscox, J.A. Localization to the nucleolus is a common feature of coronavirus nucleoproteins, and the protein may disrupt host cell division. J. Virol. 2001, 75, 9345–9356. [Google Scholar] [CrossRef]
- Rowland, R.R.; Chauhan, V.; Fang, Y.; Pekosz, A.; Kerrigan, M.; Burton, M.D. Intracellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein: Absence of nucleolar accumulation during infection and after expression as a recombinant protein in vero cells. J. Virol. 2005, 79, 11507–11512. [Google Scholar]
- Chen, H.; Wurm, T.; Britton, P.; Brooks, G.; Hiscox, J.A. Interaction of the coronavirus nucleoprotein with nucleolar antigens and the host cell. J. Virol. 2002, 76, 5233–5250. [Google Scholar] [CrossRef]
- Lai, F.W.; Stephenson, K.B.; Mahony, J.; Lichty, B.D. Human coronavirus OC43 nucleocapsid protein binds microRNA 9 and potentiates NF-kappaB activation. J. Virol. 2014, 88, 54–65. [Google Scholar] [CrossRef]
- Timani, K.A.; Liao, Q.; Ye, L.; Zeng, Y.; Liu, J.; Zheng, Y.; Yang, X.; Lingbao, K.; Gao, J.; Zhu, Y. Nuclear/nucleolar localization properties of C-terminal nucleocapsid protein of SARS coronavirus. Virus Res. 2005, 114, 23–34. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Wang, Q.; Liu, X.; Li, X.; Li, P.; Ma, Q.; Cao, C. The nucleocapsid protein of severe acute respiratory syndrome coronavirus inhibits cell cytokinesis and proliferation by interacting with translation elongation factor 1alpha. J. Virol. 2008, 82, 6962–6971. [Google Scholar] [CrossRef]
- Marra, M.A.; Jones, S.J.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The Genome sequence of the SARS-associated coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef]
- De Haan, C.A.; Rottier, P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar] [CrossRef]
- Robbins, S.G.; Frana, M.F.; McGowan, J.J.; Boyle, J.F.; Holmes, K.V. RNA-binding proteins of coronavirus MHV: Detection of monomeric and multimeric N protein with an RNA overlay-protein blot assay. Virology 1986, 150, 402–410. [Google Scholar] [CrossRef]
- Baric, R.S.; Nelson, G.W.; Fleming, J.O.; Deans, R.J.; Keck, J.G.; Casteel, N.; Stohlman, S.A. Interactions between coronavirus nucleocapsid protein and viral RNAs: Implications for viral transcription. J. Virol. 1988, 62, 4280–4287. [Google Scholar]
- Narayanan, K.; Kim, K.H.; Makino, S. Characterization of N protein self-association in coronavirus ribonucleoprotein complexes. Virus Res. 2003, 98, 131–140. [Google Scholar] [CrossRef]
- Risco, C.; Antón, I.M.; Enjuanes, L.; Carrascosa, J.L. The transmissible gastroenteritis coronavirus contains a spherical core shell consisting of M and N proteins. J. Virol. 1996, 70, 4773–4777. [Google Scholar]
- Escors, D.; Ortego, J.; Laude, H.; Enjuanes, L. The membrane M protein carboxy terminus binds to transmissible gastroenteritis coronavirus core and contributes to core stability. J. Virol. 2001, 75, 1312–1324. [Google Scholar] [CrossRef]
- Kuo, L.; Masters, P.S. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J. Virol. 2002, 76, 4987–4999. [Google Scholar] [CrossRef]
- Macneughton, M.R.; Davies, H.A. Ribonucleoprotein-like structures from coronavirus particles. J. Gen. Virol. 1978, 39, 545–549. [Google Scholar] [CrossRef]
- Chen, H.; Gill, A.; Dove, B.K.; Emmett, S.R.; Kemp, C.F.; Ritchie, M.A.; Dee, M.; Hiscox, J.A. Mass spectroscopic characterization of the coronavirus infectious bronchitis virus nucleoprotein and elucidation of the role of phosphorylation in RNA binding by using surface plasmon resonance. J. Virol. 2005, 79, 1164–1179. [Google Scholar] [CrossRef]
- Davies, H.A.; Dourmashkin, R.R.; Macnaughton, M.R. Ribonucleoprotein of Avian Infectious Bronchitis Virus. J. Gen. Virol. 1981, 53, 67–74. [Google Scholar] [CrossRef]
- De Haan, C.A.; Kuo, L.; Masters, P.S.; Vennema, H.; Rottier, P.J. Coronavirus particle assembly: Primary structure requirements of the membrane protein. J. Virol. 1998, 72, 6838–6850. [Google Scholar]
- De Haan, C.A.; Smeets, M.; Vernooij, F.; Vennema, H.; Rottier, P.J. Mapping of the coronavirus membrane protein domains involved in interaction with the spike protein. J. Virol. 1999, 73, 7441–7452. [Google Scholar]
- Simons, K.; Garoff, H. The budding mechanisms of enveloped animal viruses. J. Gen. Virol. 1980, 50, 1–21. [Google Scholar] [CrossRef]
- Lopez, S.; Yao, J.S.; Kuhn, R.J.; Strauss, E.G.; Strauss, J.H. Nucleocapsid-glycoprotein interactions required for assembly of alphaviruses. J. Virol. 1994, 68, 1316–1323. [Google Scholar]
- Suomalainen, M.; Liljestrom, P.; Garoff, H. Spike protein-nucleocapsid interactions drive the budding of alphaviruses. J. Virol. 1992, 66, 4737–4747. [Google Scholar]
- Sturman, L.S.; Holmes, K.V.; Behnke, J. Isolation of coronavirus envelope glycoproteins and interaction with the viral nucleocapsid. J. Virol. 1980, 33, 449–462. [Google Scholar]
- Narayanan, K.; Maeda, A.; Maeda, J.; Makino, S. Characterization of the coronavirus M protein and nucleocapsid interaction in infected cells. J. Virol. 2000, 74, 8127–8134. [Google Scholar] [CrossRef]
- Stephens, E.B.; Compans, R.W. Assembly of animal viruses at cellular membranes. Annu. Rev. Microbiol. 1988, 42, 489–516. [Google Scholar] [CrossRef]
- Pettersson, R.F. Protein localization and virus assembly at intracellular membranes. Curr. Top. Microbiol. Immunol. 1991, 170, 67–106. [Google Scholar]
- Tooze, J.; Tooze, S.; Warren, G. Replication of coronavirus MHV-A59 in sac- cells: Determination of the first site of budding of progeny virions. Eur. J. Cell Biol. 1984, 33, 281–293. [Google Scholar]
- Fosmire, J.A.; Hwang, K.; Makino, S. Identification and characterization of a coronavirus packaging signal. J. Virol. 1992, 66, 3522–3530. [Google Scholar]
- Van der Most, R.G.; Bredenbeek, P.J.; Spaan, W.J. A domain at the 3' end of the polymerase gene is essential for encapsidation of coronavirus defective interfering RNAs. J. Virol. 1991, 65, 3219–3226. [Google Scholar]
- Woo, K.; Joo, M.; Narayanan, K.; Kim, K.H.; Makino, S. Murine coronavirus packaging signal confers packaging to nonviral RNA. J. Virol. 1997, 71, 824–827. [Google Scholar]
- Narayanan, K.; Chen, C.J.; Maeda, J.; Makino, S. Nucleocapsid-independent specific viral RNA packaging via viral envelope protein and viral RNA signal. J. Virol. 2003, 77, 2922–2927. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; Tong, S.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef]
- Luo, Y.; Batalao, A.; Zhou, H.; Zhu, L. Mammalian two-hybrid system: A complementary approach to the yeast two-hybrid system. BioTechniques 1997, 22, 350–352. [Google Scholar]
- Klumperman, J.; Locker, J.K.; Meijer, A.; Horzinek, M.C.; Geuze, H.J.; Rottier, P.J. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol 1994, 68, 6523–6534. [Google Scholar]
- Vennema, H.; Godeke, G.J.; Rossen, J.W.; Voorhout, W.F.; Horzinek, M.C.; Opstelten, D.J.; Rottier, P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996, 15, 2020–2028. [Google Scholar]
- Corse, E.; Machamer, C.E. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 2000, 74, 4319–4326. [Google Scholar] [CrossRef]
- Boursnell, M.E.; Binns, M.M.; Brown, T.D. Sequencing of coronavirus IBV genomic RNA: Three open reading frames in the 5' 'unique' region of mRNA D. J. Gen. Virol. 1985, 66, 2253–2258. [Google Scholar] [CrossRef]
- Baudoux, P.; Carrat, C.; Besnardeau, L.; Charley, B.; Laude, H. Coronavirus pseudoparticles formed with recombinant M and E proteins induce alpha interferon synthesis by leukocytes. J. Virol. 1998, 72, 8636–8643. [Google Scholar]
- Ruch, T.R.; Machamer, C.E. The coronavirus E protein: Assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef]
- Boscarino, J.A.; Logan, H.L.; Lacny, J.J.; Gallagher, T.M. Envelope protein palmitoylations are crucial for murine coronavirus assembly. J. Virol. 2008, 82, 2989–2999. [Google Scholar] [CrossRef]
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.; et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 2008, 82, 11318–11330. [Google Scholar] [CrossRef]
- Ruch, T.R.; Machamer, C.E. The hydrophobic domain of infectious bronchitis virus E protein alters the host secretory pathway and is important for release of infectious virus. J. Virol. 2011, 85, 675–685. [Google Scholar] [CrossRef]
- Cologna, R.; Hogue, B.G. Coronavirus nucleocapsid protein. RNA interactions. Adv. Exp. Med. Biol. 1998, 440, 355–359. [Google Scholar] [CrossRef]
- Brayton, P.R.; Lai, M.M.; Patton, C.D.; Stohlman, S.A. Characterization of two RNA polymerase activities induced by mouse hepatitis virus. J. Virol. 1982, 42, 847–853. [Google Scholar]
- Spaan, W.J.; Rottier, P.J.; Horzinek, M.C.; van der Zeijst, B.A. Isolation and identification of virus-specific mRNAs in cells infected with mouse hepatitis virus (MHV-A59). Virology 1981, 108, 424–434. [Google Scholar] [CrossRef]
- Almazan, F.; Galan, C.; Enjuanes, L. The nucleoprotein is required for efficient coronavirus genome replication. J. Virol. 2004, 78, 12683–12688. [Google Scholar] [CrossRef]
- Schelle, B.; Karl, N.; Ludewig, B.; Siddell, S.G.; Thiel, V. Selective replication of coronavirus genomes that express nucleocapsid protein. J. Virol. 2005, 79, 6620–6630. [Google Scholar] [CrossRef]
- Thiel, V.; Herold, J.; Schelle, B.; Siddell, S.G. Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 2001, 75, 6676–6681. [Google Scholar] [CrossRef]
- Casais, R.; Thiel, V.; Siddell, S.G.; Cavanagh, D.; Britton, P. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 2001, 75, 12359–12369. [Google Scholar] [CrossRef]
- Yount, B.; Denison, M.R.; Weiss, S.R.; Baric, R.S. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 2002, 76, 11065–11078. [Google Scholar] [CrossRef]
- Yount, B.; Curtis, K.M.; Baric, R.S. Strategy for systematic assembly of large RNA and DNA genomes: Transmissible gastroenteritis virus model. J. Virol. 2000, 74, 10600–10611. [Google Scholar] [CrossRef]
- Yount, B.; Curtis, K.M.; Fritz, E.A.; Hensley, L.E.; Jahrling, P.B.; Prentice, E.; Denison, M.R.; Geisbert, T.W.; Baric, R.S. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2003, 100, 12995–3000. [Google Scholar] [CrossRef]
- Denison, M.R.; Spaan, W.J.; van der Meer, Y.; Gibson, C.A.; Sims, A.C.; Prentice, E.; Lu, X.T. The putative helicase of the coronavirus mouse hepatitis virus is processed from the replicase gene polyprotein and localizes in complexes that are active in viral RNA synthesis. J. Virol. 1999, 73, 6862–6871. [Google Scholar]
- Sims, A.C.; Ostermann, J.; Denison, M.R. Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. J. Virol. 2000, 74, 5647–5654. [Google Scholar] [CrossRef]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weber, F.; Kochs, G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef]
- Van der Meer, Y.; Snijder, E.J.; Dobbe, J.C.; Schleich, S.; Denison, M.R.; Spaan, W.J.; Locker, J.K. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 1999, 73, 7641–7657. [Google Scholar]
- Masters, P.S.; Koetzner, C.A.; Kerr, C.A.; Heo, Y. Optimization of targeted RNA recombination and mapping of a novel nucleocapsid gene mutation in the coronavirus mouse hepatitis virus. J. Virol. 1994, 68, 328–337. [Google Scholar]
- Hurst, K.R.; Koetzner, C.A.; Masters, P.S. Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase-transcriptase complex. J. Virol. 2013, 87, 9159–9172. [Google Scholar] [CrossRef]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A contemporary view of coronavirus transcription. J. Virol. 2007, 81, 20–29. [Google Scholar] [CrossRef]
- Zust, R.; Miller, T.B.; Goebel, S.J.; Thiel, V.; Masters, P.S. Genetic interactions between an essential 3' cis-acting RNA pseudoknot, replicase gene products, and the extreme 3' end of the mouse coronavirus genome. J. Virol. 2008, 82, 1214–1228. [Google Scholar] [CrossRef]
- Graham, R.L.; Denison, M.R. Replication of murine hepatitis virus is regulated by papain-like proteinase 1 processing of nonstructural proteins 1, 2, and 3. J. Virol. 2006, 80, 11610–11620. [Google Scholar] [CrossRef]
- Cristofari, G.; Darlix, J.L. The ubiquitous nature of RNA chaperone proteins. Prog. Nucleic Acid Res. Mol. Biol. 2002, 72, 223–268. [Google Scholar] [CrossRef]
- Herschlag, D. RNA chaperones and the RNA folding problem. J. Biol. Chem. 1995, 270, 20871–20874. [Google Scholar] [CrossRef]
- Chen, C.J.; Sugiyama, K.; Kubo, H.; Huang, C.; Makino, S. Murine coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J. Virol. 2004, 78, 10410–10419. [Google Scholar] [CrossRef]
- Chen, C.J.; Makino, S. Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 2004, 78, 5658–5669. [Google Scholar] [CrossRef]
- Ikeda, K.; Monden, T.; Kanoh, T.; Tsujie, M.; Izawa, H.; Haba, A.; Ohnishi, T.; Sekimoto, M.; Tomita, N.; Shiozaki, H.; et al. Extraction and analysis of diagnostically useful proteins from formalin-fixed, paraffin-embedded tissue sections. J. Histochem. Cytochem. 1998, 46, 397–403. [Google Scholar] [CrossRef]
- Hobbs, W.E., 2nd.; DeLuca, N.A. A. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 1999, 73, 8245–8255. [Google Scholar]
- Lomonte, P.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein Vmw110 inhibits progression of cells through mitosis and from G(1) into S phase of the cell cycle. J. Virol. 1999, 73, 9456–9467. [Google Scholar]
- Wiebusch, L.; Hagemeier, C. Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G(1). J. Virol. 1999, 73, 9274–9283. [Google Scholar]
- Lu, M.; Shenk, T. Human cytomegalovirus UL69 protein induces cells to accumulate in G1 phase of the cell cycle. J. Virol. 1999, 73, 676–683. [Google Scholar]
- Cayrol, C.; Flemington, E.K. The Epstein-Barr virus bZIP transcription factor Zta causes G0/G1 cell cycle arrest through induction of cyclin-dependent kinase inhibitors. EMBO J. 1996, 15, 2748–2759. [Google Scholar]
- Xu, L.H.; Huang, M.; Fang, S.G.; Liu, D.X. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase delta. J. Biol. Chem. 2011, 286, 39546–39559. [Google Scholar]
- Surjit, M.; Liu, B.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J. Biol. Chem. 2006, 281, 10669–10681. [Google Scholar] [CrossRef]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef]
- Shieh, Y.C.; Baric, R.S.; Woods, J.W.; Calci, K.R. Molecular surveillance of enterovirus and norwalk-like virus in oysters relocated to a municipal-sewage-impacted gulf estuary. Appl. Environ. Microbiol. 2003, 69, 7130–7136. [Google Scholar] [CrossRef]
- Raaben, M.; Groot Koerkamp, M.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell. Microbiol. 2007, 9, 2218–2229. [Google Scholar] [CrossRef]
- Dalton, K.; Casais, R.; Shaw, K.; Stirrups, K.; Evans, S.; Britton, P.; Brown, T.D.; Cavanagh, D. cis-acting sequences required for coronavirus infectious bronchitis virus defective-RNA replication and packaging. J. Virol. 2001, 75, 125–133. [Google Scholar] [CrossRef]
- Hollfelder, F.; Herschlag, D. The nature of the transition state for enzyme-catalyzed phosphoryl transfer. Hydrolysis of O-aryl phosphorothioates by alkaline phosphatase. Biochemistry 1995, 34, 12255–12264. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X. The nucleocapsid protein of coronavirus mouse hepatitis virus interacts with the cellular heterogeneous nuclear ribonucleoprotein A1 in vitro and in vivo. Virology 1999, 265, 96–109. [Google Scholar] [CrossRef]
- Menendez, A.; Chow, K.C.; Pan, O.C.; Scott, J.K. Human immunodeficiency virus type 1-neutralizing monoclonal antibody 2F5 is multispecific for sequences flanking the DKW core epitope. J. Mol. Biol. 2004, 338, 311–327. [Google Scholar] [CrossRef]
- Gross, S.R.; Kinzy, T.G. Translation elongation factor 1A is essential for regulation of the actin cytoskeleton and cell morphology. Nat. Struct. Mol. Biol. 2005, 12, 772–778. [Google Scholar] [CrossRef]
- Kurasawa, Y.; Watanabe, Y.; Numata, O. Characterization of F-actin bundling activity of Tetrahymena elongation factor 1 alpha investigated with rabbit skeletal muscle actin. Zool. Sci. 1996, 13, 371–375. [Google Scholar] [CrossRef]
- Numata, O.; Gonda, K.; Watanabe, A.; Kurasawa, Y. Cytokinesis in Tetrahymena: Determination of division plane and organization of contractile ring. Microsc. Res. Tech. 2000, 49, 127–135. [Google Scholar] [CrossRef]
- Chow, K.C.; Hsiao, C.H.; Lin, T.Y.; Chen, C.L.; Chiou, S.H. Detection of severe acute respiratory syndrome-associated coronavirus in pneumocytes of the lung. Am. J. Clin. Pathol. 2004, 121, 574–580. [Google Scholar] [CrossRef]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Surjit, M.; Liu, B.; Jameel, S.; Chow, V.T.; Lal, S.K. The SARS coronavirus nucleocapsid protein induces actin reorganization and apoptosis in COS-1 cells in the absence of growth factors. Biochem. J. 2004, 383, 13–18. [Google Scholar] [CrossRef]
- Nakanaga, K.; Yamanouchi, K.; Fujiwara, K. Protective effect of monoclonal antibodies on lethal mouse hepatitis virus infection in mice. J. Virol. 1986, 59, 168–171. [Google Scholar]
- Spiegel, M.; Pichlmair, A.; Martinez-Sobrido, L.; Cros, J.; Garcia-Sastre, A.; Haller, O.; Weber, F. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol 2005, 79, 2079–2086. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-beta response by targeting initial step of IFN-beta induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef]
- Fang, S.; Xu, L.; Huang, M.; Qisheng Li, F.; Liu, D.X. Identification of two ATR-dependent phosphorylation sites on coronavirus nucleocapsid protein with nonessential functions in viral replication and infectivity in cultured cells. Virology 2013, 444, 225–232. [Google Scholar] [CrossRef]
- Jayaram, J.; Youn, S.; Collisson, E.W. The virion N protein of infectious bronchitis virus is more phosphorylated than the N protein from infected cell lysates. Virology 2005, 339, 127–135. [Google Scholar] [CrossRef]
- Yan, X.; Hao, Q.; Mu, Y.; Timani, K.A.; Ye, L.; Zhu, Y.; Wu, J. Nucleocapsid protein of SARS-CoV activates the expression of cyclooxygenase-2 by binding directly to regulatory elements for nuclear factor-kappa B and CCAAT/enhancer binding protein. Int. J. Biochem. Cell Biol. 2006, 38, 1417–1428. [Google Scholar] [CrossRef]
- He, R.; Leeson, A.; Andonov, A.; Li, Y.; Bastien, N.; Cao, J.; Osiowy, C.; Dobie, F.; Cutts, T.; Ballantine, M.; et al. Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 2003, 311, 870–876. [Google Scholar] [CrossRef]
- Berry, M.; Manasse, T.-L.; Tan, Y.J.; Fielding, B.C. Characterisation of human coronavirus-NL63 nucleocapsid protein. Afr. J. Biotechnol. 2012, 11, 13962–13968. [Google Scholar]
- Surjit, M.; Lal, S.K. The SARS-CoV nucleocapsid protein: A protein with multifarious activities. Infect. Genet. Evol. 2008, 8, 397–405. [Google Scholar] [CrossRef]
- Masters, P.S.; Sturman, L.S. Background paper. Functions of the coronavirus nucleocapsid protein. Adv. Exp. Med. Biol 1990, 276, 235–238. [Google Scholar] [CrossRef]
- Nguyen, V.P.; Hogue, B.G. Protein interactions during coronavirus assembly. J. Virol. 1997, 71, 9278–9284. [Google Scholar]
- Opstelten, D.J.; Raamsman, M.J.; Wolfs, K.; Horzinek, M.C.; Rottier, P.J. Envelope glycoprotein interactions in coronavirus assembly. J. Cell Biol. 1995, 131, 339–349. [Google Scholar] [CrossRef]
- Rowles, D.L.; Terhune, S.S.; Cristea, I.M. Discovery of host-viral protein complexes during infection. Methods Mol. Biol. 2013, 1064, 43–70. [Google Scholar]
- Luo, C.; Luo, H.; Zheng, S.; Gui, C.; Yue, L.; Yu, C.; Sun, T.; He, P.; Chen, J.; Shen, J.; et al. Nucleocapsid protein of SARS coronavirus tightly binds to human cyclophilin A. Biochem. Biophys. Res. Commun. 2004, 321, 557–565. [Google Scholar] [CrossRef]
- Wang, Q.; Li, C.; Zhang, Q.; Wang, T.; Li, J.; Guan, W.; Yu, J.; Liang, M.; Li, D. Interactions of SARS coronavirus nucleocapsid protein with the host cell proteasome subunit p42. Virol. J. 2010, 7, 99. [Google Scholar] [CrossRef]
- Lv, M.; Chen, J.; Shi, H.; Chen, X.; Fan, X.; Shen, S.; Feng, L. Co-localization analysis between porcine epidemic diarrhea virus nucleocapsid protein and nucleolar phosphoprotein B23.1. Wei Sheng Wu Xue Bao 2011, 51, 643–647. [Google Scholar]
- Zeng, Y.; Ye, L.; Zhu, S.; Zheng, H.; Zhao, P.; Cai, W.; Su, L.; She, Y.; Wu, Z. The nucleocapsid protein of SARS-associated coronavirus inhibits B23 phosphorylation. Biochem. Biophys. Res. Commun. 2008, 369, 287–291. [Google Scholar] [CrossRef]
- Zhao, X.; Nicholls, J.M.; Chen, Y.G. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J. Biol. Chem. 2008, 283, 3272–3280. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Zhang, R.W.; Chang, W.S.; Wang, Y.Y. Cxcl16 interact with SARS-CoV N protein in and out cell. Virol. Sin. 2010, 25, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.Y.; Li, H.C.; Chen, C.Y.; Yang, C.H.; Lee, S.K.; Wang, C.W.; Ma, H.C.; Juang, Y.L.; Lo, S.Y. SARS-CoV nucleocapsid protein interacts with cellular pyruvate kinase protein and inhibits its activity. Arch. Virol. 2012, 157, 635–645. [Google Scholar] [CrossRef]
- Emmott, E.; Munday, D.; Bickerton, E.; Britton, P.; Rodgers, M.A.; Whitehouse, A.; Zhou, E.M.; Hiscox, J.A. The cellular interactome of the coronavirus infectious bronchitis virus nucleocapsid protein and functional implications for virus biology. J. Virol. 2013, 87, 9486–9500. [Google Scholar] [CrossRef]
- Meyniel-Schicklin, L.; de Chassey, B.; Andre, P.; Lotteau, V. Viruses and interactomes in translation. Mol. Cell. Proteomics 2012, 11, M111.014738. [Google Scholar] [CrossRef]
- Leung, D.T.; Tam, F.C.; Ma, C.H.; Chan, P.K.; Cheung, J.L.; Niu, H.; Tam, J.S.; Lim, P.L. Antibody response of patients with severe acute respiratory syndrome (SARS) targets the viral nucleocapsid. J. Infect. Dis. 2004, 190, 379–386. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, J.H.; Hung, C.F.; Peng, S.; Roden, R.; Wang, M.C.; Viscidi, R.; Tsai, Y.C.; He, L.; Chen, P.J.; et al. Generation and characterization of DNA vaccines targeting the nucleocapsid protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004, 78, 4638–4645. [Google Scholar] [CrossRef]
- Buchholz, U.J.; Bukreyev, A.; Yang, L.; Lamirande, E.W.; Murphy, B.R.; Subbarao, K.; Collins, P.L. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc. Natl. Acad. Sci. USA 2004, 101, 9804–9809. [Google Scholar]
- Liang, F.Y.; Lin, L.C.; Ying, T.H.; Yao, C.W.; Tang, T.K.; Chen, Y.W.; Hou, M.H. Immunoreactivity characterisation of the three structural regions of the human coronavirus OC43 nucleocapsid protein by Western blot: Implications for the diagnosis of coronavirus infection. J. Virol. Methods 2013, 187, 413–420. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McBride, R.; Van Zyl, M.; Fielding, B.C. The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses 2014, 6, 2991-3018. https://doi.org/10.3390/v6082991
McBride R, Van Zyl M, Fielding BC. The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses. 2014; 6(8):2991-3018. https://doi.org/10.3390/v6082991
Chicago/Turabian StyleMcBride, Ruth, Marjorie Van Zyl, and Burtram C. Fielding. 2014. "The Coronavirus Nucleocapsid Is a Multifunctional Protein" Viruses 6, no. 8: 2991-3018. https://doi.org/10.3390/v6082991
APA StyleMcBride, R., Van Zyl, M., & Fielding, B. C. (2014). The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses, 6(8), 2991-3018. https://doi.org/10.3390/v6082991