1. Introduction
DNA sequencing is a routine procedure in many wet laboratories. This sequencing may include direct, Sanger sequencing, or any of the many “next generation” sequencing technologies. Many sequence assembly software programs are available [
1,
2,
3,
4,
5,
6]. Most of these programs assemble short reads of next-generation sequencing data, with a small number assembling a query sequence against a reference sequence (mapping assembly). Some of these programs are only available commercially, and some are only available on specific operating system platforms. Comprehensive, integrated bioinformatics software solutions, which may also include such functionality, are typically very expensive and their usage is restricted to licensed workstations only. Such software suites are often complex, requiring training and high levels of computer proficiency to operate effectively. In addition, installing and using many of the available programs can be difficult, may require technical expertise, access to a specific operating system platform or expensive hardware. In resource-limited settings, where capacity and finances are generally limited, the purchase and use of such software is not possible. To address some of these issues, we have developed an online, web-based, sequence assembly tool to easily assemble overlapping long PCR amplicons as sequenced by direct, Sanger sequencing technology. A web-based tool requires no installation and can be used via any web-browser from any operating system platform. No specialist technical skills are required to use the tool, which was developed and tested extensively using hepatitis B virus (HBV) sequence data.
HBV has a partially double-stranded circular DNA genome, which, depending on genotype, ranges in length from 3182 to 3248 nucleotides. By convention, the
EcoRI restriction enzyme cleavage site (G
|AATTC), located within the surface gene, is denoted as nucleotide position 1. The genome contains four partially overlapping reading frames and codes for seven proteins. Nine distinct HBV genotypes are known, with up to 32 subgenotypes described to date [
7,
8]. Sequence heterogeneity is common in HBV, as the viral polymerase lacks proof-reading ability [
9,
10]. Furthermore, variant strains, which exhibit insertions, deletions or SNPs, are commonly encountered and reported. In addition, recombination of large or small regions between two or more variants has also been reported [
11].
Sequence data from either the surface (S) gene (approximately 1200 nucleotides) or the entire genome are essential to determine the viral genotype or subgenotype, to characterize the virus, and to identify indels and SNPs. Such data are routinely obtained by single or nested PCR amplification of the regions in question [
12,
13], followed by direct sequencing (typically in the forward direction only) using a number of internal sequencing primers. The resulting amplicons are typically between 500 and 800 nucleotides in length. The HBV genome is circular, but the resulting sequence is a linear fragment. Assembling these fragments into a complete gene or genome has previously been undertaken manually. The chromatogram (trace) file for each fragment is viewed [
14,
15] and checked. The poor quality ends are trimmed manually. Sequence data for each fragment is imported into an editor, such as GeneDoc [
16], and each fragment is slid until it overlaps with another fragment. The complete sequence is then constructed by either entering the sequence of bases or by editing a reference sequence. This process is extremely time-consuming, repetitive and error-prone.
We describe here the implementation of a genome-agnostic, web-based, assembly tool, which has been developed using HBV sequence data.
3. Results and Discussion
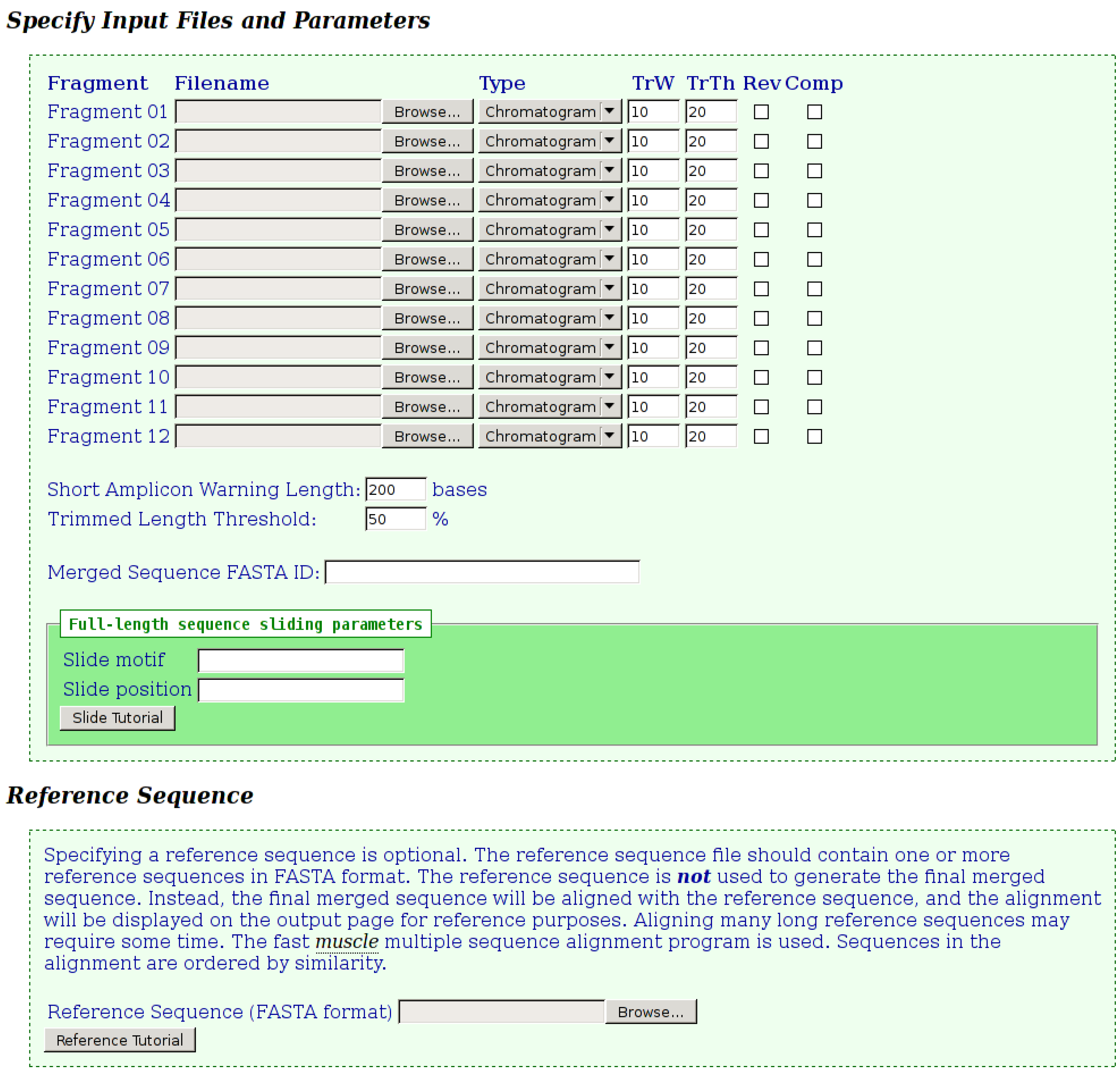
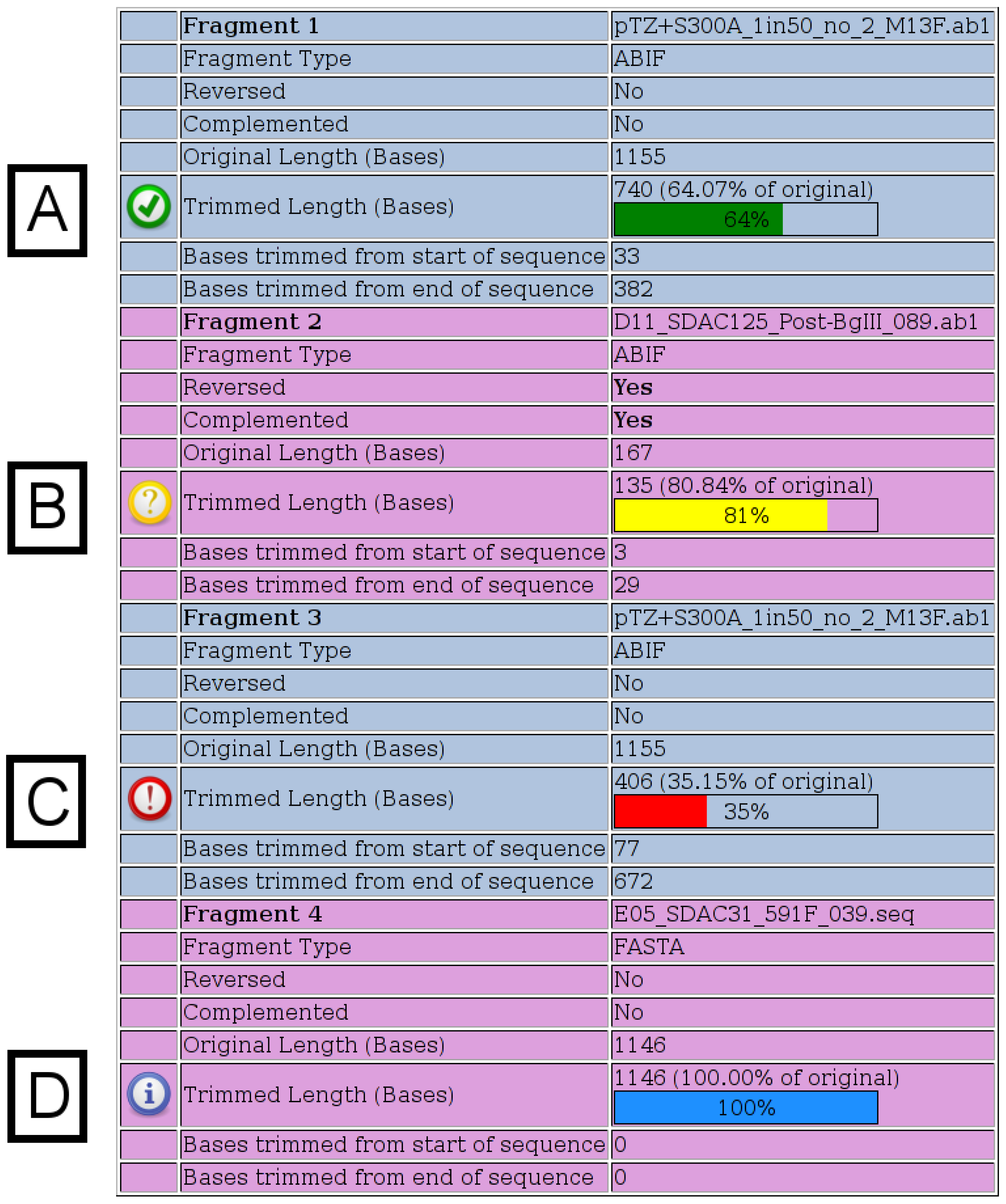
Once the merges have run, detailed output is provided to the user as shown in
Figure 2,
Figure 3,
Figure 4. The first section of output (
Figure 2) displays details for each fragment submitted. When chromatograms are submitted, the number of bases, which were trimmed from each end, is shown, whereas when FASTA files are submitted, no trimming is performed and a value of “100%” is shown. The various possible notification icons are described in
Figure 2. Data from fragments flagged with a yellow or red icon are not excluded from the merge, but the user should check the final merged sequence carefully.
The additional sections of output are shown in
Figure 3,
Figure 4. In
Figure 3A, the final merged (assembled) sequence is shown and can be downloaded as a file in FASTA format. The length is displayed, as well as any sliding parameters, which were provided. Detailed output of each of the successive merges, as generated by the
merger program, is provided in a table (
Figure 3B). If a reference sequence file was specified, the merged sequence aligned against the reference sequence/s is displayed (
Figure 3C). The alignment is intended to be used as a quick check to validate the success and/or accuracy of the merge. If no merge is possible (because of insufficient areas of overlap between two sequences), the
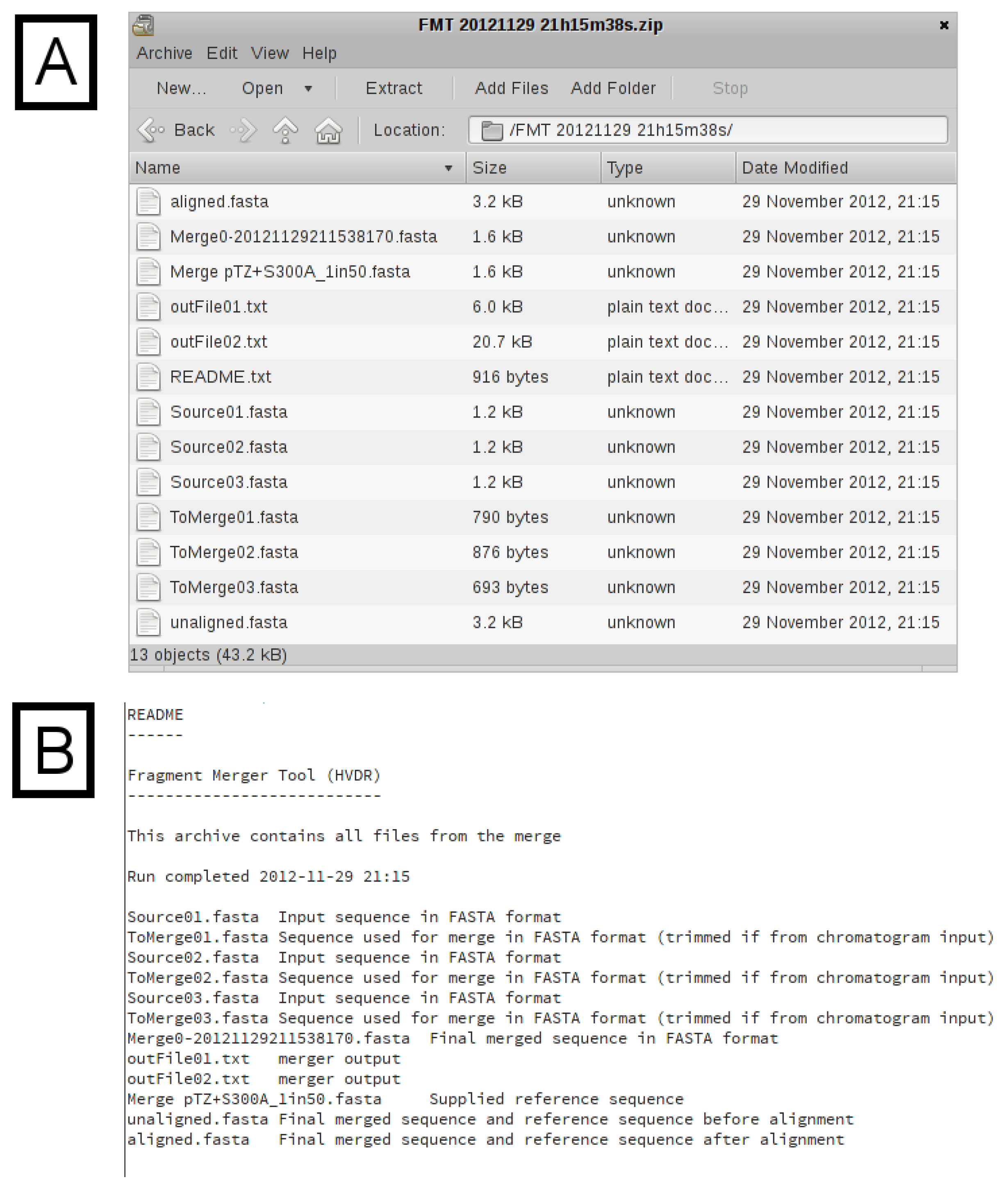
merger program will simply concatenate the two input sequences, with only one overlapping nucleotide. It is therefore important that the final sequence is checked carefully, preferably against known reference sequences. If the reference sequence file contains several sequences, additional time will be required to generate the alignment. Typically, one or two reference sequences should be sufficient. A download hyperlink to a ZIP archive file containing all input and output files (excluding chromatograms) is provided. The archive (
Figure 4A) contains the untrimmed input sequence data, the trimmed input sequence data (files starting with the name “ToMerge”), the final merged sequence (a file starting with the name “Merge0”), the reference sequence file, the unaligned merged and reference sequences, the aligned merged and reference sequences, the output text files from the
merger program for each merge (files starting with the name “outFile”) and a “README” text file describing each of the files, for reference (
Figure 4B).
The time taken for the Python CGI script to execute was calculated by subtracting the timestamp when the script completed from the timestamp when the script started. The average execution time, from 195 runs of the tool over several months, merging three overlapping fragments from chromatogram input data, was 0.48±0.12 seconds.
The tool has been used extensively to assemble the complete surface gene of HBV from three overlapping fragments. Although the HBV genome is circular, sequence data (either from direct
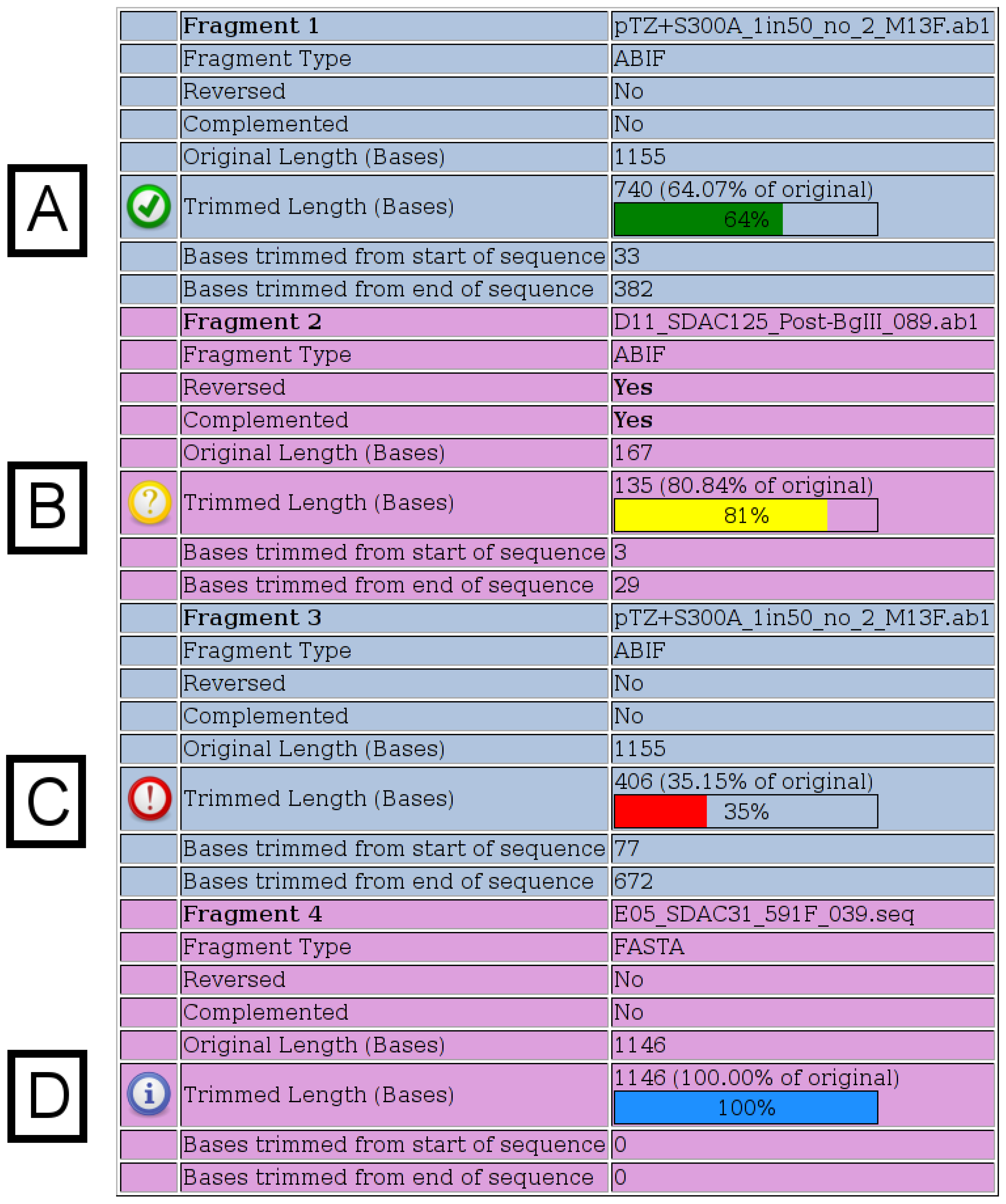
Figure 2.
Fragment Details. Details for each fragment submitted are shown on the output page. All possible notification icons are shown here. (A) A green icon indicates that the chromatogram has been trimmed, but is not shorter than any of the specified thresholds. (B) A yellow icon indicates that the trimmed chromatogram is shorter than the specified warning length, which has a default value of 200. (C) A red icon indicates that the trimmed chromatogram is shorter than the specified percentage of its original length, which has a default value of 50%. (D) A blue icon indicates that a FASTA file was specified, in which case, no trimming is performed.
Figure 2.
Fragment Details. Details for each fragment submitted are shown on the output page. All possible notification icons are shown here. (A) A green icon indicates that the chromatogram has been trimmed, but is not shorter than any of the specified thresholds. (B) A yellow icon indicates that the trimmed chromatogram is shorter than the specified warning length, which has a default value of 200. (C) A red icon indicates that the trimmed chromatogram is shorter than the specified percentage of its original length, which has a default value of 50%. (D) A blue icon indicates that a FASTA file was specified, in which case, no trimming is performed.
sequencing or from sequencing of clones) is linear. The tool can therefore be used with both circular and linear sequences. The tool has also been used to assemble entire HBV genomes from 6 or 7 overlapping fragments. In this case, fragments in both the forward and reverse direction were used.
Figure 3.
Additional Output. The additional sections of the output page are shown. (A) The final merged sequence in FASTA format is displayed and can be downloaded. (B) The detailed output of each of the successive merges is provided in a table. The alignment of the two fragments for each merge, and their score (as generated by the merger program), are provided. Since the fragments are merged in order sequentially, the expectation is that the end of the first fragment in each merge will overlap with the start of the next fragment. If this is the case, a green table cell with the word “Correct” is shown under the “Orientation” column. If the merge has occurred in the other orientation, the cell is shaded red with the word “Check” displayed. The final column provides a hyperlink to the full, detailed output for each merge, as generated by the merger program. This output includes a table detailing any conflicts, which the merger program detected, between the two sequences, and the base, which was used in the output sequence. It is advisable to check this detailed output before continuing to use the final merged sequence in any downstream applications or analyses. (C) The alignment of the merged sequence against the reference sequence(s) is shown. Conserved loci across all sequences are indicated with a “|” character, mismatches are indicated with a space character, and the total number of matches and mismatches is shown. (D) An archive containing all input and output files (excluding chromatograms) can be downloaded. All the files in the archive are in a folder named with the date and time the merge was executed.
Figure 3.
Additional Output. The additional sections of the output page are shown. (A) The final merged sequence in FASTA format is displayed and can be downloaded. (B) The detailed output of each of the successive merges is provided in a table. The alignment of the two fragments for each merge, and their score (as generated by the merger program), are provided. Since the fragments are merged in order sequentially, the expectation is that the end of the first fragment in each merge will overlap with the start of the next fragment. If this is the case, a green table cell with the word “Correct” is shown under the “Orientation” column. If the merge has occurred in the other orientation, the cell is shaded red with the word “Check” displayed. The final column provides a hyperlink to the full, detailed output for each merge, as generated by the merger program. This output includes a table detailing any conflicts, which the merger program detected, between the two sequences, and the base, which was used in the output sequence. It is advisable to check this detailed output before continuing to use the final merged sequence in any downstream applications or analyses. (C) The alignment of the merged sequence against the reference sequence(s) is shown. Conserved loci across all sequences are indicated with a “|” character, mismatches are indicated with a space character, and the total number of matches and mismatches is shown. (D) An archive containing all input and output files (excluding chromatograms) can be downloaded. All the files in the archive are in a folder named with the date and time the merge was executed.
![Viruses 05 00824 g003]()
The tool has been designed to assist users in processing sequence data and requires that the user exercise discretion when submitting data and interpreting the results. Poor quality data in chromatograms
Figure 4.
Archive Contents. (A) A list of the files in the archive. (B) The “README.txt” file from the downloaded archive. This file provides the filename and a description for all of the files in the archive.
Figure 4.
Archive Contents. (A) A list of the files in the archive. (B) The “README.txt” file from the downloaded archive. This file provides the filename and a description for all of the files in the archive.
could result in false indels in the final sequence. The tool does not disambiguate any ambiguous bases in chromatogram data, nor does it search for, or remove, any vector-specific or other primer sequences. If this is required, a FASTA file of the edited data, without the primer sequences, should be submitted to the tool. This is a not a mapping assembler and does not make use of a reference sequence for assembly.

{kind=link}
{kind=link}
{kind=link}
{kind=link}