Evidence for Retrovirus and Paramyxovirus Infection of Multiple Bat Species in China

Abstract
:1. Introduction
2. Experimental
2.1. Ethics Statement
2.2. Sample Collection and Viral Nucleic Acids Preparation
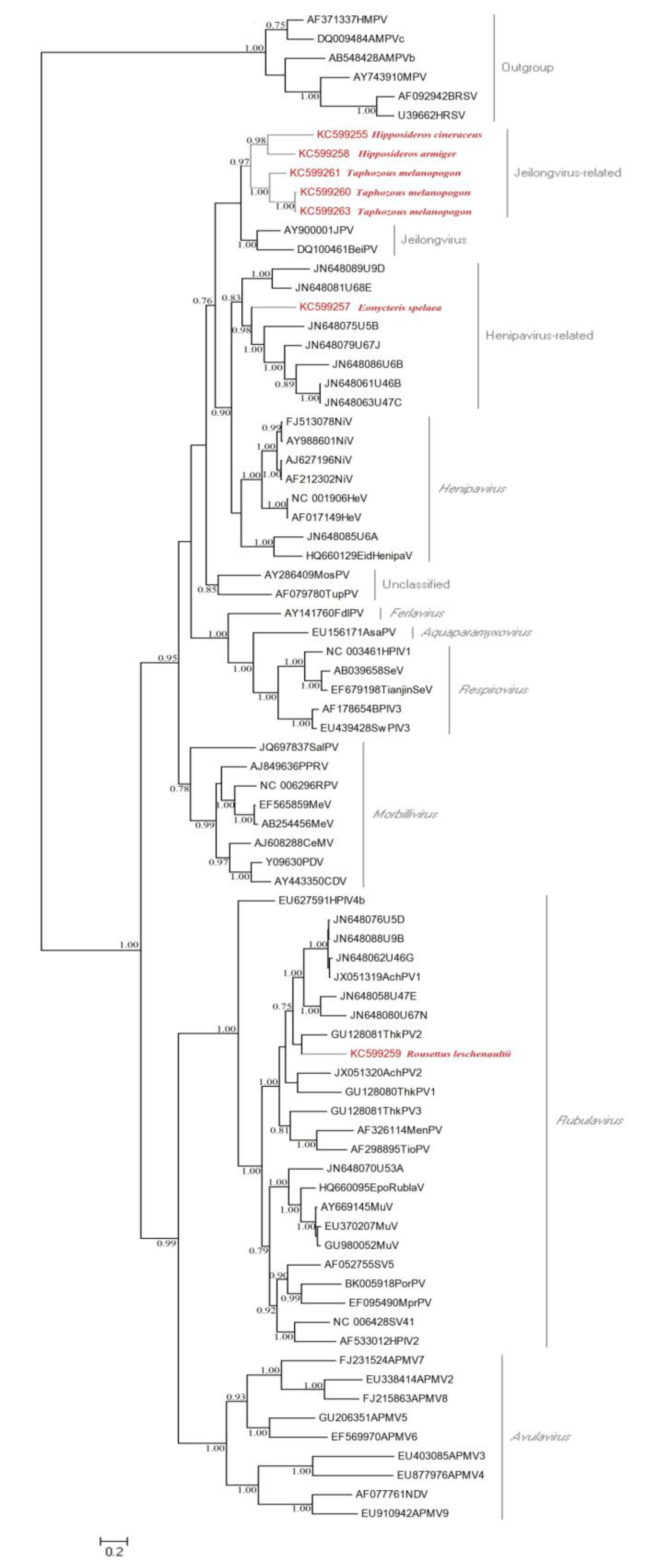
2.3. Screening of Paramyxoviruses and Phylogenetic Analysis
2.4. High-throughput Sequencing and Pathogen Analysis
2.5. Identification of Viral Homologous Sequences
2.6. Phylogenetic Analysis of Viral Sequences
2.7. Nucleotide Sequence Accession Numbers
3. Results
3.1. Detection of Paramyxoviruses
3.2. In-Depth Analysis of Pathogens by Illumina High-throughput Sequencing
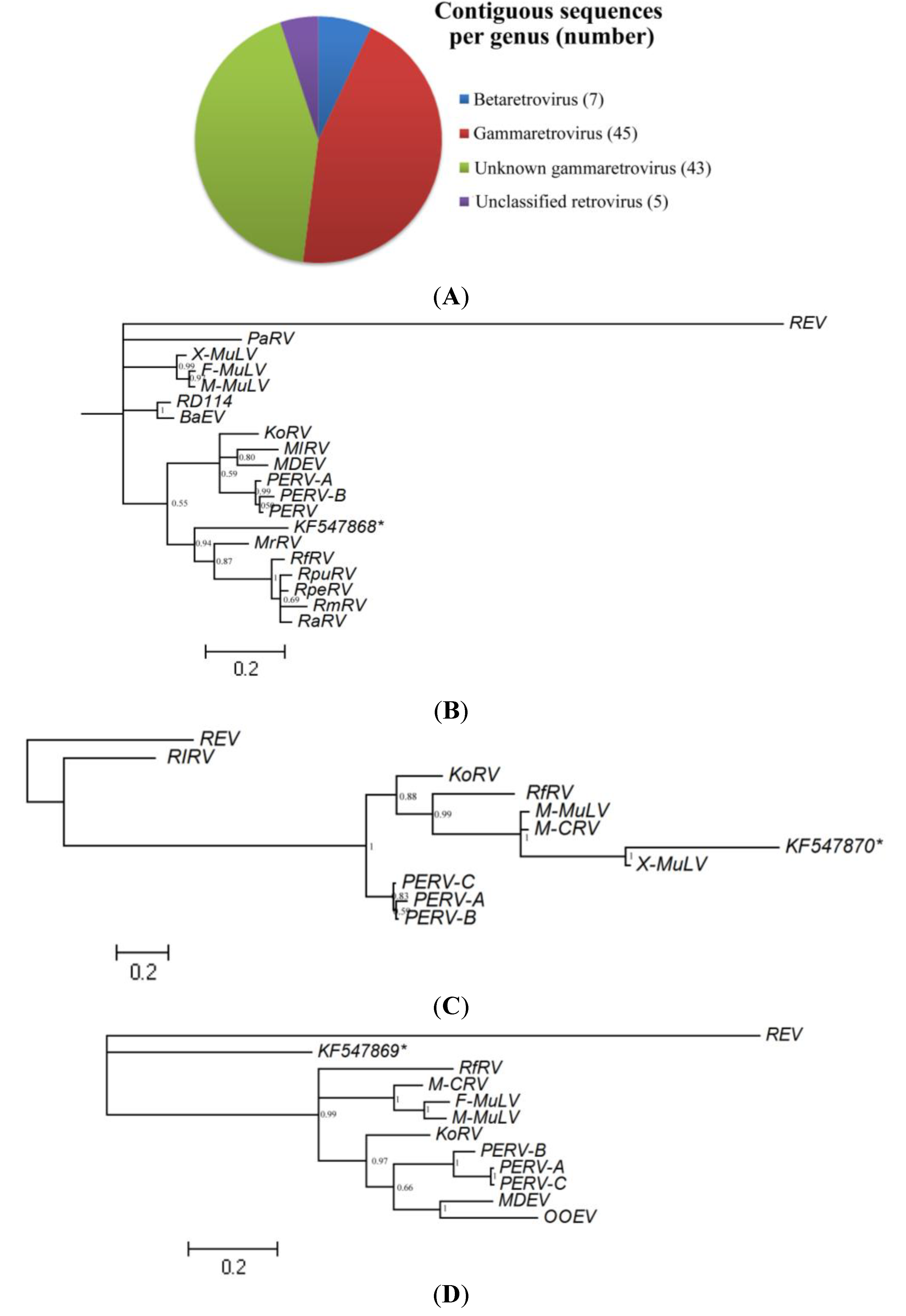
3.2.1. Assembled Contigs and BLASTx Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clade | Family | Genus | Virus Name | Contigs |
|---|---|---|---|---|
| Vertebrate virus | Retroviridae | Gammaretrovirus | Human endogenous retrovirus | 24 |
| Murine leukemia virus | 5 | |||
| Spleen necrosis virus | 4 | |||
| Moloney murine leukemia virus | 3 | |||
| Porcine endogenous retrovirus | 2 | |||
| Woolly monkey sarcoma virus | 2 | |||
| Friend murine leukemia virus | 1 | |||
| Feline leukemia virus | 1 | |||
| Xenotropic Murine Leukemia Virus | 1 | |||
| Chick syncytial virus | 1 | |||
| Gammaretrovirus RfRV/China/2011 | 1 | |||
| Unknown gammaretrovirus | Reticuloendotheliosis virus | 40 | ||
| Bat gammaretrovirus | 1 | |||
| Baboon endogenous virus strain M7 | 1 | |||
| Rousettus leschenaultii retrovirus | 1 | |||
| Betaretrovirus | Ovine enzootic nasal tumor virus (ENTV) | 2 | ||
| Jaagsiekte sheep retrovirus | 2 | |||
| Squirrel monkey retrovirus | 2 | |||
| Simian endogenous retrovirus | 1 | |||
| Unclassified | Simian retrovirus | 4 | ||
| Multiple sclerosis associated retrovirus | 1 | |||
| Polyomaviridae | Polyomavirus | STL polyomavirus | 1 | |
| Hamster polyomavirus | 1 | |||
| Merkel cell polyomavirus | 1 | |||
| Insect virus | Solenopsis invicta virus 3 | 1 | ||
| Mycovirus | Grapevine partitivirus | 1 | ||
| Phage | Myoviridae | T4-like virus | Acinetobacter phage Ac42 | 1 |
| Myoviridae | T4-like virus | Acinetobacter phage Acj61 | 1 | |
| Myoviridae | T4-like virus | Acinetobacter phage Acj9 | 2 | |
| Myoviridae | T4-like virus | Aeromonas phage 44RR2.8t | 1 | |
| Myoviridae | T4-like virus | Aeromonas phage Aeh1 | 1 | |
| Myoviridae | T4-like virus | Aeromonas phage Aes508 | 1 | |
| Myoviridae | T4-like virus | Enterobacteria phage Bp7 | 1 | |
| Myoviridae | T4-like virus | Enterobacteria phage JSE | 1 | |
| Myoviridae | T4-like virus | Enterobacteria phage Phi1 | 1 | |
| Myoviridae | T4-like virus | Enterobacteria phage RB69 | 1 | |
| Myoviridae | unclassified Myoviridae | Salmonella phage STML-198 | 1 | |
| Myoviridae | unclassified Myoviridae | Yersinia phage phiR1-RT | 1 |
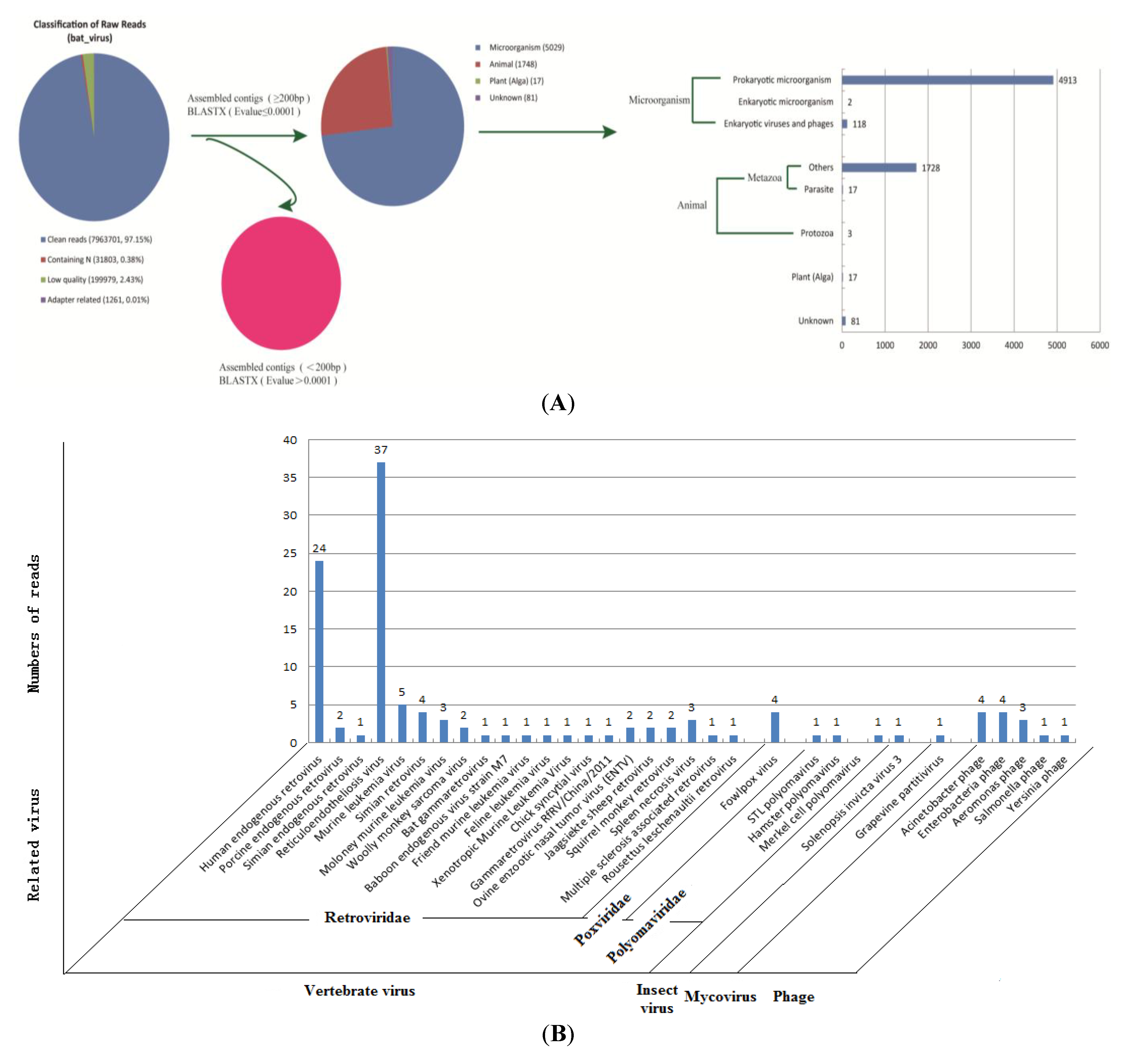
3.2.2. Identification of Bacteriophage Sequences
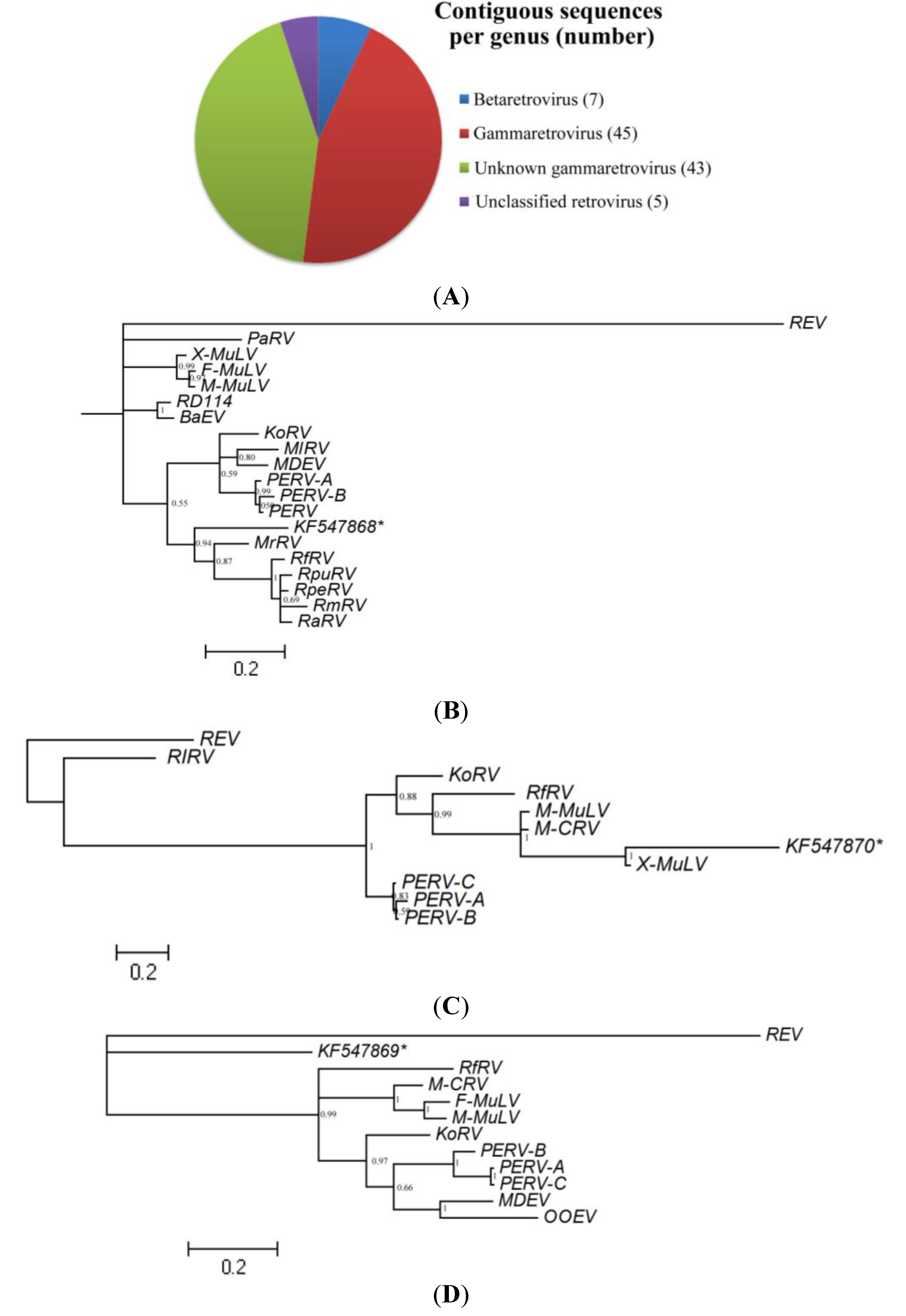
3.2.3. Analysis of Vertebrate Viral Sequences by Family
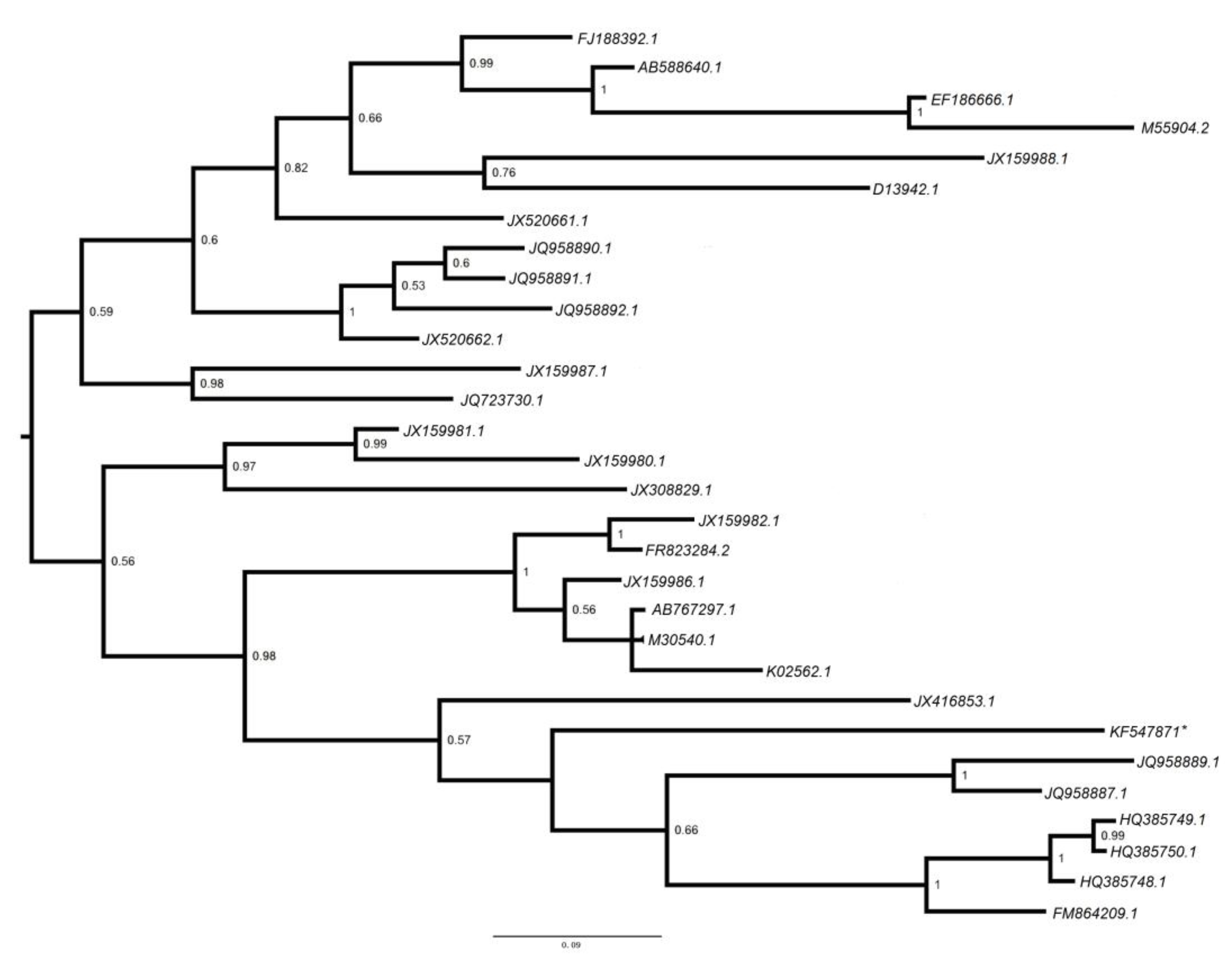
3.2.4. Retroviridae
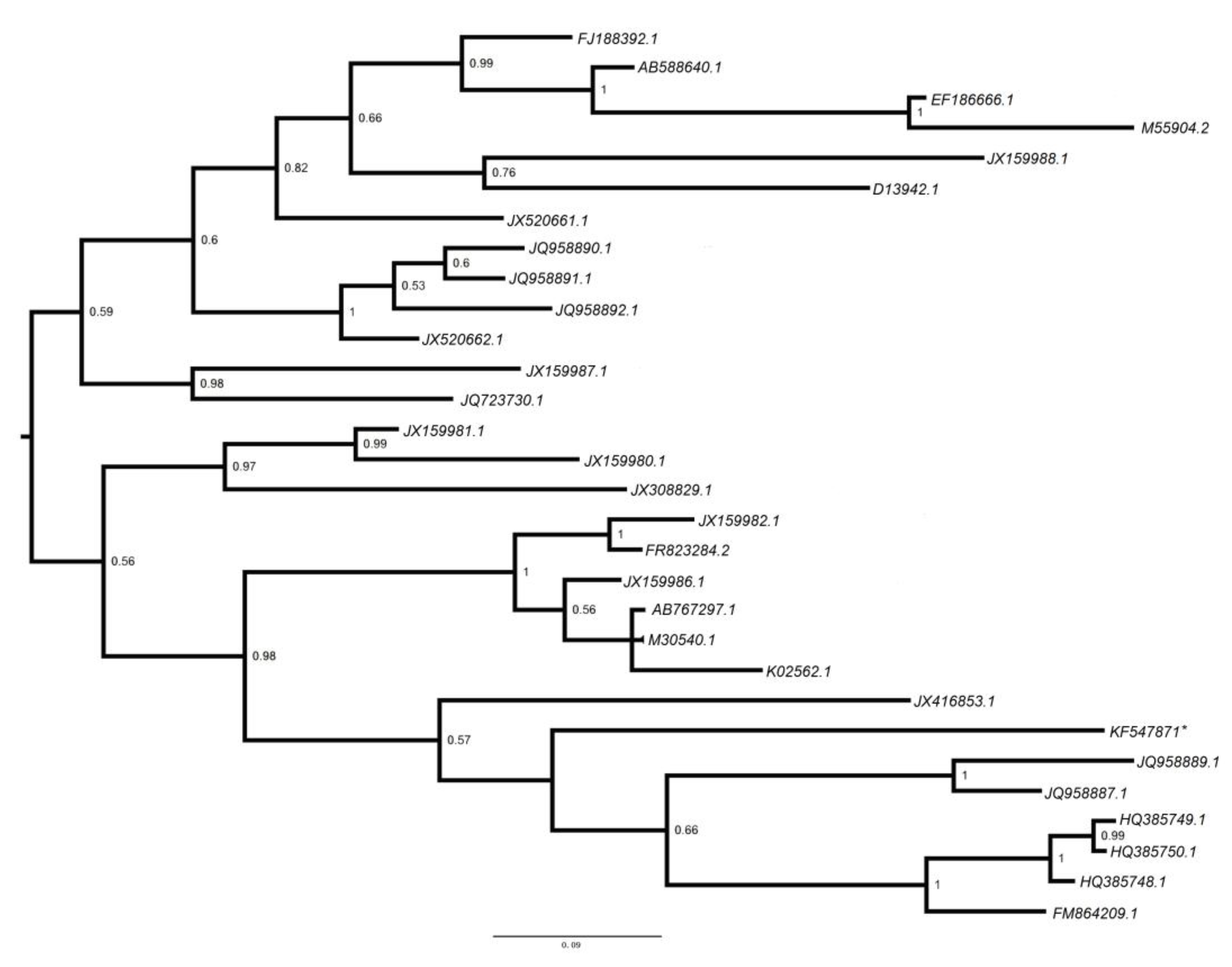
3.2.5. Polyomaviridae
4. Discussion
5. Conclusions
Supplementary Files
Acknowledgment
Author Contributions
Conflicts of Interest
References and Notes
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: A comprehensive experimental study of virus transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951. [Google Scholar] [CrossRef]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Delicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.R.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L. Isolation of genetically diverse Marburg viruses from Egyptian fruit bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Muller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Rasche, A.; Yordanov, S.; Seebens, A. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Quan, P.-L.; Firth, C.; Conte, J.M.; Williams, S.H.; Zambrana-Torrelio, C.M.; Anthony, S.J.; Ellison, J.A.; Gilbert, A.T.; Kuzmin, I.V.; Niezgoda, M. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc. Natl. Acad. Sci. USA 2013, 110, 8194–8199. [Google Scholar] [CrossRef]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Epstein, J.H.; Prakash, V.; Smith, C.S.; Daszak, P.; McLaughlin, A.B.; Meehan, G.; Field, H.E.; Cunningham, A.A. Henipavirus infection in fruit bats (Pteropus giganteus), India. Emerg. Infect. Dis. 2008, 14, 1309–1311. [Google Scholar] [CrossRef]
- King, A.; Davies, P.; Lawrie, A. The rabies viruses of bats. Vet. Microbiol. 1990, 23, 165–174. [Google Scholar] [CrossRef]
- McCall, B.J.; Epstein, J.H.; Neill, A.S.; Heel, K.; Field, H.; Barrett, J.; Smith, G.A.; Selvey, L.A.; Rodwell, B.; Lunt, R. Potential exposure to Australian bat lyssavirus, Queensland, 1996–1999. Emerg. Infect. Dis. 2000, 6, 259–264. [Google Scholar] [CrossRef]
- Slenczka, W.G. The Marburg virus outbreak of 1967 and subsequent episodes. Curr. Top. Microbiol. Immunol. 1999, 235, 49–75. [Google Scholar]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef]
- Wang, Y.X. A Complete Checklist of Mammal Species and Subspecies in China: A Taxonomic and Geographic Reference; China Forestry Press: Beijing, China, 2003. [Google Scholar]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef]
- Baker, K.S.; Todd, S.; Marsh, G.A.; Crameri, G.; Barr, J.; Kamins, A.O.; Peel, A.J.; Yu, M.; Hayman, D.T.; Nadjm, B.; et al. Novel, potentially zoonotic paramyxoviruses from the African straw-colored fruit bat Eidolon helvum. J. Virol. 2013, 87, 1348–1358. [Google Scholar] [CrossRef]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef]
- Djikeng, A.; Kuzmickas, R.; Anderson, N.G.; Spiro, D.J. Metagenomic analysis of RNA viruses in a fresh water lake. PLoS One 2009, 4, e7264. [Google Scholar] [CrossRef]
- Ng, T.F.; Manire, C.; Borrowman, K.; Langer, T.; Ehrhart, L.; Breitbart, M. Discovery of a novel single-stranded DNA virus from a sea turtle fibropapilloma by using viral metagenomics. J. Virol. 2009, 83, 2500–2509. [Google Scholar] [CrossRef]
- Victoria, J.G.; Kapoor, A.; Dupuis, K.; Schnurr, D.P.; Delwart, E.L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog. 2008, 4, e1000163. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic analysis of the viromes of three North American bat species: Viral diversity among different bat species that share a common habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef]
- Blinkova, O.; Kapoor, A.; Victoria, J.; Jones, M.; Wolfe, N.; Naeem, A.; Shaukat, S.; Sharif, S.; Alam, M.M.; Angez, M.; et al. Cardioviruses are genetically diverse and cause common enteric infections in South Asian children. J. Virol. 2009, 83, 4631–4641. [Google Scholar] [CrossRef]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar]
- Baker, K.S.; Leggett, R.M.; Bexfield, N.H.; Alston, M.; Daly, G.; Todd, S.; Tachedjian, M.; Holmes, C.E.G.; Crameri, S.; Wang, L.-F. Metagenomic study of the viruses of African straw-colored fruit bats: Detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology 2013, 441, 95–106. [Google Scholar] [CrossRef]
- He, B.; Li, Z.; Yang, F.; Zheng, J.; Feng, Y.; Guo, H.; Li, Y.; Wang, Y.; Su, N.; Zhang, F.; et al. Virome profiling of bats from myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses. PLoS One 2013, 8, e61950. [Google Scholar]
- Tong, S.; Chern, S.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5, molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Li, Z.; Yu, M.; Zhang, H.; Magoffin, D.E.; Jack, P.J.; Hyatt, A.; Wang, H.Y.; Wang, L.F. Beilong virus, a novel paramyxovirus with the largest genome of non-segmented negative-stranded RNA viruses. Virology 2006, 346, 219–228. [Google Scholar] [CrossRef]
- Hayward, J.A.; Tachedjian, M.; Cui, J.; Field, H.; Holmes, E.C.; Wang, L.F.; Tachedjian, G. Identification of diverse full-length endogenous betaretroviruses in megabats and microbats. Retrovirology 2013, 10. [Google Scholar] [CrossRef]
- Cui, J.; Tachedjian, M.; Wang, L.; Tachedjian, G.; Wang, L.-F.; Zhang, S. Discovery of retroviral homologs in bats: Implications for the origin of mammalian gammaretroviruses. J. Virol. 2012, 86, 4288–4293. [Google Scholar] [CrossRef]
- Cui, J.; Tachedjian, G.; Tachedjian, M.; Holmes, E.C.; Zhang, S.; Wang, L.-F. Identification of diverse groups of endogenous gammaretroviruses in mega-and microbats. J. Gen. Virol. 2012, 93, 2037–2045. [Google Scholar] [CrossRef]
- Halpin, K.; Hyatt, A.D.; Plowright, R.K.; Epstein, J.H.; Daszak, P.; Field, H.E.; Wang, L.; Daniels, P.W. Emerging viruses: Coming in on a wrinkled wing and a prayer. Clin. Infect. Dis. 2007, 44, 711–717. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Hickey, A.C.; Zhang, Y.; Li, Y.; Wu, Y.; Zhang, H.; Yuan, J.; Han, Z.; McEachern, J.; et al. Antibodies to Nipah or Nipah-like viruses in bats, China. Emerg. Infect. Dis. 2008, 14, 1974–1976. [Google Scholar] [CrossRef]
- Baker, K.S.; Todd, S.; Marsh, G.; Fernandez-Loras, A.; Suu-Ire, R.; Wood, J.L.N.; Wang, L.-F.; Murcia, P.R.; Cunningham, A.A. Co-circulation of diverse paramyxoviruses in an urban African fruit bat population. J. Gen. Virol. 2012, 93, 850–856. [Google Scholar] [CrossRef]
- Streicker, D.G.; Turmelle, A.S.; Vonhof, M.J.; Kuzmin, I.V.; McCracken, G.F.; Rupprecht, C.E. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 2010, 329, 676–679. [Google Scholar] [CrossRef]
- Chua, K.B.; Goh, K.J.; Wong, K.T.; Kamarulzaman, A.; Tan, P.S.; Ksiazek, T.G.; Zaki, S.R.; Paul, G.; Lam, S.K.; Tan, C.T. Fatal encephalitis due to Nipah virus among pig-farmers in Malaysia. Lancet 1999, 354, 1257–1259. [Google Scholar] [CrossRef]
- Luby, S.P.; Hossain, M.J.; Gurley, E.S.; Ahmed, B.-N.; Banu, S.; Khan, S.U.; Homaira, N.; Rota, P.A.; Rollin, P.E.; Comer, J.A. Recurrent zoonotic transmission of Nipah virus into humans, Bangladesh, 2001–2007. Emerg. Infect. Dis. 2009, 15. [Google Scholar] [CrossRef]
- Luby, S.P.; Rahman, M.; Hossain, M.J.; Blum, L.S.; Husain, M.M.; Gurley, E.; Khan, R.; Ahmed, B.-N.; Rahman, S.; Nahar, N. Foodborne transmission of Nipah virus, Bangladesh. Emerg. Infect. Dis. 2006, 12. [Google Scholar] [CrossRef]
- Kunin, V.; Copeland, A.; Lapidus, A.; Mavromatis, K.; Hugenholtz, P. A bioinformatician’s guide to metagenomics. Microbiol. Mol. Biol. Rev. 2008, 72, 557–578. [Google Scholar] [CrossRef]
- Afonso, C.L.; Tulman, E.R.; Lu, Z.; Zsak, L.; Kutish, G.F.; Rock, D.L. The genome of fowlpox virus. J. Virol. 2000, 74, 3815–3831. [Google Scholar] [CrossRef]
- Singh, P.; Schnitzlein, W.M.; Tripathy, D.N. Reticuloendotheliosis virus sequences within the genomes of field strains of fowlpox virus display variability. J. Virol. 2003, 77, 5855–5862. [Google Scholar] [CrossRef]
- Weiss, R.A. The discovery of endogenous retroviruses. Retrovirology 2006, 3, e67. [Google Scholar] [CrossRef]
- Niewiadomska, A.M.; Gifford, R.J. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol. 2013, 11, e1001642. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yuan, L.; Li, M.; Li, L.; Monagin, C.; Chmura, A.A.; Schneider, B.S.; Epstein, J.H.; Mei, X.; Shi, Z.; Daszak, P.; et al. Evidence for Retrovirus and Paramyxovirus Infection of Multiple Bat Species in China. Viruses 2014, 6, 2138-2154. https://doi.org/10.3390/v6052138
Yuan L, Li M, Li L, Monagin C, Chmura AA, Schneider BS, Epstein JH, Mei X, Shi Z, Daszak P, et al. Evidence for Retrovirus and Paramyxovirus Infection of Multiple Bat Species in China. Viruses. 2014; 6(5):2138-2154. https://doi.org/10.3390/v6052138
Chicago/Turabian StyleYuan, Lihong, Min Li, Linmiao Li, Corina Monagin, Aleksei A. Chmura, Bradley S. Schneider, Jonathan H. Epstein, Xiaolin Mei, Zhengli Shi, Peter Daszak, and et al. 2014. "Evidence for Retrovirus and Paramyxovirus Infection of Multiple Bat Species in China" Viruses 6, no. 5: 2138-2154. https://doi.org/10.3390/v6052138
APA StyleYuan, L., Li, M., Li, L., Monagin, C., Chmura, A. A., Schneider, B. S., Epstein, J. H., Mei, X., Shi, Z., Daszak, P., & Chen, J. (2014). Evidence for Retrovirus and Paramyxovirus Infection of Multiple Bat Species in China. Viruses, 6(5), 2138-2154. https://doi.org/10.3390/v6052138