Interferon Induction by RNA Viruses and Antagonism by Viral Pathogens


Abstract
:
1. Introduction
2. Interferons and Their Classification

{kind=link}
{kind=link}
{kind=link}
| Type | Subtypes | Receptors |
|---|---|---|
| Type I | IFN-α (13subtypes) | IFNAR1 and IFNAR2 |
| IFN-β, IFN-ε, IFN-κ and IFN-ω | ||
| IFN-δ (swine), IFN-τ (ruminant) and IFN-ζ (mice) | ||
| Type II | IFN-γ | IFNGR1 and IFNGR2 |
| Type III | IFN-λ (1-4) | IFRL1 and IL-10R2 |
3. Toll-Like Receptor Pathway
3.1. Discovery of Toll in Drosophila
3.2. Discovery of Toll-Like Receptors in Mammalian Hosts
3.3. TLR Ligands
| TLRs | Adaptor(s) | Ligand [Ref] |
|---|---|---|
| TLR1 | MyD88 | Peptidoglycan/lipoproteins [58,59] |
| TLR2 | MyD88 | Peptidoglycan/lipoproteins [58,59,60] |
| TLR3 | TRIF | dsRNA [61] |
| TLR4 | MyD88/TRIF/TRAM/TICAM | LPS [56] |
| TLR5 | MyD88 | Bacterial flagellin [62] |
| TLR6 | MyD88 | Lipopeptide [58] |
| TLR7 | MyD88 | ssRNA [63,64] |
| TLR8 | MyD88 | ssRNA [65] |
| TLR9 | MyD88 | CpG DNA [66] |
| TLR10 | MyD88 | Unknown |
| TLR11 | MyD88 | Unknown |
| TLR12 | MyD88 | Unknown |
| TLR13 | MyD88 | Unknown |
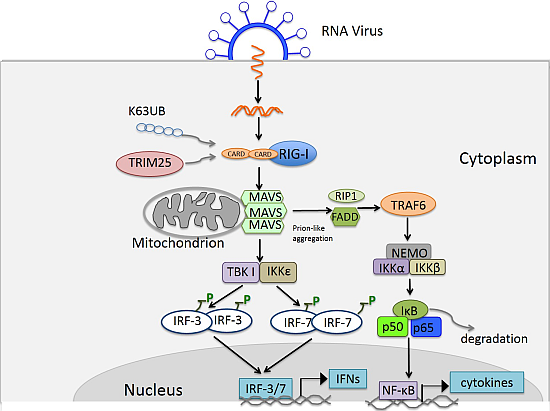
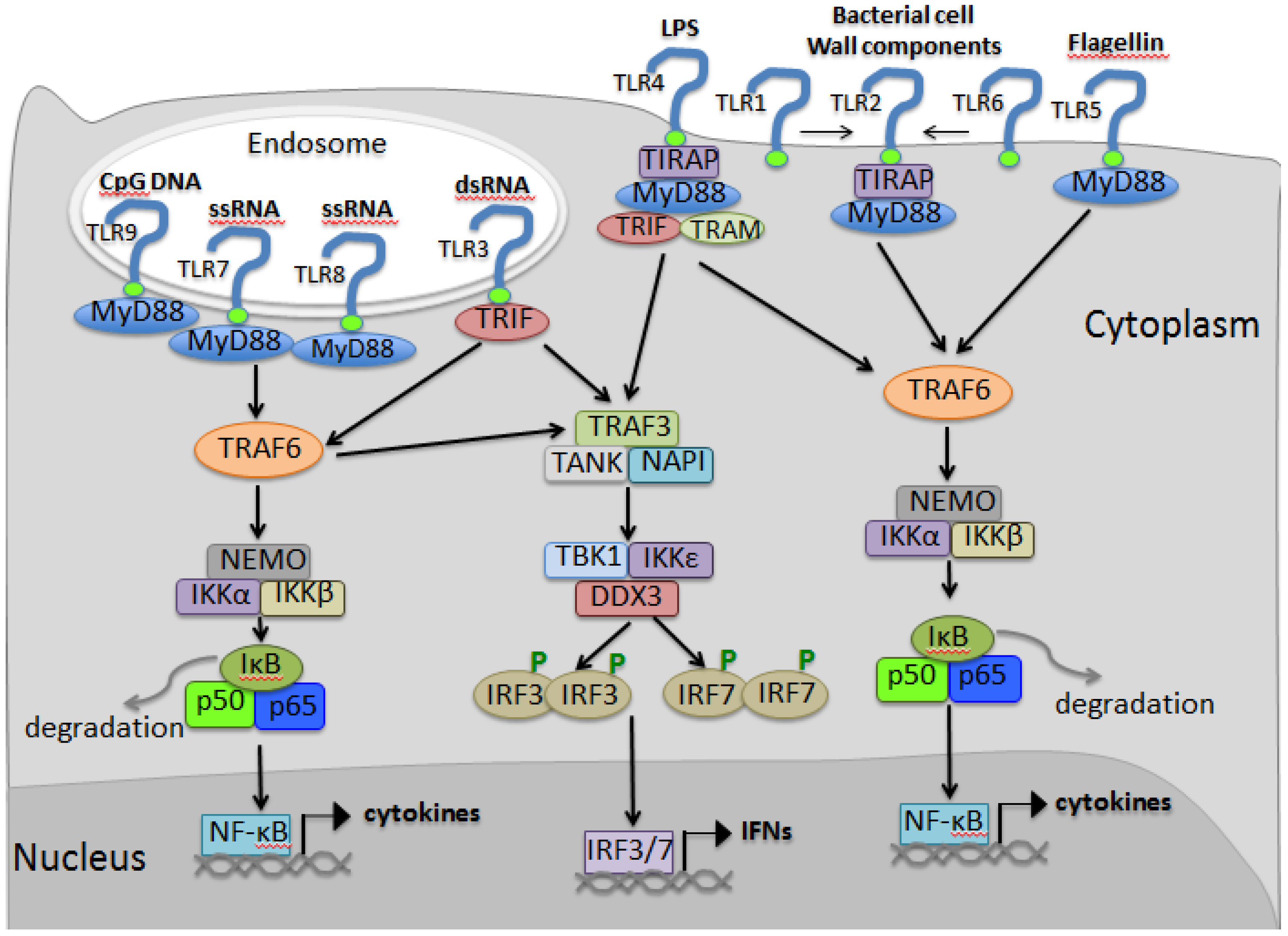
3.4. TLR Signaling
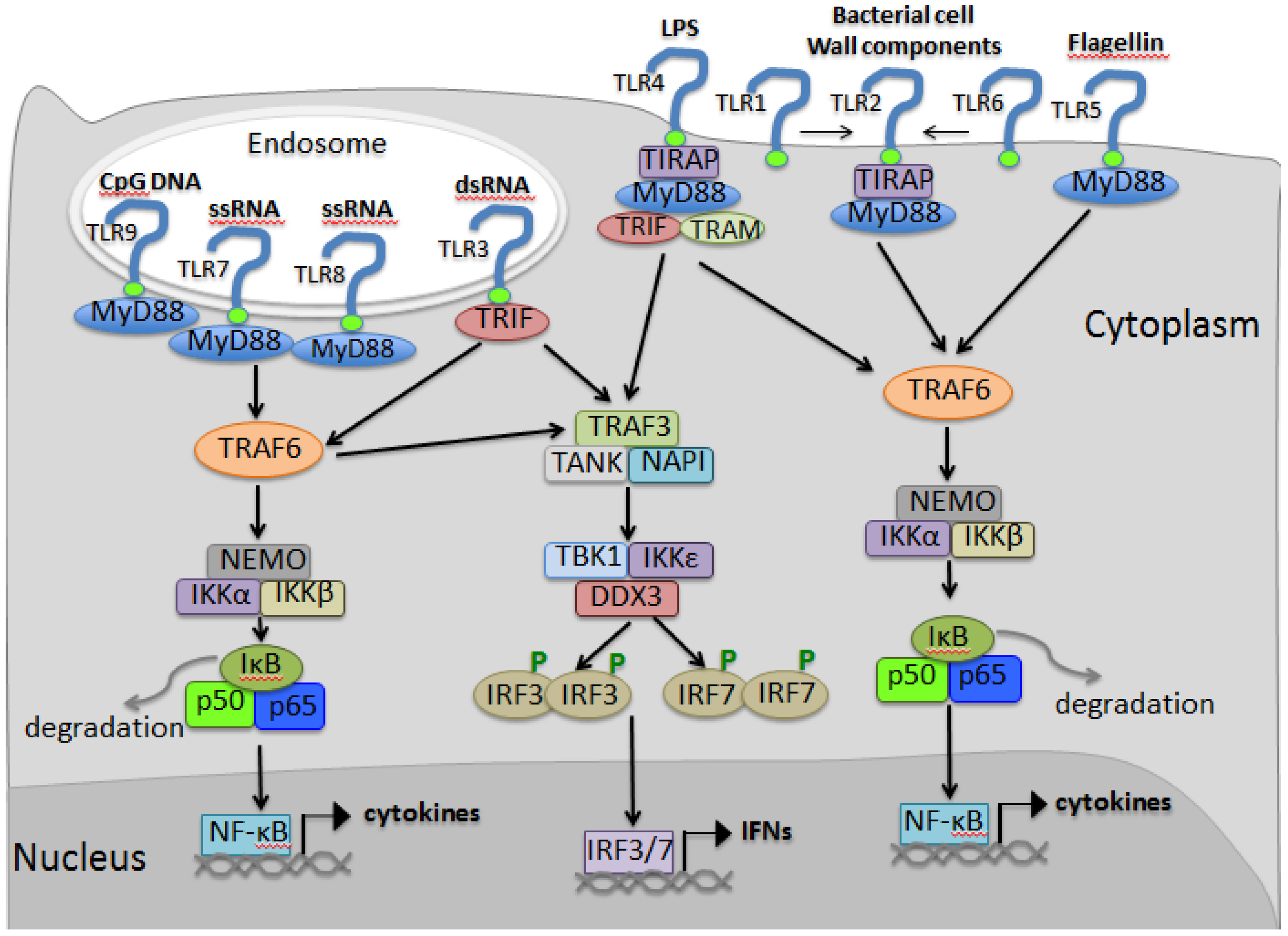
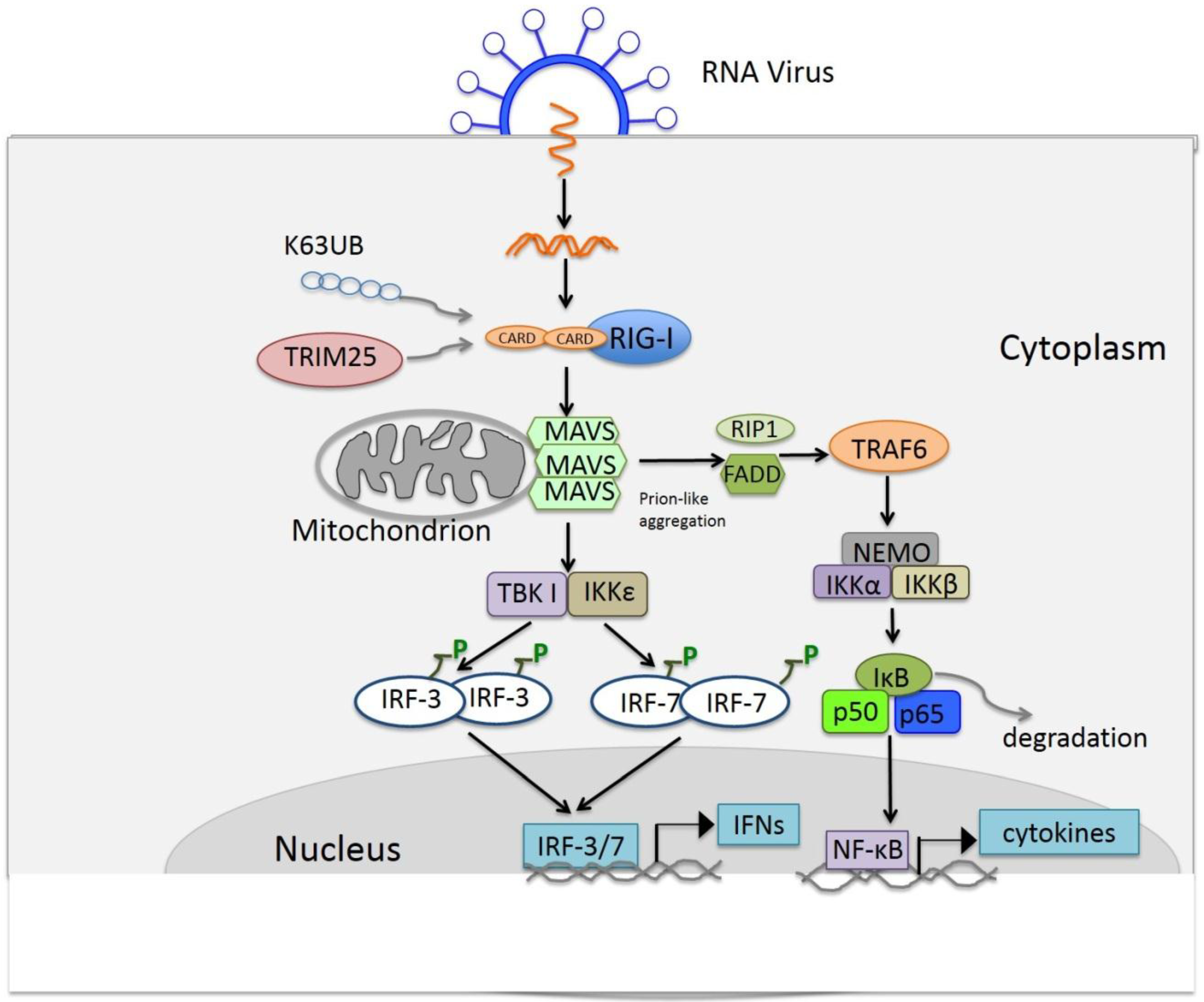
4. RIG-I Like Receptor Pathway
4.1. Discovery of the RIG-I Like Receptor
4.2. Ligands of RLRs
4.3. Signaling of RLRs
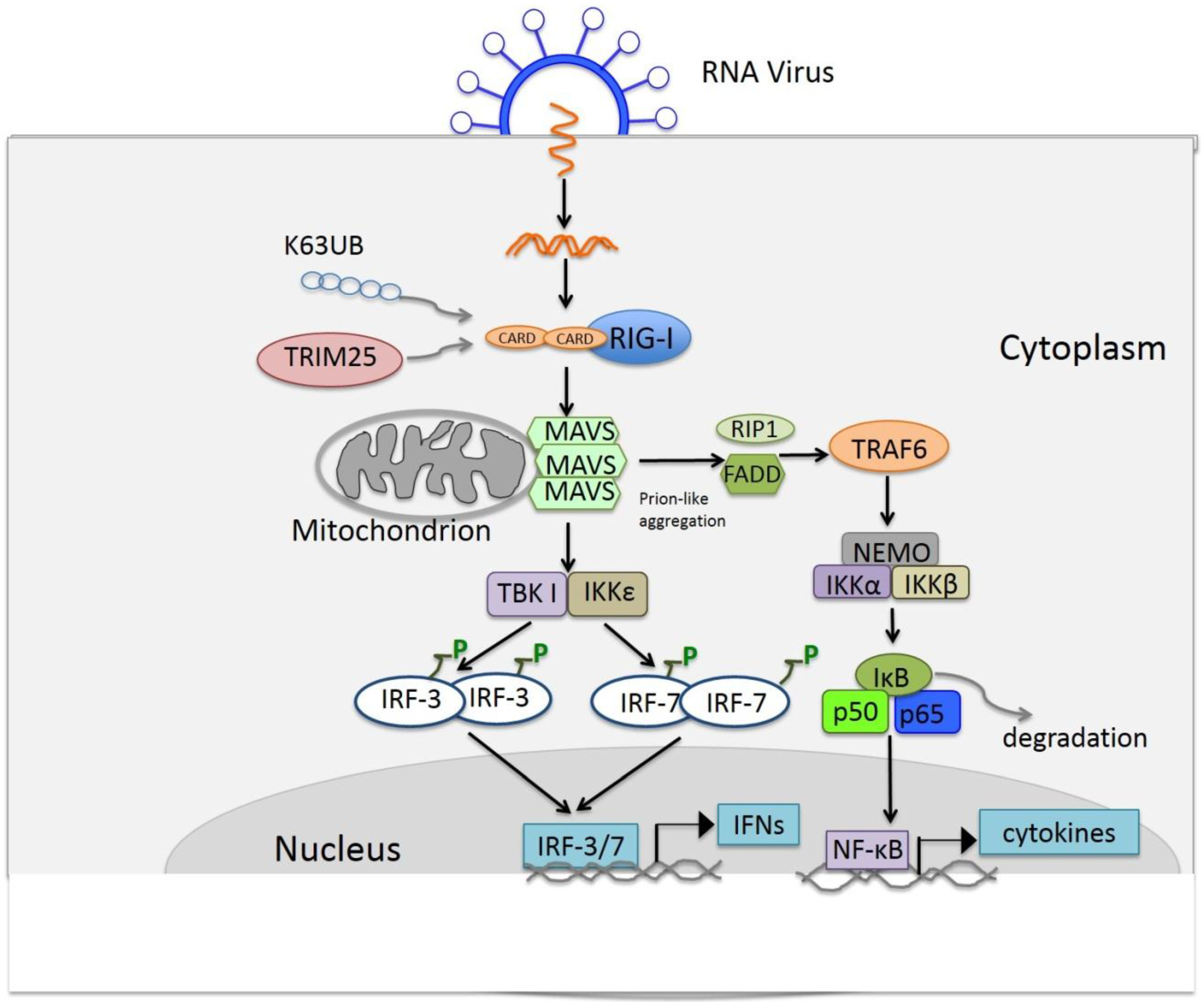
5. Other Functions of TLRs and RLRs
6. Viral Antagonism of IFN Induction
6.1. Antagonism via Direct Interaction with Host Molecules in the IFN Induction Pathway
6.2. Antagonism via Degradation or Cleavage of Host Molecules in the IFN Induction Pathway
6.3. Antagonism IFN Induction via Inhibiting IRF-3 Activation or Function
6.4. Antagonism of IFN Induction via Virally-Encoded Deubiquitinases
6.5. Antagonism via Viral Homologues of Host Molecules
| Mechanism | Target | Example of viral antagonist [Ref] |
|---|---|---|
| Binding to host molecules | RIG-I | NS1 of influenza A [149] |
| V of paramyxoviruses [150] | ||
| MDA-5 | NS1 of respiratory syncytial virus [151] | |
| MAVS | γ134.5 of HSV [152] | |
| TBK1 and iKKε | VP35 of Ebola virus [153] | |
| IRF3 | E6 of Human papillomavirus 16 [155] | |
| Degradation or cleavage of host molecules | RIG-I | 3C protease of EMCV [156] |
| TRIF MAVS | HAV 3CD [157], coxsackievirus B 3C [158] | |
| IRF3 | Npro of CSFV [160] | |
| Inhibition of IRF3 phosphorylation | IRF3 | HCV NS3/4A protease, PRRSV nsp1β, HBV polymerase Varicella-Zoster virus, IE62 protein, HSV ICP0 protein [161,162,163,166,167] |
| Virally-encoded deubiquitinases | RIG-I, TBK1, TRAF6 and TRAF3 | PCP 2 of arterivirus [172], nsp2 of PRRSV [173,174], Lpro of FMDV [175], PCP of HEV [176] |
| Viral homologues of host molecules | TIR domain, IRF | A46R and A52R of vaccinia [178,179] viral-encoded IRFs of KSHV [180,181] |
Acknowledgments
Author Contributions
Conflict of Interest
References
- Lengyel, P. Biochemistry of interferons and their actions. Annu. Rev. Biochem. 1982, 51, 251–282. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Rubinstein, S.; Familletti, P.C.; Miller, R.S.; Waldman, A.A.; Pestka, S. Human leukocyte interferon: Production, purification to homogeneity, and initial characterization. Proc. Natl. Acad. Sci. USA 1979, 76, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef] [PubMed]
- Havell, E.A.; Berman, B.; Ogburn, C.A.; Berg, K.; Paucker, K.; Vilcek, J. Two antigenically distinct species of human interferon. Proc. Natl. Acad. Sci. USA 1975, 72, 2185–2187. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Uze, G.; Schreiber, G.; Piehler, J.; Pellegrini, S. The receptor of the type I interferon family. Curr. Top. Microbiol. Immunol. 2007, 316, 71–95. [Google Scholar] [PubMed]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 2004, 22, 929–979. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Sheikh, F.; Kotenko, S.V.; Dickensheets, H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J. Leukoc. Biol 2004, 76, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Langer, J.A. Full house: 12 receptors for 27 cytokines. Int. Immunopharmacol. 2004, 4, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. Interferons and viral infections. Biofactors 2009, 35, 14–20. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [PubMed]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef] [PubMed]
- Moraga, I.; Harari, D.; Schreiber, G.; Uze, G.; Pellegrini, S. Receptor density is key to the alpha2/beta interferon differential activities. Mol. Cell. Biol. 2009, 29, 4778–4787. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Roisman, L.C.; Jaks, E.; Gavutis, M.; Piehler, J.; van der Heyden, J.; Uze, G.; Schreiber, G. Inquiring into the differential action of interferons (IFNs): An IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol. Cell. Biol. 2006, 26, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Kalie, E.; Jaitin, D.A.; Podoplelova, Y.; Piehler, J.; Schreiber, G. The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J. Biol. Chem. 2008, 283, 32925–32936. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.; Ozmen, L.; Novelli, F.; Geuna, M.; Palestro, G.; Forni, G.; Garotta, G. Distribution of interferon-gamma receptor in human tissues. Eur. J. Immunol. 1992, 22, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.R.; Windsor, W.T.; Nagabhushan, T.L.; Lundell, D.J.; Lunn, C.A.; Zauodny, P.J.; Narula, S.K. Crystal structure of a complex between interferon-gamma and its soluble high-affinity receptor. Nature 1995, 376, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Lew, D.J.; Cheng, Y.S.; Levy, D.E.; Darnell, J.E., Jr. Interactions of alpha- and gamma-interferon in the transcriptional regulation of the gene encoding a guanylate-binding protein. EMBO J. 1989, 8, 2009–2014. [Google Scholar] [PubMed]
- Lew, D.J.; Decker, T.; Darnell, J.E., Jr. Alpha interferon and gamma interferon stimulate transcription of a single gene through different signal transduction pathways. Mol. Cell. Biol. 1989, 9, 5404–5411. [Google Scholar] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Garcia-Sastre, A. Induction of type I interferon by RNA viruses: Cellular receptors and their substrates. Amino Acids 2010, 38, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Belvin, M.P.; Anderson, K.V. A conserved signaling pathway: The Drosophila toll-dorsal pathway. Annu. Rev. Cell Dev. Biol. 1996, 12, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Sarkar, D.; Su, Z.Z.; Barber, G.N.; DeSalle, R.; Racaniello, V.R.; Fisher, P.B. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: Key regulators of innate immunity. Pharmacol. Ther. 2009, 124, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Dushay, M.S.; Eldon, E.D. Drosophila immune responses as models for human immunity. Am. J. Hum. Genet. 1998, 62, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Takahashi, Y.; Putnam, F.W. Periodicity of leucine and tandem repetition of a 24-amino acid segment in the primary structure of leucine-rich alpha 2-glycoprotein of human serum. Proc. Natl. Acad. Sci. USA 1985, 82, 1906–1910. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Keith, F.J. Drosophila Toll and IL-1 receptor. Nature 1991, 351, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.S.; Hudson, K.L.; Lin, T.Y.; Anderson, K.V. Dominant and recessive mutations define functional domains of Toll, a transmembrane protein required for dorsal-ventral polarity in the Drosophila embryo. Genes Dev. 1991, 5, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Morisato, D.; Anderson, K.V. The spatzle gene encodes a component of the extracellular signaling pathway establishing the dorsal-ventral pattern of the Drosophila embryo. Cell 1994, 76, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Rosetto, M.; Engstrom, Y.; Baldari, C.T.; Telford, J.L.; Hultmark, D. Signals from the IL-1 receptor homolog, Toll, can activate an immune response in a Drosophila hemocyte cell line. Biochem. Biophys. Res. Commun. 1995, 209, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol 1989, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rock, F.L.; Hardiman, G.; Timans, J.C.; Kastelein, R.A.; Bazan, J.F. A family of human receptors structurally related to Drosophila Toll. Proc. Natl. Acad. Sci. USA 1998, 95, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Beachy, P.A. Expression of a novel Toll-like gene spans the parasegment boundary and contributes to hedgehog function in the adult eye of Drosophila. Mech. Dev. 1994, 47, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Kawai, T.; Sanjo, H.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Takeda, K.; Akira, S. TLR6: A novel member of an expanding toll-like receptor family. Gene 1999, 231, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.H.; Ulevitch, R.J. Cloning and characterization of a sub-family of human toll-like receptors: hTLR7, hTLR8 and hTLR9. Eur. Cytokine Netw. 2000, 11, 372–378. [Google Scholar]
- Chuang, T.; Ulevitch, R.J. Identification of hTLR10: A novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta 2001, 1518, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Lauw, F.N.; Caffrey, D.R.; Golenbock, D.T. Of mice and man: TLR11 (finally) finds profilin. Trends Immunol. 2005, 26, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, G.; Hayden, M.S.; Greenblatt, M.B.; Bussey, C.; Flavell, R.A.; Ghosh, S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science 2004, 303, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef]
- Hasan, U.; Chaffois, C.; Gaillard, C.; Saulnier, V.; Merck, E.; Tancredi, S.; Guiet, C.; Briere, F.; Vlach, J.; Lebecque, S.; et al. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J. Immunol. 2005, 174, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Roach, J.C.; Glusman, G.; Rowen, L.; Kaur, A.; Purcell, M.K.; Smith, K.D.; Hood, L.E.; Aderem, A. The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 9577–9582. [Google Scholar] [CrossRef] [PubMed]
- Baoprasertkul, P.; Xu, P.; Peatman, E.; Kucuktas, H.; Liu, Z. Divergent Toll-like receptors in catfish (Ictalurus punctatus): TLR5S, TLR20, TLR21. Fish Shellfish Immunol. 2007, 23, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Palti, Y. Toll-like receptors in bony fish: From genomics to function. Dev. Comp. Immunol. 2011, 35, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. N. Y. Acad. Sci. 2008, 1143, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997, 91, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.B.; Mark, M.R.; Gray, A.; Huang, A.; Xie, M.H.; Zhang, M.; Goddard, A.; Wood, W.I.; Gurney, A.L.; Godowski, P.J. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 1998, 395, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Sultzer, B.M. Genetic control of leucocyte responses to endotoxin. Nature 1968, 219, 1253–1254. [Google Scholar] [CrossRef] [PubMed]
- Rosenstreich, D.L.; Glode, L.M.; Mergenhagen, S.E. Action of endotoxin on lymphoid cells. J. Infect. Dis. 1977, 136, S239–S245. [Google Scholar] [CrossRef]
- Apte, R.N.; Ascher, O.; Pluznik, D.H. Genetic analysis of generation of serum interferon by bacterial lipopolysaccharide. J. Immunol. 1977, 119, 1898–1902. [Google Scholar] [PubMed]
- Michalek, S.M.; Moore, R.N.; McGhee, J.R.; Rosenstreich, D.L.; Mergenhagen, S.E. The primary role of lymphoreticular cells in the mediation of host responses to bacterial endotoxim. J. Infect. Dis. 1980, 141, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [PubMed]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Farhat, K.; Riekenberg, S.; Heine, H.; Debarry, J.; Lang, R.; Mages, J.; Buwitt-Beckmann, U.; Roschmann, K.; Jung, G.; Wiesmuller, K.H.; et al. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J. Leukoc. Biol. 2008, 83, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 1999, 274, 17406–17409. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis E Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [PubMed]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Forsbach, A.; Nemorin, J.G.; Montino, C.; Muller, C.; Samulowitz, U.; Vicari, A.P.; Jurk, M.; Mutwiri, G.K.; Krieg, A.M.; Lipford, G.B.; et al. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol. 2008, 180, 3729–3738. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Mulla, M.J.; Myrtolli, K.; Tadesse, S.; Stanwood, N.L.; Gariepy, A.; Guller, S.; Norwitz, E.R.; Abrahams, V.M. Cutting-edge report: TLR10 plays a role in mediating bacterial peptidoglycan-induced trophoblast apoptosis. Am. J. Reprod. Immunol. 2013, 69, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M. Master sensors of pathogenic RNA—RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A. Targeting Toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Gong, M.; Cepika, A.M.; Xu, Z.; Tripodo, C.; Bennett, L.; Crain, C.; Quartier, P.; Cush, J.J.; Pascual, V.; et al. RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 2013, 210, 2903–2919. [Google Scholar] [CrossRef] [PubMed]
- Pisetsky, D.S. The origin and properties of extracellular DNA: From PAMP to DAMP. Clin. Immunol. 2012, 144, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Moynagh, P.N. TLR signalling and activation of IRFs: Revisiting old friends from the NF-kappaB pathway. Trends Immunol. 2005, 26, 469–476. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Koblansky, A.A.; Ghosh, S. Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 2006, 22, 409–437. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Bhatia, D.; Chang, Q.; Castranova, V. Finding NEMO by K63-linked polyubiquitin chain. Cell Death Differ. 2006, 13, 1835–1838. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Xu, M.; Chen, Z.J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007, 26, 3214–3226. [Google Scholar] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Trinchieri, G.; Liu, Y.J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sugiyama, M.; Yamamoto, M.; Watanabe, Y.; Kawai, T.; Takeda, K.; Akira, S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 2003, 171, 4304–4310. [Google Scholar] [CrossRef] [PubMed]
- Sasai, M.; Oshiumi, H.; Matsumoto, M.; Inoue, N.; Fujita, F.; Nakanishi, M.; Seya, T. Cutting Edge: NF-kappaB-activating kinase-associated protein 1 participates in TLR3/Toll-IL-1 homology domain-containing adapter molecule-1-mediated IFN regulatory factor 3 activation. J. Immunol. 2005, 174, 27–30. [Google Scholar] [CrossRef]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Sasai, M.; Shida, K.; Fujita, T.; Matsumoto, M.; Seya, T. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-beta. J. Biol. Chem. 2003, 278, 49751–49762. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Horng, T.; Barton, G.M.; Flavell, R.A.; Medzhitov, R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 2002, 420, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Linder, P.; Lasko, P.F.; Ashburner, M.; Leroy, P.; Nielsen, P.J.; Nishi, K.; Schnier, J.; Slonimski, P.P. Birth of the D-E-A-D box. Nature 1989, 337, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989, 17, 4713–4730. [Google Scholar] [CrossRef] [PubMed]
- Matsumiya, T.; Stafforini, D.M. Function and regulation of retinoic acid-inducible gene-I. Crit. Rev. Immunol. 2010, 30, 489–513. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Aratani, S.; Nakajima, T.; Carlson, M.; Matsumiya, T.; Tanji, K.; Ookawa, K.; Yoshida, H.; Tsuchida, S.; McIntyre, T.M.; et al. Retinoic acid-inducible gene-I is induced in endothelial cells by LPS and regulates expression of COX-2. Biochem. Biophys. Res. Commun. 2002, 292, 274–279. [Google Scholar]
- Zhang, X.; Wang, C.; Schook, L.B.; Hawken, R.J.; Rutherford, M.S. An RNA helicase, RHIV -1, induced by porcine reproductive and respiratory syndrome virus (PRRSV) is mapped on porcine chromosome 10q13. Microb. Pathog. 2000, 28, 267–278. [Google Scholar] [PubMed]
- Liu, T.X.; Zhang, J.W.; Tao, J.; Zhang, R.B.; Zhang, Q.H.; Zhao, C.J.; Tong, J.H.; Lanotte, M.; Waxman, S.; Chen, S.J.; et al. Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood 2000, 96, 1496–1504. [Google Scholar] [PubMed]
- Monastyrskaia, G.S.; Kostina, M.B.; Filiukova, O.B.; Protopopova, E.V.; Konovalova, S.N.; Kachko, A.V.; Nikolaev, L.G.; Loktev, V.B.; Sverdlov, E.D. Activation of the RIG-I gene, coding for DEXH/D-protein in infection of RH cells by tick-borne encephalitis virus. Bioorg. Khim. 2004, 30, 146–150. [Google Scholar] [PubMed]
- Cui, X.F.; Imaizumi, T.; Yoshida, H.; Borden, E.C.; Satoh, K. Retinoic acid-inducible gene-I is induced by interferon-gamma and regulates the expression of interferon-gamma stimulated gene 15 in MCF-7 cells. Biochem. Cell Biol. 2004, 82, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef]
- Huang, F.; Adelman, J.; Jiang, H.; Goldstein, N.I.; Fisher, P.B. Differentiation induction subtraction hybridization (DISH): A strategy for cloning genes displaying differential expression during growth arrest and terminal differentiation. Gene 1999, 236, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Adelman, J.; Jiang, H.; Goldstein, N.I.; Fisher, P.B. Identification and temporal expression pattern of genes modulated during irreversible growth arrest and terminal differentiation in human melanoma cells. Oncogene 1999, 18, 3546–3552. [Google Scholar] [PubMed]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Shillingford, J.M.; Smith, G.H.; Grimm, S.L.; Wagner, K.U.; Oka, T.; Rosen, J.M.; Robinson, G.W.; Hennighausen, L. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar epithelium. J. Cell Biol. 2001, 155, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Li, M.; Walton, K.D.; Sun, K.; Hanover, J.A.; Furth, P.A.; Hennighausen, L. The Stat3/5 locus encodes novel endoplasmic reticulum and helicase-like proteins that are preferentially expressed in normal and neoplastic mammary tissue. Genomics 2001, 78, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [PubMed]
- Kim, D.H.; Longo, M.; Han, Y.; Lundberg, P.; Cantin, E.; Rossi, J.J. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat. Biotechnol. 2004, 22, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Strahle, L.; Garcin, D.; Kolakofsky, D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 2006, 351, 101–111. [Google Scholar] [CrossRef]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Hartmann, G. The Chase for the RIG-I Ligand-Recent Advances. Mol. Ther. 2010, 18, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:Polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 601–610. [Google Scholar] [CrossRef]
- Luthra, P.; Sun, D.; Silverman, R.H.; He, B. Activation of IFN-β; expression by a viral mRNA through RNase L and MDA5. Proc. Natl. Acad. Sci. USA 2011, 108, 2118–2123. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Horvath, C.M. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol. 2006, 80, 12332–12342. [Google Scholar] [CrossRef] [PubMed]
- Rothenfusser, S.; Goutagny, N.; DiPerna, G.; Gong, M.; Monks, B.G.; Schoenemeyer, A.; Yamamoto, M.; Akira, S.; Fitzgerald, K.A. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 2005, 175, 5260–5268. [Google Scholar] [PubMed]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ranjith-Kumar, C.T.; Brooks, M.T.; Dharmaiah, S.; Herr, A.B.; Kao, C.; Li, P. The RIG-I-like receptor LGP2 recognizes the termini of double-stranded RNA. J. Biol. Chem. 2009, 284, 13881–13891. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, T.; Valdes, M.; Elsby, R.; Kakuta, S.; Caceres, G.; Saijo, S.; Iwakura, Y.; Barber, G.N. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 2007, 178, 6444–6455. [Google Scholar] [CrossRef] [PubMed]
- Suthar, M.S.; Ramos, H.J.; Brassil, M.M.; Netland, J.; Chappell, C.P.; Blahnik, G.; McMillan, A.; Diamond, M.S.; Clark, E.A.; Bevan, M.J.; et al. The RIG-I-like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity 2012, 37, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Yoneyama, M.; Nishihori, T.; Hirai, R.; Kumeta, H.; Narita, R.; Gale, M., Jr.; Inagaki, F.; Fujita, T. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol. Cell 2008, 29, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S.M.; Tenoever, B.R.; Maniatis, T. Connecting mitochondria and innate immunity. Cell 2005, 122, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Robek, M.D. Peroxisomal MAVS activates IRF1-mediated IFN-lambda production. Nat. Immunol. 2014, 15, 700–701. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Thomas, E.; Barber, G.N. A FADD-dependent innate immune mechanism in mammalian cells. Nature 2004, 432, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 51–61. [Google Scholar] [CrossRef]
- Tu, D.; Zhu, Z.; Zhou, A.Y.; Yun, C.H.; Lee, K.E.; Toms, A.V.; Li, Y.; Dunn, G.P.; Chan, E.; Thai, T.; et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 2013, 3, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jia, X.; Xue, Q.; Dou, Z.; Ma, Y.; Zhao, Z.; Jiang, Z.; He, B.; Jin, Q.; Wang, J. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc. Natl. Acad. Sci. USA 2014, 111, E245–E254. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Akira, S. TLR signalling and the function of dendritic cells. Chem. Immunol. Allergy 2005, 86, 120–135. [Google Scholar] [PubMed]
- Reis E Sousa, C. Toll-like receptors and dendritic cells: For whom the bug tolls. Semin. Immunol. 2004, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Sabroe, I.; Dower, S.K.; Whyte, M.K. The role of Toll-like receptors in the regulation of neutrophil migration, activation, and apoptosis. Clin. Infect. Dis. 2005, 41 (Suppl. 7), S421–S426. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grumont, R.J.; Banerjee, A. Regulating B-cell activation and survival in response to TLR signals. Immunol. Cell Biol. 2007, 85, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Raghavan, B.; Muller, V.; Vogl, T.; Fejer, G.; Tchaptchet, S.; Keck, S.; Kalis, C.; Nielsen, P.J.; Galanos, C.; et al. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat. Immunol. 2010, 11, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Tidswell, M.; Tillis, W.; Larosa, S.P.; Lynn, M.; Wittek, A.E.; Kao, R.; Wheeler, J.; Gogate, J.; Opal, S.M. Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit. Care Med. 2010, 38, 72–83. [Google Scholar] [PubMed]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef]
- Rolls, A.; Shechter, R.; London, A.; Ziv, Y.; Ronen, A.; Levy, R.; Schwartz, M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 2007, 9, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sayed, N.; Hunter, A.; Au, K.F.; Wong, W.H.; Mocarski, E.S.; Pera, R.R.; Yakubov, E.; Cooke, J.P. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 2012, 151, 547–558. [Google Scholar] [CrossRef]
- Zhang, N.N.; Shen, S.H.; Jiang, L.J.; Zhang, W.; Zhang, H.X.; Sun, Y.P.; Li, X.Y.; Huang, Q.H.; Ge, B.X.; Chen, S.J.; et al. RIG-I plays a critical role in negatively regulating granulocytic proliferation. Proc. Natl. Acad. Sci. USA 2008, 105, 10553–10558. [Google Scholar] [CrossRef] [PubMed]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef] [PubMed]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; San Juan-Vergara, H.; Mohapatra, S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS One 2012, 7, e29386. [Google Scholar] [CrossRef] [PubMed]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.C.; Cardenas, W.B.; Basler, C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 2009, 83, 3069–3077. [Google Scholar] [PubMed]
- Anglero-Rodriguez, Y.I.; Pantoja, P.; Sariol, C.A. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IkappaB kinase epsilon interaction. Clin. Vaccine Immunol. 2014, 21, 29–38. [Google Scholar] [PubMed]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Papon, L.; Oteiza, A.; Imaizumi, T.; Kato, H.; Brocchi, E.; Lawson, T.G.; Akira, S.; Mechti, N. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 2009, 393, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Feng, Z.; Yamane, D.; Liang, Y.; Lanford, R.E.; Li, K.; Lemon, S.M. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 2011, 7, e1002169. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Bauhofer, O.; Summerfield, A.; Sakoda, Y.; Tratschin, J.D.; Hofmann, M.A.; Ruggli, N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 2007, 81, 3087–3096. [Google Scholar] [CrossRef] [PubMed]
- Beura, L.K.; Sarkar, S.N.; Kwon, B.; Subramaniam, S.; Jones, C.; Pattnaik, A.K.; Osorio, F.A. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J. Virol. 2010, 84, 1574–1584. [Google Scholar] [PubMed]
- Foy, E.; Li, K.; Wang, C.; Sumpter, R., Jr.; Ikeda, M.; Lemon, S.M.; Gale, M., Jr. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chen, J.; Wu, M.; Chen, H.; Kato, N.; Yuan, Z. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J. Gen. Virol. 2010, 91, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M.; Baran, M.; Bowie, A.G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008, 27, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Soulat, D.; Burckstummer, T.; Westermayer, S.; Goncalves, A.; Bauch, A.; Stefanovic, A.; Hantschel, O.; Bennett, K.L.; Decker, T.; Superti-Furga, G. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008, 27, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Sommer, M.; Che, X.; White, K.; Ruyechan, W.T.; Arvin, A.M. Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: A novel mechanism of IRF3 inhibition among herpesviruses. J. Virol. 2010, 84, 9240–9253. [Google Scholar] [CrossRef] [PubMed]
- Melroe, G.T.; Silva, L.; Schaffer, P.A.; Knipe, D.M. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-beta induction. Virology 2007, 360, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Xu, M.; Liu, S.; Sun, L.; Chen, Z.J. Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol. Cell 2009, 36, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.P.; Li, S.; Zhong, B.; Li, Y.; Yan, J.; Li, Q.; Teng, C.; Shu, H.B. Virus-triggered ubiquitination of TRAF3/6 by cIAP1/2 is essential for induction of interferon-beta (IFN-beta) and cellular antiviral response. J. Biol. Chem. 2010, 285, 9470–9476. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Q.; Chen, W.; Wang, C. Dynamic regulation of innate immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Factor Rev. 2013, 24, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Van Kasteren, P.B.; Bailey-Elkin, B.A.; James, T.W.; Ninaber, D.K.; Beugeling, C.; Khajehpour, M.; Snijder, E.J.; Mark, B.L.; Kikkert, M. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc. Natl. Acad. Sci. USA 2013, 110, E838–E847. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zheng, Z.; Zhou, P.; Zhang, B.; Shi, Z.; Hu, Q.; Wang, H. The cysteine protease domain of porcine reproductive and respiratory syndrome virus non-structural protein 2 antagonizes interferon regulatory factor 3 activation. J. Gen. Virol. 2010, 91, 2947–2958. [Google Scholar] [CrossRef]
- Sun, Z.; Chen, Z.; Lawson, S.R.; Fang, Y. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J. Virol. 2010, 84, 7832–7846. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Li, P.; Sun, L.; Fan, J.; Zhang, Q.; Luo, R.; Liu, X.; Li, K.; Chen, H.; et al. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 2011, 85, 3758–3766. [Google Scholar]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.J. Hepatitis E Virus Inhibits Type I Interferon Induction by ORF1 Product. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [PubMed]
- Kubota, T.; Matsuoka, M.; Chang, T.H.; Tailor, P.; Sasaki, T.; Tashiro, M.; Kato, A.; Ozato, K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 2008, 283, 25660–25670. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.; Kiss-Toth, E.; Symons, J.A.; Smith, G.L.; Dower, S.K.; O’Neill, L.A. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 10162–10167. [Google Scholar] [CrossRef] [PubMed]
- Stack, J.; Haga, I.R.; Schroder, M.; Bartlett, N.W.; Maloney, G.; Reading, P.C.; Fitzgerald, K.A.; Smith, G.L.; Bowie, A.G. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 2005, 201, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Damania, B. The viral interferon regulatory factors of KSHV: Immunosuppressors or oncogenes? Front. Immunol. 2011, 2, e19. [Google Scholar] [CrossRef]
- Lee, H.R.; Kim, M.H.; Lee, J.S.; Liang, C.; Jung, J.U. Viral interferon regulatory factors. J. Interferon Cytokine Res. 2009, 29, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Genin, P.; Mamane, Y.; Sgarbanti, M.; Battistini, A.; Harrington, W.J., Jr.; Barber, G.N.; Hiscott, J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 2001, 20, 800–811. [Google Scholar]
- Burysek, L.; Yeow, W.S.; Lubyova, B.; Kellum, M.; Schafer, S.L.; Huang, Y.Q.; Pitha, P.M. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 1999, 73, 7334–7342. [Google Scholar] [PubMed]
- Burysek, L.; Yeow, W.S.; Pitha, P.M. Unique properties of a second human herpesvirus 8-encoded interferon regulatory factor (vIRF-2). J. Hum. Virol. 1999, 2, 19–32. [Google Scholar]
- Areste, C.; Mutocheluh, M.; Blackbourn, D.J. Identification of caspase-mediated decay of interferon regulatory factor-3, exploited by a Kaposi sarcoma-associated herpesvirus immunoregulatory protein. J. Biol. Chem. 2009, 284, 23272–23285. [Google Scholar] [CrossRef] [PubMed]
- Lubyova, B.; Kellum, M.J.; Frisancho, A.J.; Pitha, P.M. Kaposi’s sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7. J. Biol. Chem. 2004, 279, 7643–7654. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.H.; Shin, Y.C.; Gack, M.; Wu, L.; Levy, D.; Jung, J.U. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 2007, 81, 8282–8292. [Google Scholar] [CrossRef] [PubMed]
- Wies, E.; Hahn, A.S.; Schmidt, K.; Viebahn, C.; Rohland, N.; Lux, A.; Schellhorn, T.; Holzer, A.; Jung, J.U.; Neipel, F. The Kaposi’s Sarcoma-associated Herpesvirus-encoded vIRF-3 Inhibits Cellular IRF-5. J. Biol. Chem. 2009, 284, 8525–8538. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, Y.; Nan, G.; Zhang, Y.-J. Interferon Induction by RNA Viruses and Antagonism by Viral Pathogens. Viruses 2014, 6, 4999-5027. https://doi.org/10.3390/v6124999
Nan Y, Nan G, Zhang Y-J. Interferon Induction by RNA Viruses and Antagonism by Viral Pathogens. Viruses. 2014; 6(12):4999-5027. https://doi.org/10.3390/v6124999
Chicago/Turabian StyleNan, Yuchen, Guoxin Nan, and Yan-Jin Zhang. 2014. "Interferon Induction by RNA Viruses and Antagonism by Viral Pathogens" Viruses 6, no. 12: 4999-5027. https://doi.org/10.3390/v6124999
APA StyleNan, Y., Nan, G., & Zhang, Y.-J. (2014). Interferon Induction by RNA Viruses and Antagonism by Viral Pathogens. Viruses, 6(12), 4999-5027. https://doi.org/10.3390/v6124999