Lost in Transcription: Molecular Mechanisms that Control HIV Latency


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Events of Productive HIV Infection
3. Mechanisms that Enforce Entrance of HIV into Latency
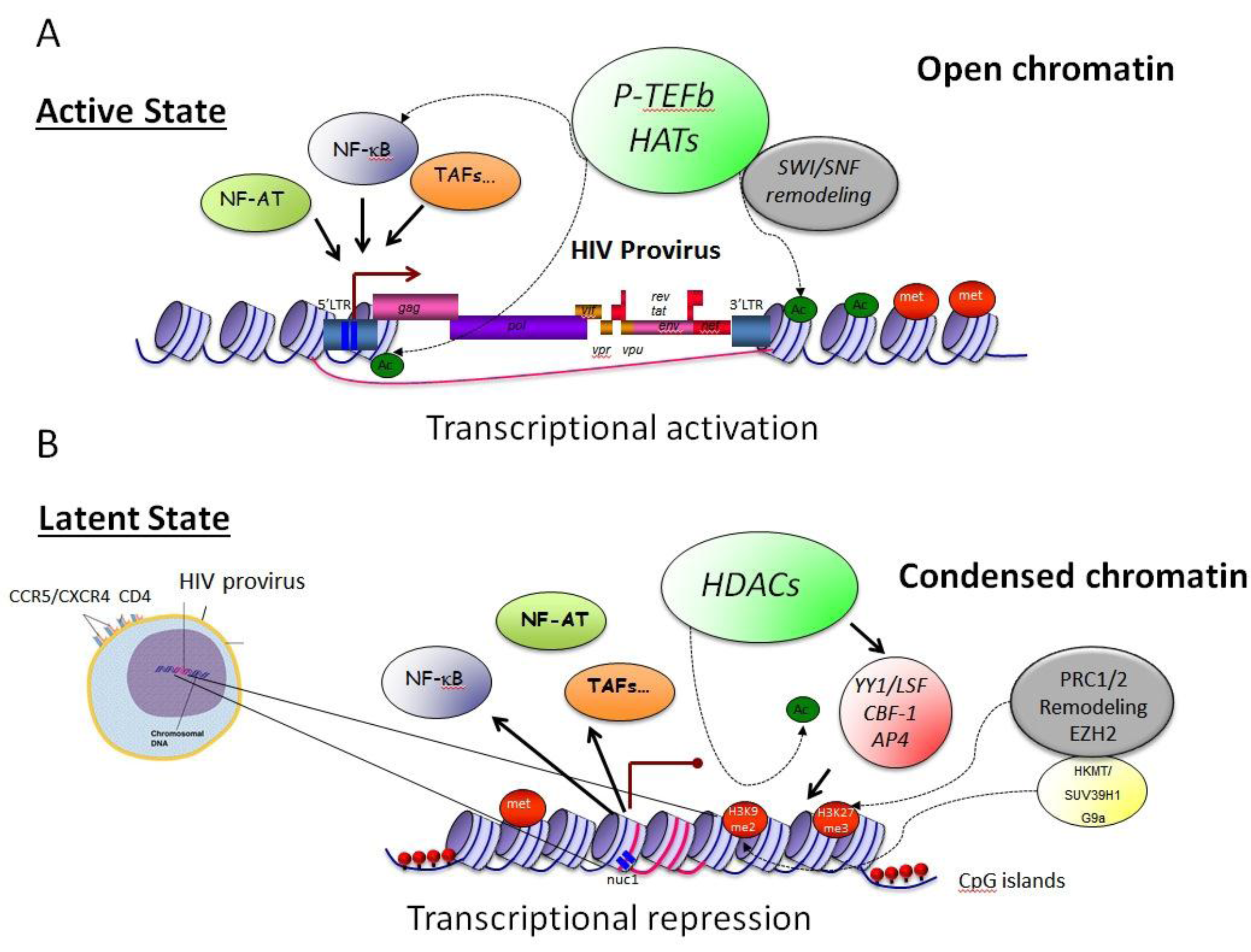
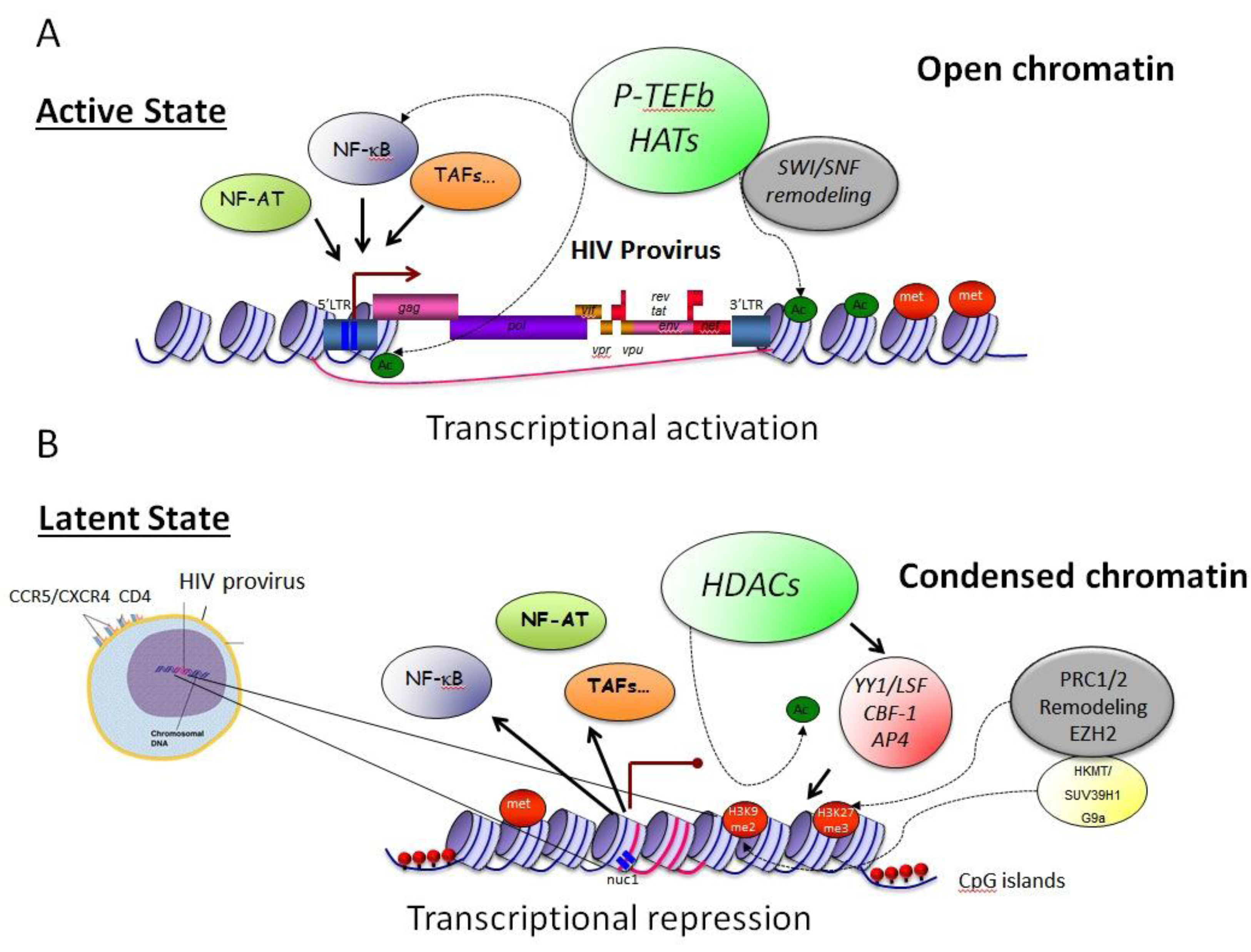
3.1. Epigenetic Constraints– Impact on Transcription Initiation
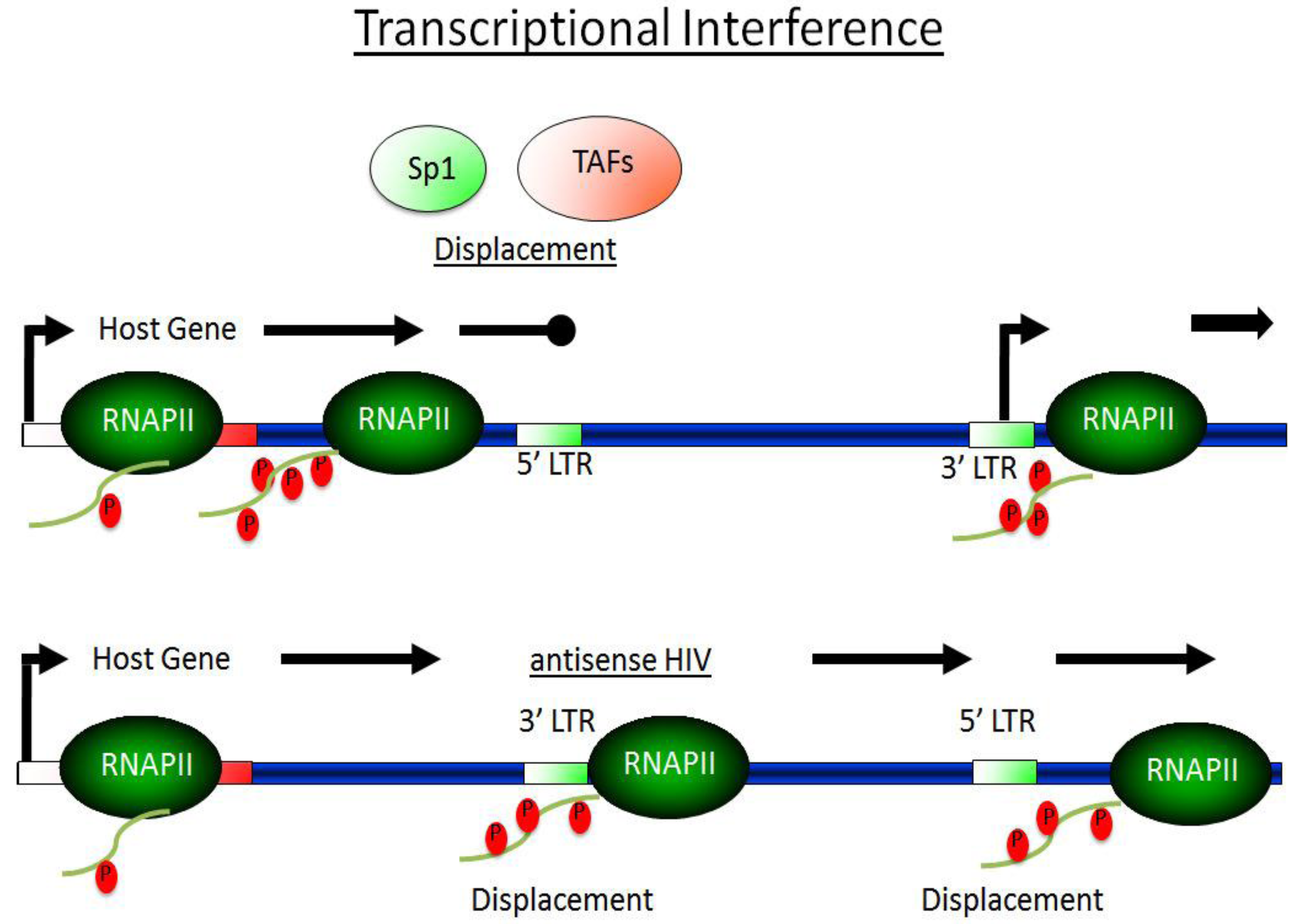
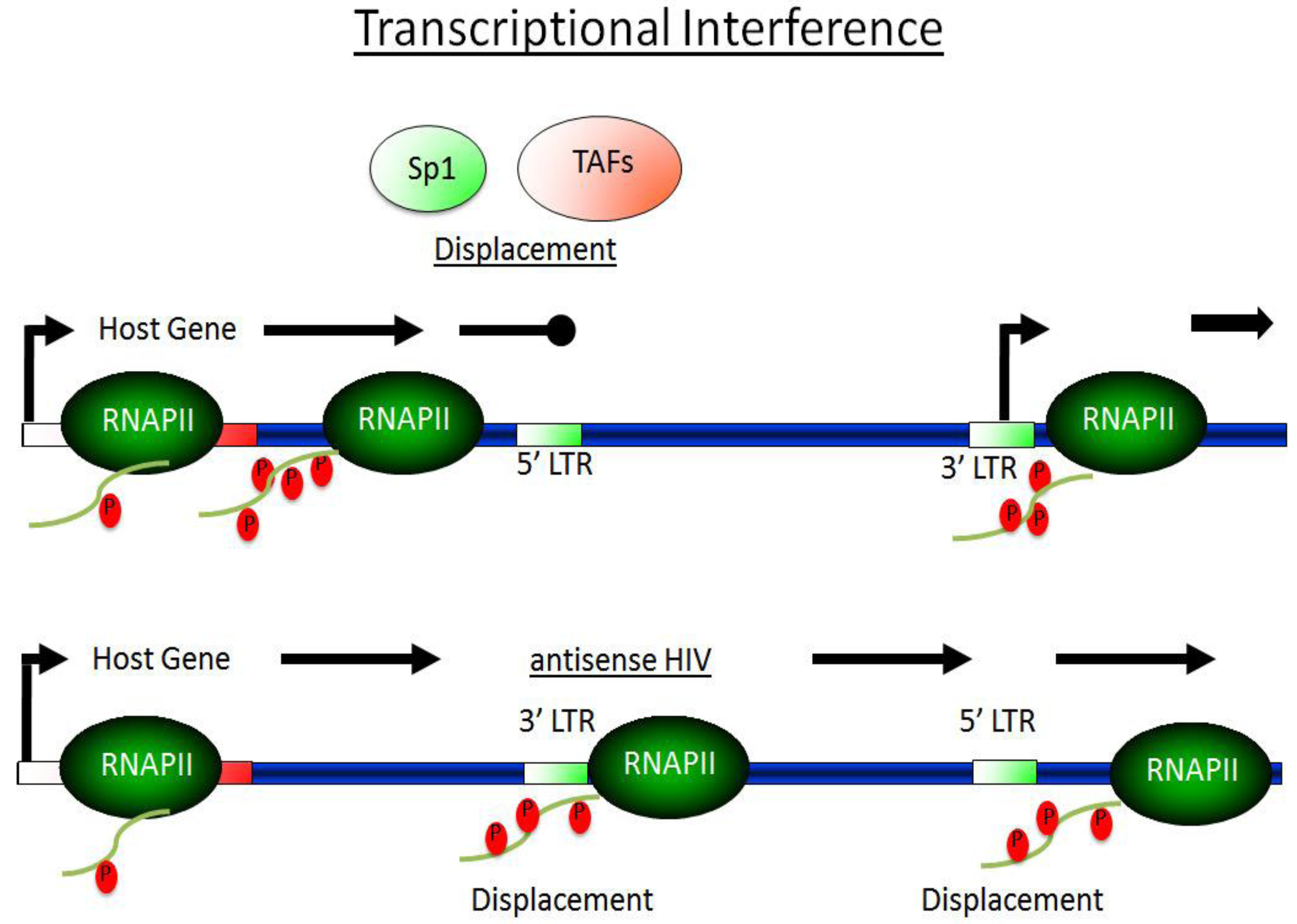
3.2. Transcriptional Interference—TI
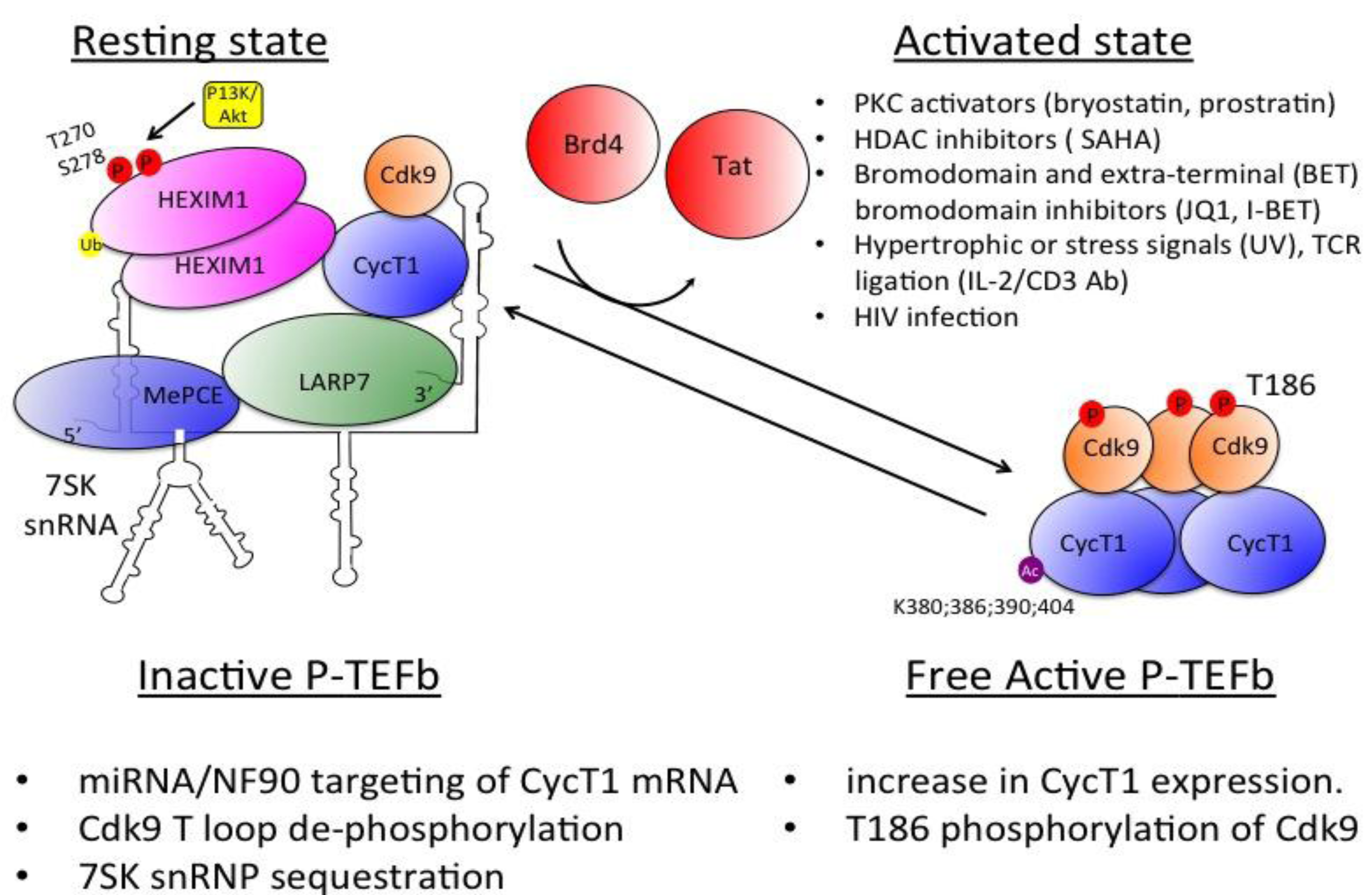
3.3 The Dynamic between Transcriptionally Active and Inactive P-TEFb Controls HIV Transcription
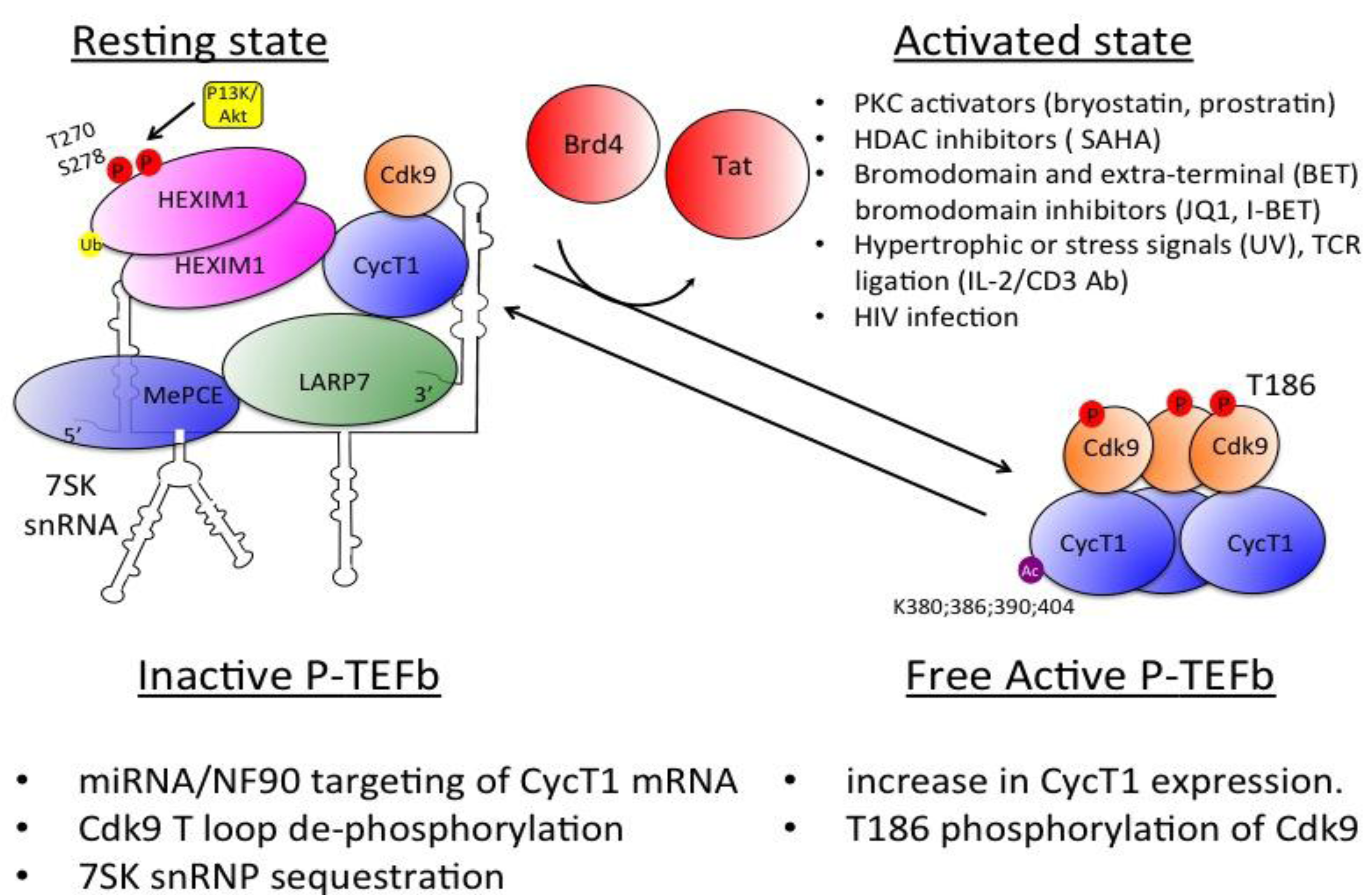
3.4 Regulation of P-TEFb Expression and Activity
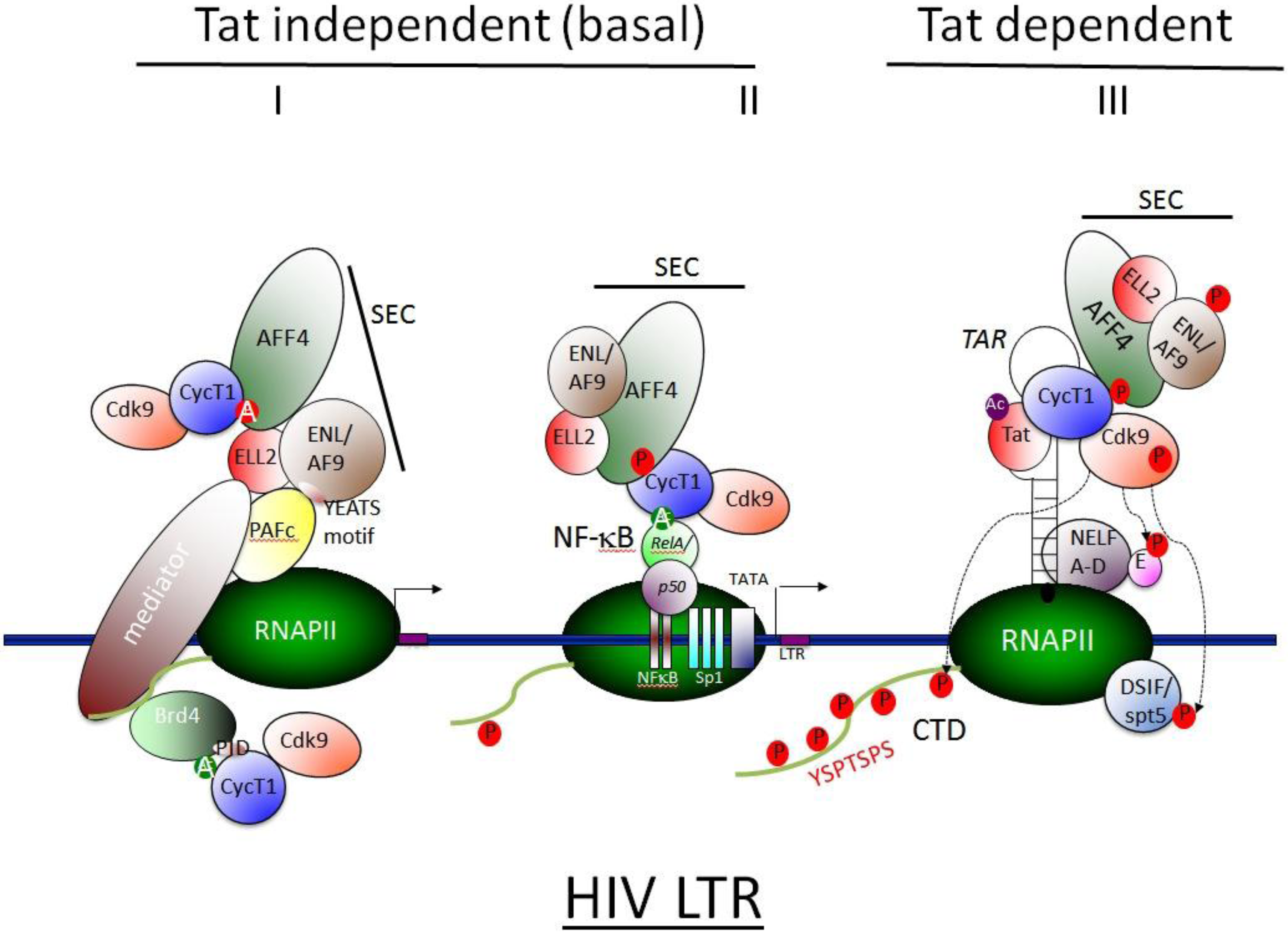
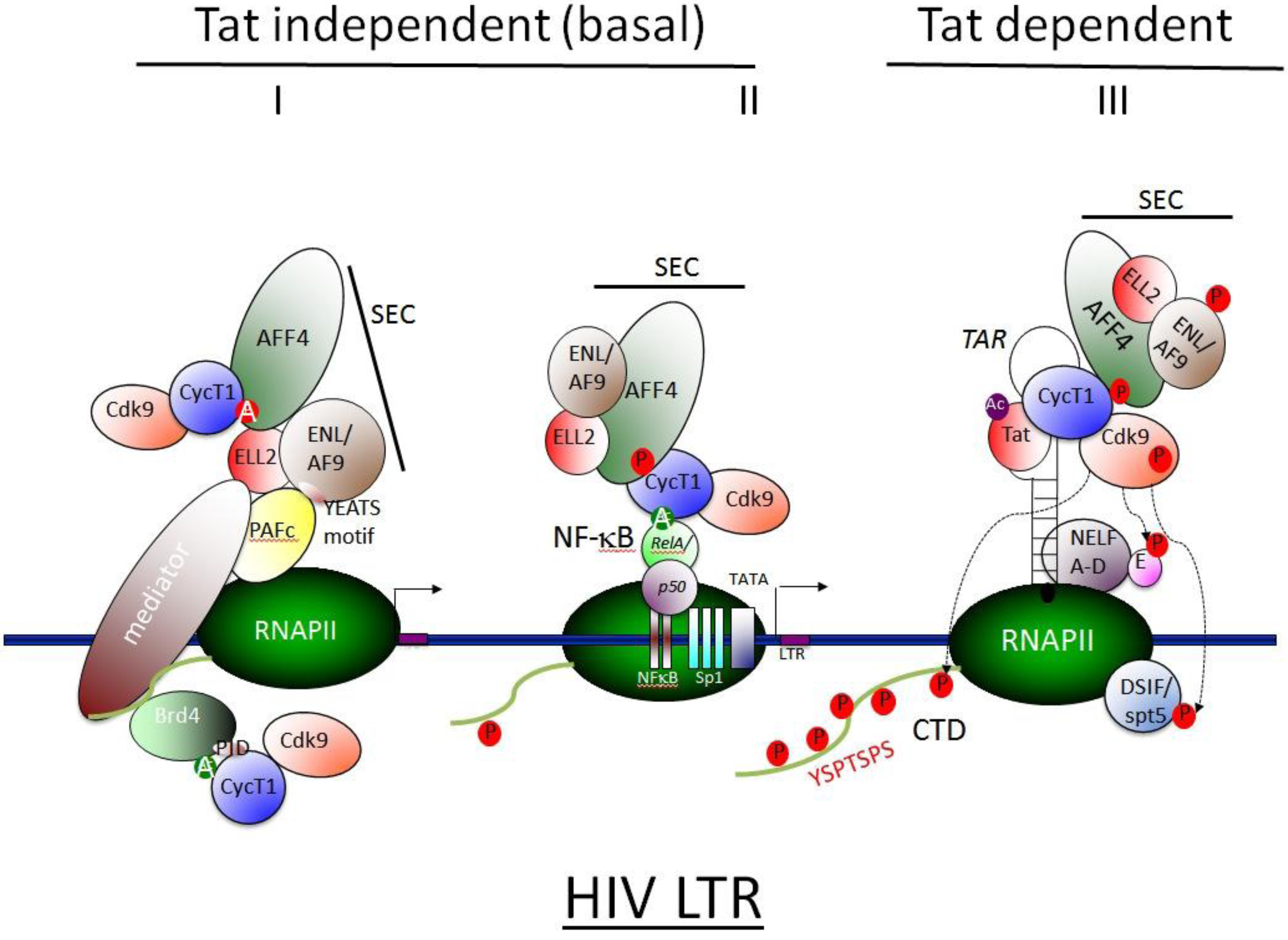
3.5 SEC Associates with HIV Tat and Activates Viral Transcription
4. Therapeutic Approaches
5. Conclusions
Acknowledgements
Conflict of Interest
References
- Lassen, K.; Han, Y.; Zhou, Y.; Siliciano, J.; Siliciano, R.F. The multifactorial nature of HIV–1 latency. Trends Mol. Med. 2004, 10, 525–531. [Google Scholar] [CrossRef]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV–1 infection. Nature 1997, 387, 183–188. [Google Scholar]
- Eisele, E.; Siliciano, R.F. Redefining the Viral Reservoirs that Prevent HIV–1 Eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef]
- Laughlin, M.A.; Zeichner, S.; Kolson, D.; Alwine, J.C.; Seshamma, T.; Pomerantz, R.J.; Gonzalez–Scarano, F. Sodium butyrate treatment of cells latently infected with HIV–1 results in the expression of unspliced viral RNA. Virology 1993, 196, 496–505. [Google Scholar] [CrossRef]
- Prins, J.M.; Jurriaans, S.; van Praag, R.M.; Blaak, H.; van Rij, R.; Schellekens, P.T.; ten Berge, I.J.; Yong, S.L.; Fox, C.H.; Roos, M.T.; et al. Immuno–activation with anti–CD3 and recombinant human IL–2 in HIV–1–infected patients on potent antiretroviral therapy. AIDS 1999, 13, 2405–2410. [Google Scholar] [CrossRef]
- Popik, W.; Pitha, P.M. Role of tumor necrosis factor alpha in activation and replication of the tat–defective human immunodeficiency virus type 1. J. Virol. 1993, 67, 1094–1099. [Google Scholar]
- Folks, T.M.; Justement, J.; Kinter, A.; Dinarello, C.A.; Fauci, A.S. Cytokine–induced expression of HIV–1 in a chronically infected promonocyte cell line. Science 1987, 238, 800–802. [Google Scholar]
- Poli, G.; Kinter, A.L.; Fauci, A.S. Interleukin 1 induces expression of the human immunodeficiency virus alone and in synergy with interleukin 6 in chronically infected U1 cells: inhibition of inductive effects by the interleukin 1 receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 108–112. [Google Scholar] [CrossRef]
- Managlia, E.Z.; Landay, A.; Al–Harthi, L. Interleukin–7 induces HIV replication in primary naive T cells through a nuclear factor of activated T cell (NFAT)–dependent pathway. Virology 2006, 350, 443–452. [Google Scholar] [CrossRef]
- Chomont, N.; El–Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine–Diab, B.; Boucher, G.; Boulasse, M.–R.; Ghatta, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S. HIV–1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Stevenson, M.; Stanwick, T.L.; Dempsey, M.P.; Lamonica, C.A. HIV–1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 1990, 9, 1551–1560. [Google Scholar]
- Bukrinsky, M.I.; Stanwick, T.L.; Dempsey, M.P.; Stevenson, M. Quiescent T lymphocytes as an inducible virus reservoir in HIV–1 infection. Science 1991, 254, 423–427. [Google Scholar]
- Luo, Z.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547. [Google Scholar] [CrossRef]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti–termination of transcription within the long terminal repeat of HIV–1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef]
- Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Heaphy, S.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A. HIV–1 tat protein stimulates transcription by binding to a U–rich bulge in the stem of the TAR RNA structure. EMBO J. 1990, 9, 4145–4153. [Google Scholar]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9–associated C–type cyclin interacts directly with HIV–1 Tat and mediates its high–affinity, loop–specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Mahmoudi, T.; Parra, M.; Vries, R.G.; Kauder, S.E.; Verrijzer, C.P.; Ott, M.; Verdin, E. The SWI/SNF chromatin–remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 2006, 281, 19960–19968. [Google Scholar]
- Ott, M.; Dorr, A.; Hetzer–Egger, C.; Kaehlcke, K.; Schnolzer, M.; Henklein, P.; Cole, P.; Zhou, M.M.; Verdin, E. Tat acetylation: a regulatory switch between early and late phases in HIV transcription elongation. Novartis Found. Symp. 2004, 259, 182–193; discussion 193–186, 223–185. [Google Scholar] [CrossRef]
- Dorr, A.; Kiermer, V.; Pedal, A.; Rackwitz, H.R.; Henklein, P.; Schubert, U.; Zhou, M.M.; Verdin, E.; Ott, M. Transcriptional synergy between Tat and PCAF is dependent on the binding of acetylated Tat to the PCAF bromodomain. EMBO J. 2002, 21, 2715–2723. [Google Scholar] [CrossRef]
- Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV–1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar]
- Boulanger, M.C.; Liang, C.; Russell, R.S.; Lin, R.; Bedford, M.T.; Wainberg, M.A.; Richard, S. Methylation of Tat by PRMT6 regulates human immunodeficiency virus type 1 gene expression. J. Virol. 2005, 79, 124–131. [Google Scholar]
- Treand, C.; du Chene, I.; Bres, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S.; et al. Requirement for SWI/SNF chromatin–remodeling complex in Tat–mediated activation of the HIV–1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Wada, T.; Watanabe, D.; Takagi, T.; Hasegawa, J.; Handa, H. Structure and function of the human transcription elongation factor DSIF. J. Biol. Chem. 1999, 274, 8085–8092. [Google Scholar]
- Narita, T.; Yamaguchi, Y.; Yano, K.; Sugimoto, S.; Chanarat, S.; Wada, T.; Kim, D.–K.; Hasegawa, J.; Omori, M.; Inukai, N.; et al. Human transcription elongation factor NELF: identification of novel subunits and reconstitution of the functionally active complex. Mol. Cell. Biol. 2003, 23, 1863–1873. [Google Scholar] [CrossRef]
- Marciniak, R.A.; Calnan, B.J.; Frankel, A.D.; Sharp, P.A. HIV–1 Tat protein trans–activates transcription in vitro. Cell 1990, 63, 791–802. [Google Scholar] [CrossRef]
- Mancebo, H.S.; Lee, G.; Flygare, J.; Tomassini, J.; Luu, P.; Zhu, Y.; Peng, J.; Blau, C.; Hazuda, D.; Price, D.; et al. P–TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes. Dev. 1997, 11, 2633–2644. [Google Scholar] [CrossRef]
- Zhu, Y.; Pe'ery, T.; Peng, J.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendt, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P–TEFb is required for HIV–1 tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632. [Google Scholar] [CrossRef]
- Kim, H.; Erickson, B.; Luo, W.; Seward, D.; Graber, J.H.; Pollock, D.D.; Megee, P.C.; Bentley, D.L. Gene–specific RNA polymerase II phosphorylation and the CTD code. Nat. Struct. Mol. Biol. 2010, 17, 1279–1286. [Google Scholar] [CrossRef]
- Lenasi, T.; Peterlin, B.M.; Dovc, P. Distal regulation of alternative splicing by splicing enhancer in equine beta–casein intron 1. RNA 2006, 12, 498–507. [Google Scholar] [CrossRef]
- Gu, B.; Eick, D.; Bensaude, O. CTD serine–2 plays a critical role in splicing and termination factor recruitment to RNA polymerase II in vivo. Nucleic. Acids. Res. 2013, 41((3)), 1591–1603. [Google Scholar]
- Marshall, N.F.; Peng, J.; Xie, Z.; Price, D.H. Control of RNA polymerase II elongation potential by a novel carboxyl–terminal domain kinase. J. Biol. Chem. 1996, 271, 27176–27183. [Google Scholar] [CrossRef]
- Marshall, N.F.; Price, D.H. Purification of P–TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef]
- Marciniak, R.A.; Sharp, P.A. HIV–1 Tat protein promotes formation of more–processive elongation complexes. EMBO J. 1991, 10, 4189–4196. [Google Scholar]
- Yang, X.; Herrmann, C.H.; Rice, A.P. The human immunodeficiency virus Tat proteins specifically associate with TAK in vivo and require the carboxyl–terminal domain of RNA polymerase II for function. J. Virol. 1996, 70, 4576–4584. [Google Scholar]
- Gold, M.O.; Yang, X.; Herrmann, C.H.; Rice, A.P. PITALRE, the catalytic subunit of TAK, is required for human immunodeficiency virus Tat transactivation in vivo. J. Virol. 1998, 72, 4448–4453. [Google Scholar]
- Yang, X.; Gold, M.O.; Tang, D.N.; Lewis, D.E.; Aguilar–Cordova, E.; Rice, A.P.; Herrmann, C.H. TAK, an HIV Tat–associated kinase, is a member of the cyclin–dependent family of protein kinases and is induced by activation of peripheral blood lymphocytes and differentiation of promonocytic cell lines. Proc. Natl. Acad. Sci. U S A 1997, 94, 12331–12336. [Google Scholar]
- Herrmann, C.H.; Rice, A.P. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl–terminal domain of the large subunit of RNA polymerase II: candidate for a Tat cofactor. J. Virol. 1995, 69, 1612–1620. [Google Scholar]
- Kephart, D.D.; Marshall, N.F.; Price, D.H. Stability of Drosophila RNA polymerase II elongation complexes in vitro. Mol. Cell. Biol. 1992, 12, 2067–2077. [Google Scholar]
- Alonso, A.; Cujec, T.P.; Peterlin, B.M. Effects of human chromosome 12 on interactions between Tat and TAR of human immunodeficiency virus type 1. J. Virol. 1994, 68, 6505–6513. [Google Scholar]
- Alonso, A.; Derse, D.; Peterlin, B.M. Human chromosome 12 is required for optimal interactions between Tat and TAR of human immunodeficiency virus type 1 in rodent cells. J. Virol. 1992, 66, 4617–4621. [Google Scholar]
- Garber, M.E.; Wei, P.; KewalRamani, V.N.; Mayall, T.P.; Herrmann, C.H.; Rice, A.P.; Littman, D.R.; Jones, K.A. The interaction between HIV–1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998, 12, 3512–3527. [Google Scholar] [CrossRef]
- De Luca, A.; De Falco, M.; Baldi, A.; Paggi, M.G. Cyclin T: three forms for different roles in physiological and pathological functions. J. Cell. Physiol. 2003, 194, 101–107. [Google Scholar] [CrossRef]
- Sung, T.L.; Rice, A.P. Effects of prostratin on Cyclin T1/P–TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology 2006, 3, 66. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Yu, W.; Rice, A.P. Limited redundancy in genes regulated by Cyclin T2 and Cyclin T1. BMC Res. Notes 2011, 4, 260. [Google Scholar] [CrossRef]
- Kohoutek, J.; Li, Q.; Blazek, D.; Luo, Z.; Jiang, H.; Peterlin, B.M. Cyclin T2 is essential for mouse embryogenesis. Mol. Cell. Biol. 2009, 29, 3280–3285. [Google Scholar] [CrossRef]
- De Luca, A.; Tosolini, A.; Russo, P.; Severino, A.; Baldi, A.; et al. Cyclin T2a gene maps on human chromosome 2q21. J. Histochem. Cytochem. 2001, 49, 693–698. [Google Scholar] [CrossRef]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef]
- Kohoutek, J.; Blazek, D. Cyclin K goes with Cdk12 and Cdk13. Cell Div. 2012, 7, 12. [Google Scholar] [CrossRef]
- Blazek, D. The cyclin K/Cdk12 complex: an emerging new player in the maintenance of genome stability. Cell Cycle 2012, 11, 1049–1050. [Google Scholar] [CrossRef]
- Bartkowiak, B.; Liu, P.; Phatnani, H.P.; Fuda, N.J.; Cooper, J.J.; Price, D.H.; Adelman, K.; Lis, J.T.; Greenleaf, A.L. CDK12 is a transcription elongation–associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes. Dev. 2010, 24, 2303–2316. [Google Scholar] [CrossRef]
- Dai, Q.; Lei, T.; Zhao, C.; Zhong, J.; Tang, Y.Z.; Chen, B.; Yang, J.; Li, C.; Wang, S.; Song, X.; Li, L.; Li, Q. Cyclin K–containing kinase complexes maintain self–renewal in murine embryonic stem cells. J. Biol. Chem. 2012, 287, 25344–25352. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P–TEFb. Mol. Cell. 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Adelman, K.; Lis, J.T. Promoter–proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef]
- Core, L.J.; Lis, J.T. Transcription regulation through promoter–proximal pausing of RNA polymerase II. Science 2008, 319, 1791–1792. [Google Scholar] [CrossRef]
- Kanazawa, S.; Okamoto, T.; Peterlin, B.M. Tat competes with CIITA for the binding to P–TEFb and blocks the expression of MHC class II genes in HIV infection. Immunity 2000, 12, 61–70. [Google Scholar] [CrossRef]
- Kanazawa, S.; Soucek, L.; Evan, G.; Okamoto, T.; Peterlin, B.M. c–Myc recruits P–TEFb for transcription, cellular proliferation and apoptosis. Oncogene 2003, 22, 5707–5711. [Google Scholar] [CrossRef]
- Kornblihtt, A.R.; de la Mata, M.; Fededa, J.P.; Munoz, M.J.; Nogues, G. Multiple links between transcription and splicing. RNA 2004, 10, 1489–1498. [Google Scholar] [CrossRef]
- Glover–Cutter, K.; Larochelle, S.; Erickson, B.; Zhang, C.; Shokat, K.; Fisher, R.P.; Bentley, D.L. TFIIH–associated Cdk7 kinase functions in phosphorylation of C–terminal domain Ser7 residues, promoter–proximal pausing, and termination by RNA polymerase II. Mol. Cell. Biol. 2009, 29, 5455–5464. [Google Scholar] [CrossRef]
- Cho, E.J.; Takagi, T.; Moore, C.R.; Buratowski, S. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy–terminal domain. Genes Dev. 1997, 11, 3319–3326. [Google Scholar] [CrossRef]
- Wen, Y.; Shatkin, A.J. Transcription elongation factor hSPT5 stimulates mRNA capping. Genes Dev. 1999, 13, 1774–1779. [Google Scholar] [CrossRef]
- Ahn, S.H.; Kim, M.; Buratowski, S. Phosphorylation of serine 2 within the RNA polymerase II C–terminal domain couples transcription and 3' end processing. Mol. Cell. 2004, 13, 67–76. [Google Scholar] [CrossRef]
- Barboric, M.; Lenasi, T.; Chen, H.; Johansen, E.B.; Guo, S.; Peterlin, B.M. 7SK snRNP/P–TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc. Natl. Acad. Sci. U S A 2009, 106, 7798–7803. [Google Scholar]
- Pirngruber, J.; Shchebet, A.; Schreiber, L.; Shema, E.; Minsky, N.; Chapman, R.D.; Eick, D.; Aylon, Y.; Oren, M.; Johnsen, S.A. CDK9 directs H2B monoubiquitination and controls replication–dependent histone mRNA 3'–end processing. EMBO Rep. 2009, 10, 894–900. [Google Scholar] [CrossRef]
- Lenasi, T.; Peterlin, B.M.; Barboric, M. Cap–binding protein complex links pre–mRNA capping to transcription elongation and alternative splicing through positive transcription elongation factor b (P–TEFb). J. Biol. Chem. 2011, 286, 22758–22768. [Google Scholar]
- Bres, V.; Gomes, N.; Pickle, L.; Jones, K.A. A human splicing factor, SKIP, associates with P–TEFb and enhances transcription elongation by HIV–1 Tat. Genes Dev. 2005, 19, 1211–1226. [Google Scholar] [CrossRef]
- Berro, R.; Kehn, K.; de la Fuente, C.; Wade, J.; Colberg–Poley, A.M.; Berro, R. Acetylated Tat regulates human immunodeficiency virus type 1 splicing through its interaction with the splicing regulator p32. J. Virol. 2006, 80, 3189–3204. [Google Scholar] [CrossRef]
- Bohne, J.; Krausslich, H.G. Mutation of the major 5' splice site renders a CMV–driven HIV–1 proviral clone Tat–dependent: connections between transcription and splicing. FEBS Lett. 2004, 563, 113–118. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Coronel, E.; Ho, C.K.; Shuman, S.; Rana, T.M. HIV–1 Tat protein interacts with mammalian capping enzyme and stimulates capping of TAR RNA. J. Biol. Chem. 2001, 276, 12959–12966. [Google Scholar]
- Chiu, Y.L.; Ho, C.K.; Saha, N.; Schwer, B.; Shuman, S.; Rana, T.M. Tat stimulates cotranscriptional capping of HIV mRNA. Mol. Cell. 2002, 10, 585–597. [Google Scholar] [CrossRef]
- Tahirov, T.H.; Babayeva, N.D.; Varzavand, K.; Cooper, J.J.; Sedore, S.C.; Price, D.H. Crystal structure of HIV–1 Tat complexed with human P–TEFb. Nature 2010, 465, 747–751. [Google Scholar] [CrossRef]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; et al. Identification of a reservoir for HIV–1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV–1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar]
- Bintu, L.; Ishibashi, T.; Dangkulwanich, M.; Wu, Y.Y.; Lubkowska, L.; Kashlev, M.; Bustamante, C. Nucleosomal elements that control the topography of the barrier to transcription. Cell 151, 2012, 738–749. [Google Scholar]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV–1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar]
- Mbonye, U.; Karn, J. Control of HIV latency by epigenetic and non–epigenetic mechanisms. Curr. HIV Res. 2011, 9, 554–567. [Google Scholar] [CrossRef]
- Coiras, M.; Lopez–Huertas, M.R.; Perez–Olmeda, M.; Alcami, J. Understanding HIV–1 latency provides clues for the eradication of long–term reservoirs. Nat. Rev. Microbiol. 2009, 7, 798–812. [Google Scholar] [CrossRef]
- du Chene, I.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; et al. Suv39H1 and HP1gamma are responsible for chromatin–mediated HIV–1 transcriptional silencing and post–integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin–modifying enzymes by CTIP2 promotes HIV–1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV–1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef]
- Keedy, K.S.; Archin, N.M.; Gates, A.T.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J. Virol. 2009, 83, 4749–4756. [Google Scholar] [CrossRef]
- Margolis, D.M.; Somasundaran, M.; Green, M.R. Human transcription factor YY1 represses human immunodeficiency virus type 1 transcription and virion production. J. Virol. 1994, 68, 905–910. [Google Scholar]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P–TEFb restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef]
- Tyagi, M.; Karn, J. CBF–1 promotes transcriptional silencing during the establishment of HIV–1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV–1 latency by cytosine methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef]
- Williams, S.A.; Kwon, H.; Chen, L.F.; Greene, W.C. Sustained induction of NF–kappa B is required for efficient expression of latent human immunodeficiency virus type 1. J. Virol. 2007, 81, 6043–6056. [Google Scholar] [CrossRef]
- Bosque, A.; Planelles, V. Induction of HIV–1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T cell activation factor NF–ATc positively regulates HIV–1 replication and gene expression in T cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef]
- Bednarik, D.P.; Cook, J.A.; Pitha, P.M. Inactivation of the HIV LTR by DNA CpG methylation: evidence for a role in latency. EMBO J. 1990, 9, 1157–1164. [Google Scholar]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef]
- Han, Y.; Lassen, K.; Monie, D.; Sedaghat, A.R.; Shimoji, S.; et al. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV–1)–infected individuals carry integrated HIV–1 genomes within actively transcribed host genes. J. Virol. 2004, 78, 6122–6133. [Google Scholar] [CrossRef]
- Shan, L.; Yang, H.C.; Rabi, S.A.; Bravo, H.C.; Shroff, N.S.; Irizarry, R.A.; Zhang, H.; Margolick, J.B.; Siliciano, J.D.; Siliciano, R.F. Influence of host gene transcription level and orientation on HIV–1 latency in a primary–cell model. J. Virol. 2011, 85, 5384–5393. [Google Scholar] [CrossRef]
- Michel, F.; Crucifix, C.; Granger, F.; Eiler, S.; Mouscadet, J.F.; et al. Structural basis for HIV–1 DNA integration in the human genome, role of the LEDGF/P75 cofactor. EMBO J. 2009, 28, 980–991. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV–1 integration in the human genome favors active genes and local hotspots. Cell 110, 2002, 521–529. [Google Scholar]
- Jordan, A.; Defechereux, P.; Verdin, E. The site of HIV–1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 2001, 20, 1726–1738. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Bisgrove, D.; Shinn, P.; Chen, H.; Hoffmann, C.; Hannenhalli, S.; Verdin, E.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Genome–wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 2005, 79, 6610–6619. [Google Scholar] [CrossRef]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome–wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring. Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar]
- Chan, J.K.; Greene, W.C. NF–kappaB/Rel: agonist and antagonist roles in HIV–1 latency. Curr. Opin. HIV AIDS 2011, 6, 12–18. [Google Scholar] [CrossRef]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.C.; O'Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, RF. Orientation–dependent regulation of integrated HIV–1 expression by host gene transcriptional readthrough. Cell. Host. Microbe. 2008, 4, 134–146. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Brogie, J.E.; Price, D.H. 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip. Rev. RNA 2012, 3((1)), 92–103. [Google Scholar] [CrossRef]
- Cho, S.; Schroeder, S.; Kaehlcke, K.; Kwon, H.S.; Pedal, A.; Herker, E.; Schnoelzer, M.; Ott, M. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P–TEFb in cells. EMBO J. 2009, 28, 1407–1417. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, T.; Price, D.H. RNA Polymerase II Elongation Control. Annu. Rev. Biochem. 2012, 81, 119–143. [Google Scholar] [CrossRef]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U S A 2003, 100, 8758–8763. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P–TEFb–interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 13690–13695. [Google Scholar]
- Chen, R.; Liu, M.; Li, H.; Xue, Y.; Ramey, W.N.; et al. PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P–TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008, 22, 1356–1368. [Google Scholar] [CrossRef]
- Chiang, C.M. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol. Rep. 2009, 1, 98. [Google Scholar]
- Ai, N.; Hu, X.; Ding, F.; Yu, B.; Wang, H.; et al. Signal–induced Brd4 release from chromatin is essential for its role transition from chromatin targeting to transcriptional regulation. Nucleic. Acids Res. 2011, 39, 9592–9604. [Google Scholar] [CrossRef]
- Schroder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; et al. Two–pronged binding with bromodomain–containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar]
- Takahashi, H.; Parmely, T.J.; Sato, S.; Tomomori–Sato, C.; Banks, C.A.; et al. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 2011, 146, 92–104. [Google Scholar] [CrossRef]
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene bookmarking accelerates the kinetics of post–mitotic transcriptional re–activation. Nat. Cell. Biol. 2011, 13, 1295–1304. [Google Scholar] [CrossRef]
- Casse, C.; Giannoni, F.; Nguyen, V.T.; Dubois, M.F.; Bensaude, O. The transcriptional inhibitors, actinomycin D and alpha–amanitin, activate the HIV–1 promoter and favor phosphorylation of the RNA polymerase II C–terminal domain. J. Biol. Chem. 1999, 274, 16097–16106. [Google Scholar]
- Michels, A.A.; Fraldi, A.; Li, Q.; Adamson, T.E.; Bonnet, F.; et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P–TEFb (CDK9/cyclin T) inhibitor. E. EMBO J. 2004, 23, 2608–2619. [Google Scholar] [CrossRef]
- Michels, A.A.; Nguyen, V.T.; Fraldi, A.; Labas, V.; Edwards, M.; Bonnet, F.; Lania, L.; Bensaude, O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription–dependent manner. Mol. Cell. Biol. 2003, 23, 4859–4869. [Google Scholar]
- Yik, J.H.; Chen, R.; Pezda, A.C.; Samford, C.S.; Zhou, Q. A human immunodeficiency virus type 1 Tat–like arginine–rich RNA–binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA–mediated inactivation of P–TEFb. Mol. Cell. Biol. 2004, 24, 5094–5105. [Google Scholar] [CrossRef]
- Budhiraja, S.; Ramakrishnan, R.; Rice, A.P. (2012) Phosphatase PPM1A negatively regulates P–TEFb function in resting CD4T+ T cells and inhibits HIV–1 gene expression. Retrovirology 52, 9. [Google Scholar]
- Chiang, K.; Sung, T.L.; Rice, A.P. Regulation of cyclin T1 and HIV–1 Replication by microRNAs in resting CD4+ T lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Dow, E.C.; Rice, A.P. Characterization of Cdk9 T–loop phosphorylation in resting and activated CD4(+) T lymphocytes. J. Leukoc. Biol. 2009, 86, 1345–1350. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Rice, A.P. Cdk9 T–loop phosphorylation is regulated by the calcium signaling pathway. J. Cell. Physiol. 2012, 227, 609–617. [Google Scholar] [CrossRef]
- Ghose, R.; Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Induction of TAK (cyclin T1/P–TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J. Virol. 2001, 75, 11336–11343. [Google Scholar] [CrossRef]
- Liou, L.Y.; Herrmann, C.H.; Rice, A.P. Human immunodeficiency virus type 1 infection induces cyclin T1 expression in macrophages. J. Virol. 2004, 78, 8114–8119. [Google Scholar] [CrossRef]
- Liou, L.Y.; Herrmann, C.H.; Rice, A.P. HIV–1 infection and regulation of Tat function in macrophages. Int. J. Biochem. Cell. Biol. 2004, 36, 1767–1775. [Google Scholar] [CrossRef]
- Sung, T.L.; Rice, A.P. miR–198 inhibits HIV–1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef]
- Yu, W.; Ramakrishnan, R.; Wang, Y.; Chiang, K.; Sung, T.L.; Rice, A.P. Cyclin T1–dependent genes in activated CD4 T and macrophage cell lines appear enriched in HIV–1 co–factors. PLoS One 2008, 3, e3146. [Google Scholar]
- Hoque, M.; Shamanna, R.A.; Guan, D.; Pe'ery, T.; Mathews, M.B. HIV–1 replication and latency are regulated by translational control of cyclin T1. J. Mol. Biol. 2011, 410, 917–932. [Google Scholar] [CrossRef]
- Fong, Y.W.; Zhou, Q. Relief of two built–In autoinhibitory mechanisms in P–TEFb is required for assembly of a multicomponent transcription elongation complex at the human immunodeficiency virus type 1 promoter. Mol. Cell. Biol. 2000, 20, 5897–5907. [Google Scholar] [CrossRef]
- Garber, M.E.; Mayall, T.P.; Suess, E.M.; Meisenhelder, J.; Thompson, N.E.; Jones, K.A. CDK9 autophosphorylation regulates high–affinity binding of the human immunodeficiency virus type 1 tat–P–TEFb complex to TAR RNA. Mol. Cell. Biol. 2000, 20, 6958–6969. [Google Scholar]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.E.; Knapp, S.; Johnson, L.N. The structure of P–TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, D.; Pierstorff, E.; Luo, K. Transcription elongation factor P–TEFb mediates Tat activation of HIV–1 transcription at multiple stages. EMBO J. 1998, 17, 3681–3691. [Google Scholar] [CrossRef]
- Vollmuth, F.; Blankenfeldt, W.; Geyer, M. Structures of the dual bromodomains of the P–TEFb–activating protein Brd4 at atomic resolution. J. Biol. Chem. 2009, 284, 36547–36556. [Google Scholar] [CrossRef]
- Liu, H.; Rice, A.P. Genomic organization and characterization of promoter function of the human CDK9 gene. Gene 2000, 252, 51–59. [Google Scholar] [CrossRef]
- Bagella, L.; Stiegler, P.; De Luca, A.; Siracusa, L.D.; Giordano, A. Genomic organization, promoter analysis, and chromosomal mapping of the mouse gene encoding Cdk9. J. Cell. Biochem. 2000, 78, 170–178. [Google Scholar]
- Herrmann, C.H.; Mancini, M.A. The Cdk9 and cyclin T subunits of TAK/P–TEFb localize to splicing factor–rich nuclear speckle regions. J. Cell. Sci. 2001, 114, 1491–1503. [Google Scholar]
- Pendergrast, P.S.; Wang, C.; Hernandez, N.; Huang, S. FBI–1 can stimulate HIV–1 Tat activity and is targeted to a novel subnuclear domain that includes the Tat–P–TEFb–containing nuclear speckles. Mol. Biol. Cell. 2002, 13, 915–929. [Google Scholar] [CrossRef]
- Liu, H.; Herrmann, C.H. Differential localization and expression of the Cdk9 42k and 55k isoforms. J. Cell. Physiol. 2005, 203, 251–260. [Google Scholar] [CrossRef]
- Chen, R.; Yang, Z.; Zhou, Q. Phosphorylated positive transcription elongation factor b (P–TEFb) is tagged for inhibition through association with 7SK snRNA. J. Biol. Chem. 2004, 279, 4153–4160. [Google Scholar] [CrossRef]
- Barboric, M.; Zhang, F.; Besenicar, M.; Plemenitas, A.; Peterlin, B.M. Ubiquitylation of Cdk9 by Skp2 facilitates optimal Tat transactivation. J. Virol. 2005, 79, 11135–11141. [Google Scholar] [CrossRef]
- Sabo, A.; Lusic, M.; Cereseto, A.; Giacca, M. Acetylation of conserved lysines in the catalytic core of cyclin–dependent kinase 9 inhibits kinase activity and regulates transcription. Mol. Cell. Biol. 2008, 28, 2201–2212. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P–TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Li, Q.; Price, J.P.; Byers, S.A.; Cheng, D.; Peng, J.; Price, D.H. Analysis of the large inactive P–TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P–TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 2005, 280, 28819–28826. [Google Scholar]
- Russo, A.A.; Jeffrey, P.D.; Pavletich, N.P. Structural basis of cyclin–dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996, 3, 696–700. [Google Scholar] [CrossRef]
- Zhou, M.; Nekhai, S.; Bharucha, D.C.; Kumar, A.; Ge, H.; Price, D.H.; Egly, J.M.; Brady, J.N. TFIIH inhibits CDK9 phosphorylation during human immunodeficiency virus type 1 transcription. J. Biol. Chem. 2001, 276, 44633–44640. [Google Scholar]
- Breuer, D.; Kotelkin, A.; Ammosova, T.; Kumari, N.; Ivanov, A.; et al. CDK2 regulates HIV–1 transcription by phosphorylation of CDK9 on serine 90. Retrovirology 2012, 9, 94. [Google Scholar] [CrossRef]
- Larochelle, S.; Amat, R.; Glover–Cutter, K.; Sansó, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin–dependent kinase control of the initiation–to–elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef]
- Ammosova, T.; Obukhov, Y.; Kotelkin, A.; Breuer, D.; Beullens, M.; Gordeuk, V.R.; Bollen, M. Nekhai, S. Protein phosphatase–1 activates CDK9 by dephosphorylating Ser175. PLoS One 2011, 6, e18985. [Google Scholar]
- Ammosova, T.; Yedavalli, V.R.; Niu, X.; Jerebtsova, M.; Van Eynde, A.; Beullens, M.; Bollen, M.; Jeang, K.T.; Nekhai, S. Expression of a protein phosphatase 1 inhibitor, cdNIPP1, increases CDK9 threonine 186 phosphorylation and inhibits HIV–1 transcription. J. Biol. Chem. 2011, 286, 3798–3804. [Google Scholar] [CrossRef]
- Wang, Y.; Dow, E.C.; Liang, Y.Y.; Ramakrishnan, R.; Liu, H.; Sung, T.L.; Lin, X.; Rice, A.P. Phosphatase PPM1A regulates phosphorylation of Thr–186 in the Cdk9 T–loop. J. Biol. Chem. 2008, 283, 33578–33584. [Google Scholar]
- Kiernan, R.E.; Emiliani, S.; Nakayama, K.; Castro, A.; Labbé, J.C.; Lorca, T.; Nakayama, Ki, K.; Benkirane, M. Interaction between cyclin T1 and SCF(SKP2) targets CDK9 for ubiquitination and degradation by the proteasome. Mol. Cell. Biol. 2001, 21, 7956–7970. [Google Scholar] [CrossRef]
- Fu, J.; Yoon, H.G.; Qin, J.; Wong, J. Regulation of P–TEFb elongation complex activity by CDK9 acetylation. Mol. Cell. Biol. 2007, 27, 4641–4651. [Google Scholar] [CrossRef]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV–1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell. 2010, 38, 439–451. [Google Scholar] [CrossRef]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV–1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV–1 transcription. Mol. Cell. 2010, 38, 428–438. [Google Scholar] [CrossRef]
- Yokoyama, A.; Lin, M.; Naresh, A.; Kitabayashi, I.; Cleary, M.L. A higher–order complex containing AF4 and ENL family proteins with P–TEFb facilitates oncogenic and physiologic MLL–dependent transcription. Cancer Cell 2010, 17, 198–212. [Google Scholar] [CrossRef]
- Shilatifard, A.; Conaway, R.C.; Conaway, J.W. The RNA polymerase II elongation complex. Annu. Rev. Biochem. 2003, 72, 693–715. [Google Scholar] [CrossRef]
- Shilatifard, A.; Lane, W.S.; Jackson, K.W.; Conaway, R.C.; Conaway, J.W. An RNA polymerase II elongation factor encoded by the human ELL gene. Science 1996, 271, 1873–1876. [Google Scholar]
- Shilatifard, A.; Duan, D.R.; Haque, D.; Florence, C.; Schubach, W.H.; Conaway, J.W.; Conaway, R.C. ELL2, a new member of an ELL family of RNA polymerase II elongation factors. Proc. Natl. Acad. Sci. U S A 1997, 94, 3639–3643. [Google Scholar]
- He, N.; Chan, C.K.; Sobhian, B.; Chou, S.; Xue, Y.; Liu, M.; Alber, T.; Benkirane, M.; Zhou, Q. Human Polymerase–Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc. Natl. Acad. Sci. U S A 2011, 108, E636–E645. [Google Scholar]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV–1–infected T cells: quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef]
- Finzi, L.; Gelles, J. Measurement of lactose repressor–mediated loop formation and breakdown in single DNA molecules. Science 1995, 267, 378–380. [Google Scholar] [CrossRef]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication–competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Chun, T.W.; Engel, D.; Mizell, S.B.; Hallahan, C.W.; Fischette, M.; et al. Effect of interleukin–2 on the pool of latently infected, resting CD4+ T cells in HIV–1–infected patients receiving highly active anti–retroviral therapy. Nat. Med. 1999, 5, 651–655. [Google Scholar] [CrossRef]
- Stellbrink, H.J.; van Lunzen, J.; Westby, M.; O'Sullivan, E.; Schneider, C.; et al. Effects of interleukin–2 plus highly active antiretroviral therapy on HIV–1 replication and proviral DNA (COSMIC trial). AIDS 2002, 16, 1479–1487. [Google Scholar] [CrossRef]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; et al. The challenge of finding a cure for HIV infection. Science 2009, 323, 1304–1307. [Google Scholar] [CrossRef]
- Scripture–Adams, D.D.; Brooks, D.G.; Korin, Y.D.; Zack, J.A. Interleukin–7 induces expression of latent human immunodeficiency virus type 1 with minimal effects on T–cell phenotype. J. Virol. 2002, 76, 13077–13082. [Google Scholar] [CrossRef]
- Wang, F.X.; Xu, Y.; Sullivan, J.; Souder, E.; Argyris, E.G.; et al. IL–7 is a potent and proviral strain–specific inducer of latent HIV–1 cellular reservoirs of infected individuals on virally suppressive HAART. J. Clin. Invest. 2005, 115, 128–137. [Google Scholar]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Fenard, D.; Bisgrove, D.; Verdin, E.; Greene, W.C. Prostratin antagonizes HIV latency by activating NF–kappaB. J. Biol. Chem. 2004, 279, 42008–42017. [Google Scholar]
- Kulkosky, J.; Culnan, D.M.; Roman, J.; Dornadula, G.; Schnell, M.; Boyd, M.R.; Pomerantz, R.J. Prostratin: activation of latent HIV–1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 2001, 98, 3006–3015. [Google Scholar] [CrossRef]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis–Cremer, N. Disulfiram reactivates latent HIV–1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef]
- Xing, S.; Bullen, C.K.; Shroff, N.S.; Shan, L.; Yang, H.C.; et al. Disulfiram reactivates latent HIV–1 in a Bcl–2–transduced primary CD4+ T cell model without inducing global T cell activation. J. Virol. 2011, 85, 6060–6064. [Google Scholar]
- Siegel, D.S.; Zhang, X.; Feinman, R.; Teitz, T.; Zelenetz, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Michaeli, J. Hexamethylene bisacetamide induces programmed cell death (apoptosis) and down–regulates BCL–2 expression in human myeloma cells. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 162–166. [Google Scholar] [CrossRef]
- Richon, V.M.; Webb, Y.; Merger, R.; Sheppard, T.; Jursic, B.; Ngo, L.; Civoli, F.; Breslow, R.; Rifkind, R.A.; Marks, P.A. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. U S A 1996, 93, 5705–5708. [Google Scholar] [CrossRef]
- Choudhary, S.K.; Archin, N.M.; Margolis, D.M. Hexamethylbisacetamide and disruption of human immunodeficiency virus type 1 latency in CD4(+) T cells. J. Infect. Dis. 2008, 197, 1162–1170. [Google Scholar] [CrossRef]
- Contreras, X.M; Barboric, M.; Lenasi, T.; Peterlin, B.M. HMBA releases P–TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 2007, 3, 1459–1469. [Google Scholar]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; Spina, C.A.; et al. Depletion of latent HIV–1 infection in vivo: a proof–of–concept study. Lancet 2005, 366, 549–555. [Google Scholar]
- Siliciano, J.D.; Lai, J.; Callender, M.; Pitt, E.; Zhang, H.; et al. Stability of the latent reservoir for HIV–1 in patients receiving valproic acid. J. Infect. Dis. 2007, 195, 833–836. [Google Scholar] [CrossRef]
- Ylisastigui, L.; Archin, N.M.; Lehrman, G.; Bosch, R.J.; Margolis, D.M. Coaxing HIV–1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS 2004, 18, 1101–1108. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long–term follow–up studies confirm the stability of the latent reservoir for HIV–1 in resting CD4+ T cells. Nat. Med 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Sagot–Lerolle, N.; Lamine, A.; Chaix, M.L.; Boufassa, F.; Aboulker, J.P.; Costagliola, D.; Goujard, C.; Pallier, C.; Delfraissy, J.F.; Lambotte, O. Prolonged valproic acid treatment does not reduce the size of latent HIV reservoir. AIDS 2008, 22, 1125–1129. [Google Scholar] [CrossRef]
- Contreras, X.; Schweneker, M.; Chen, C.S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; et al. Administration of vorinostat disrupts HIV–1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; et al. Stimulation of HIV–1–specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; et al. BET bromodomain–targeting compounds reactivate HIV from latency via a Tat–independent mechanism. Cell Cycle 2012, 12((3)), 452–462. [Google Scholar]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat–transactivation. Nucleic. Acids. Res. 2012, 41(1) , 277–287. [Google Scholar]
- Lassen, K.G.; Hebbeler, A.M.; Bhattacharyya, D.; Lobritz, M.A.; Greene, W.C. A flexible model of HIV–1 latency permitting evaluation of many primary CD4 T–cell reservoirs. PLoS One 2012, 7, e30176. [Google Scholar]
- Sahu, G.K.; Cloyd, M.W. Latent HIV in primary T lymphocytes is unresponsive to histone deacetylase inhibitors. Virol. J. 2011, 8, 400. [Google Scholar] [CrossRef]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; et al. Long–term control of HIV by CCR5 Delta32/Delta32 stem–cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Taube, R.; Peterlin, M. Lost in Transcription: Molecular Mechanisms that Control HIV Latency. Viruses 2013, 5, 902-927. https://doi.org/10.3390/v5030902
Taube R, Peterlin M. Lost in Transcription: Molecular Mechanisms that Control HIV Latency. Viruses. 2013; 5(3):902-927. https://doi.org/10.3390/v5030902
Chicago/Turabian StyleTaube, Ran, and Matija Peterlin. 2013. "Lost in Transcription: Molecular Mechanisms that Control HIV Latency" Viruses 5, no. 3: 902-927. https://doi.org/10.3390/v5030902
APA StyleTaube, R., & Peterlin, M. (2013). Lost in Transcription: Molecular Mechanisms that Control HIV Latency. Viruses, 5(3), 902-927. https://doi.org/10.3390/v5030902