Pathogenic Mechanisms Involved in the Hematological Alterations of Arenavirus-induced Hemorrhagic Fevers

Abstract
:1. Introduction
2. Hemostasis

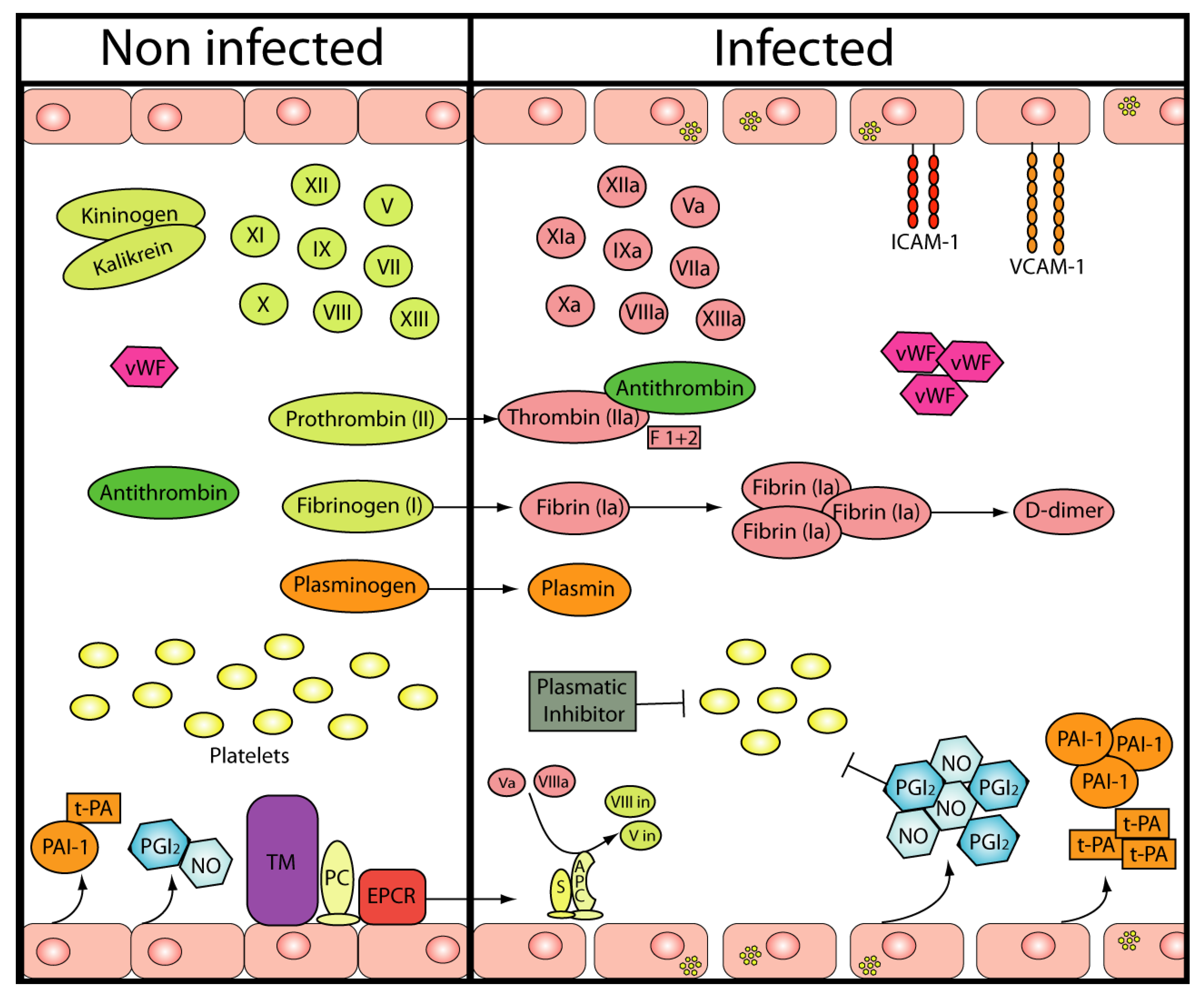{kind=link}
| Coagulation/Fibrinolysis | Platelets | ||
| Factor VIII | ⇩ | ||
| Factor IX | ⇩ | Count (in vivo and in vitro) | ⇩ |
| Factor XI | ⇩ | Function (in vivo) | Not determined |
| Factor V | ⇧ | ||
| vWF | ⇧ | ||
| Thr/AT complexes | ⇧ | Endothelium | |
| Prothrombin fragment 1+2 | ⇧ | Viral replication | Yes |
| FDPs | Not detected | Vascular lesions | No |
| AT III | = or slightly ⇩ | Cell adhesión molecules | ⇧ |
| Protein C | = or slightly ⇩ | NO | ⇧ |
| Free protein S | = or slightly ⇩ | PGI2 | ⇧ |
| t-PA | ⇧ | vWF | ⇩ |
| PAI-1 | ⇧ | ||
| D-dimer | ⇧ |
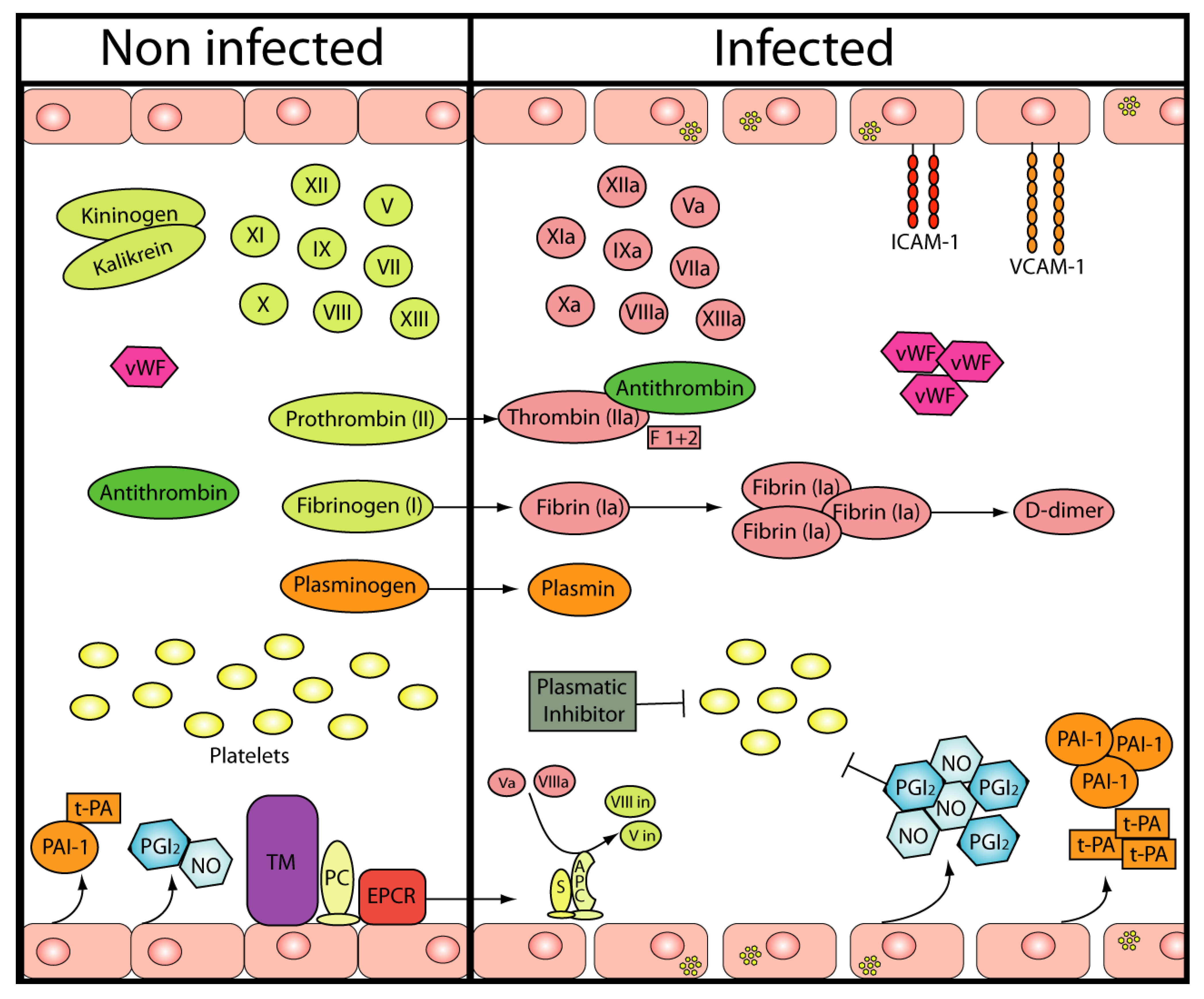
3. Endothelium
4. Platelets
Acknowledgements
References
- Salvato, M.S.; Clegg, J.C.S.; Buchmeier, M.J.; Charrel, R.N.; Gonzalez, J.P.; Lukashevich, I.S.; Peters, C.J.; Romanowski, V. Arenaviridae. In Virus taxonomy classification and nomenclature of viruses: Ninth report of the international committee on taxonomy of viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier-Academic Press: Oxford, 2011; pp. 715–724. [Google Scholar]
- Delgado, S.; Erickson, B.R.; Agudo, R.; Blair, P.J.; Vallejo, E.; Albarino, C.G.; Vargas, J.; Comer, J.A.; Rollin, P.E.; Ksiazek, T.G.; et al. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in bolivia. PLoS Pathog. 2008, 4, e1000047. [Google Scholar] [CrossRef]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of lujo virus, a new hemorrhagic fever-associated arenavirus from southern africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef]
- Charrel, R.N.; de Lamballerie, X. Zoonotic aspects of arenavirus infections. Vet. Microbiol. 2010, 140, 213–220. [Google Scholar] [CrossRef]
- Oldstone, M.B.; Campbell, K.P. Decoding arenavirus pathogenesis: Essential roles for alpha-dystroglycan-virus interactions and the immune response. Virology 2011, 411, 170–179. [Google Scholar] [CrossRef]
- Romanowski, V.; Ferrelli, L.; Pidre, M.L.; Bender, C.; Gómez, R.M. Argentine hemorrhagic fever. In Viral hemorrhagic fevers; Singh, S.K., Ruzek, D., Eds.; Taylor & Francis Group: Boca Raton, Florida, 2012; Vol. en prensa. [Google Scholar]
- Gomez, R.M.; Jaquenod de Giusti, C.; Sanchez Vallduvi, M.M.; Frik, J.; Ferrer, M.F.; Schattner, M. Junin virus. A xxi century update. Microbes Infect. 2011, 13, 303–311. [Google Scholar] [CrossRef]
- Moraz, M.L.; Kunz, S. Pathogenesis of arenavirus hemorrhagic fevers. Expert Rev. Anti Infect. Ther. 2011, 9, 49–59. [Google Scholar] [CrossRef]
- Kunz, S. The role of the vascular endothelium in arenavirus haemorrhagic fevers. Thromb. Haemost. 2009, 102, 1024–1029. [Google Scholar]
- Goeijenbier, M.; Wagenaar, J.; Goris, M.; Martina, B.; Henttonen, H.; Vaheri, A.; Reusken, C.; Hartskeerl, R.; Osterhaus, A.; Van Gorp, E. Rodent-borne hemorrhagic fevers: Under-recognized, widely spread and preventable - epidemiology, diagnostics and treatment. Crit. Rev. Microbiol. 2013, 39, 26–42. [Google Scholar] [CrossRef]
- Ambrosio, A.; Saavedra, M.; Mariani, M.; Gamboa, G.; Maiza, A. Argentine hemorrhagic fever vaccines. Hum. Vaccin. 2011, 7, 694–700. [Google Scholar]
- Heller, M.V.; Marta, R.F.; Sturk, A.; Maiztegui, J.I.; Hack, C.E.; Cate, J.W.; Molinas, F.C. Early markers of blood coagulation and fibrinolysis activation in argentine hemorrhagic fever. Thromb. Haemost. 1995, 73, 368–373. [Google Scholar]
- Molinas, F.C.; de Bracco, M.M.; Maiztegui, J.I. Coagulation studies in argentine hemorrhagic fever. J. Infect. Dis. 1981, 143, 1–6. [Google Scholar] [CrossRef]
- Molinas, F.C.; Maiztegui, J.I. Factor viii: C and factor viii r: Ag in argentine hemorrhagic fever. Thromb. Haemost. 1981, 46, 525–527. [Google Scholar]
- Molinas, F.C.; Kordich, L.; Porterie, P.; Lerer, G.; Maiztegui, J.I. Plasminogen abnormalities in patients with argentine hemorrhagic fever. Thromb. Res. 1987, 48, 713–720. [Google Scholar] [CrossRef]
- Frame, J.D.; Casals, J.; Dennis, E.A. Lassa virus antibodies in hospital personnel in western liberia. Trans. R. Soc. Trop. Med. Hyg. 1979, 73, 219–224. [Google Scholar] [CrossRef]
- Richmond, J.K.; Baglole, D.J. Lassa fever: Epidemiology, clinical features, and social consequences. BMJ 2003, 327, 1271–1275. [Google Scholar] [CrossRef]
- Bird, B.H.; Dodd, K.A.; Erickson, B.R.; Albarino, C.G.; Chakrabarti, A.K.; McMullan, L.K.; Bergeron, E.; Stroeher, U.; Cannon, D.; Martin, B.; et al. Severe hemorrhagic fever in strain 13/n guinea pigs infected with lujo virus. PLoS Negl. Trop. Dis. 2012, 6, e1801. [Google Scholar] [CrossRef]
- Peters, C.J.; Zaki, S.R. Role of the endothelium in viral hemorrhagic fevers. Crit. Care Med. 2002, 30, S268–S273. [Google Scholar]
- Fisher-Hoch, S.P.; Mitchell, S.W.; Sasso, D.R.; Lange, J.V.; Ramsey, R.; McCormick, J.B. Physiological and immunologic disturbances associated with shock in a primate model of lassa fever. J. Infect. Dis. 1987, 155, 465–474. [Google Scholar] [CrossRef]
- Gowen, B.B.; Julander, J.G.; London, N.R.; Wong, M.H.; Larson, D.; Morrey, J.D.; Li, D.Y.; Bray, M. Assessing changes in vascular permeability in a hamster model of viral hemorrhagic fever. Virol. J. 2010, 7, 240. [Google Scholar] [CrossRef]
- Walker, D.H.; McCormick, J.B.; Johnson, K.M.; Webb, P.A.; Komba-Kono, G.; Elliott, L.H.; Gardner, J.J. Pathologic and virologic study of fatal lassa fever in man. Am. J. Pathol. 1982, 107, 349–356. [Google Scholar]
- Hensley, L.E.; Smith, M.A.; Geisbert, J.B.; Fritz, E.A.; Daddario-DiCaprio, K.M.; Larsen, T.; Geisbert, T.W. Pathogenesis of lassa fever in cynomolgus macaques. Virol. J. 2011, 8, 205. [Google Scholar]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for new world haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar]
- Agrawal, S.; Anderson, P.; Durbeej, M.; van Rooijen, N.; Ivars, F.; Opdenakker, G.; Sorokin, L.M. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1007–1019. [Google Scholar] [CrossRef]
- Andrews, B.S.; Theofilopoulos, A.N.; Peters, C.J.; Loskutoff, D.J.; Brandt, W.E.; Dixon, F.J. Replication of dengue and junin viruses in cultured rabbit and human endothelial cells. Infect. Immun. 1978, 20, 776–781. [Google Scholar]
- Lukashevich, I.S.; Maryankova, R.; Vladyko, A.S.; Nashkevich, N.; Koleda, S.; Djavani, M.; Horejsh, D.; Voitenok, N.N.; Salvato, M.S. Lassa and mopeia virus replication in human monocytes/macrophages and in endothelial cells: Different effects on il-8 and tnf-alpha gene expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef]
- Gomez, R.M.; Pozner, R.G.; Lazzari, M.A.; D'Atri, L.P.; Negrotto, S.; Chudzinski-Tavassi, A.M.; Berria, M.I.; Schattner, M. Endothelial cell function alteration after junin virus infection. Thromb. Haemost. 2003, 90, 326–333. [Google Scholar]
- Brocato, R.L.; Voss, T.G. Pichinde virus induces microvascular endothelial cell permeability through the production of nitric oxide. Virol. J. 2009, 6, 162. [Google Scholar]
- Schmitz, H.; Kohler, B.; Laue, T.; Drosten, C.; Veldkamp, P.J.; Gunther, S.; Emmerich, P.; Geisen, H.P.; Fleischer, K.; Beersma, M.F.; et al. Monitoring of clinical and laboratory data in two cases of imported lassa fever. Microbes. Infect. 2002, 4, 43–50. [Google Scholar] [CrossRef]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L.; Goba, A.; Bah, A.; Peters, C.J.; Rollin, P.E. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar] [CrossRef]
- Rodrigo, W.W.; Ortiz-Riano, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martinez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa b. J. Virol. 2012, 86, 8185–8197. [Google Scholar] [CrossRef]
- Harrison, L.H.; Halsey, N.A.; McKee, K.T., Jr.; Peters, C.J.; Barrera Oro, J.G.; Briggiler, A.M.; Feuillade, M.R.; Maiztegui, J.I. Clinical case definitions for argentine hemorrhagic fever. Clin. Infect. Dis. 1999, 28, 1091–1094. [Google Scholar]
- Geisbert, T.W.; Jahrling, P.B. Exotic emerging viral diseases: Progress and challenges. Nat. Med. 2004, 10, S110–S121. [Google Scholar] [CrossRef]
- Salas, R.; de Manzione, N.; Tesh, R.B.; Rico-Hesse, R.; Shope, R.E.; Betancourt, A.; Godoy, O.; Bruzual, R.; Pacheco, M.E.; Ramos, B.; et al. Venezuelan haemorrhagic fever. Lancet 1991, 338, 1033–1036. [Google Scholar] [CrossRef]
- de Manzione, N.; Salas, R.A.; Paredes, H.; Godoy, O.; Rojas, L.; Araoz, F.; Fulhorst, C.F.; Ksiazek, T.G.; Mills, J.N.; Ellis, B.A.; et al. Venezuelan hemorrhagic fever: Clinical and epidemiological studies of 165 cases. Clin. Infect. Dis. 1998, 26, 308–313. [Google Scholar]
- Rodas, J.D.; Salvato, M.S. Tales of mice and men: Natural history of arenaviruses. Rev. Col. Cienc. Pec. 2006, 19, 382–400. [Google Scholar]
- Aguilar, P.V.; Camargo, W.; Vargas, J.; Guevara, C.; Roca, Y.; Felices, V.; Laguna-Torres, V.A.; Tesh, R.; Ksiazek, T.G.; Kochel, T.J. Reemergence of bolivian hemorrhagic fever, 2007-2008. Emerg. Infect. Dis. 2009, 15, 1526–1528. [Google Scholar] [CrossRef]
- de Bracco, M.M.; Rimoldi, M.T.; Cossio, P.M.; Rabinovich, A.; Maiztegui, J.I.; Carballal, G.; Arana, R.M. Argentine hemorrhagic fever. Alterations of the complement system and anti-junin-virus humoral response. N. Engl. J. Med. 1978, 299, 216–221. [Google Scholar] [CrossRef]
- Gallardo, F. Fiebre hemorragica argentina. Hallazgos anatomopatologicos en 10 necropsias. Medicina (Buenos Aires) 1970, 30, 77–84. [Google Scholar]
- Ponzinibbio, C.; Gonzalez, P.H.; Maiztegui, J.; Laguens, R.P. [morphological study of human bone marrow in argentinian hemorrhagic fever]. Medicina (B Aires) 1979, 39, 441–446. [Google Scholar]
- Carballal, G.; Rodriguez, M.; Frigerio, M.J.; Vasquez, C. Junin virus infection of guinea pigs: Electron microscopic studies of peripheral blood and bone marrow. J. Infect. Dis. 1977, 135, 367–373. [Google Scholar]
- Pozner, R.G.; Ure, A.E.; Jaquenod de Giusti, C.; D'Atri, L.P.; Italiano, J.E.; Torres, O.; Romanowski, V.; Schattner, M.; Gomez, R.M. Junin virus infection of human hematopoietic progenitors impairs in vitro proplatelet formation and platelet release via a bystander effect involving type i ifn signaling. PLoS Pathog. 2010, 6, e1000847. [Google Scholar] [CrossRef]
- Levis, S.C.; Saavedra, M.C.; Ceccoli, C.; Feuillade, M.R.; Enria, D.A.; Maiztegui, J.I.; Falcoff, R. Correlation between endogenous interferon and the clinical evolution of patients with argentine hemorrhagic fever. J. Interferon. Res. 1985, 5, 383–389. [Google Scholar] [CrossRef]
- Dejean, C.B.; Oubina, J.R.; Carballal, G.; Teyssie, A.R. Circulating interferon in the guinea pig infected with the xj, prototype junin virus strain. J. Med. Virol. 1988, 24, 97–99. [Google Scholar] [CrossRef]
- Cummins, D.; Fisher-Hoch, S.P.; Walshe, K.J.; Mackie, I.J.; McCormick, J.B.; Bennett, D.; Perez, G.; Farrar, B.; Machin, S.J. A plasma inhibitor of platelet aggregation in patients with lassa fever. Br. J. Haematol. 1989, 72, 543–548. [Google Scholar]
- Cummins, D.; Molinas, F.C.; Lerer, G.; Maiztegui, J.I.; Faint, R.; Machin, S.J. A plasma inhibitor of platelet aggregation in patients with argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 1990, 42, 470–475. [Google Scholar]
- Marta, R.F.; Heller, M.V.; Maiztegui, J.I.; Molinas, F.C. Further studies on the plasma inhibitor of platelet activation in argentine hemorrhagic fever. Thromb. Haemost. 1993, 69, 526–527. [Google Scholar]
- Goerge, T.; Ho-Tin-Noe, B.; Carbo, C.; Benarafa, C.; Remold-O'Donnell, E.; Zhao, B.Q.; Cifuni, S.M.; Wagner, D.D. Inflammation induces hemorrhage in thrombocytopenia. Blood 2008, 111, 4958–4964. [Google Scholar] [CrossRef]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Whitmire, J.K.; Marchese, P.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets prevent ifn-alpha/beta-induced lethal hemorrhage promoting ctl-dependent clearance of lymphocytic choriomeningitis virus. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 629–634. [Google Scholar]
- Negrotto, S.; De Giusti, C.J.; Lapponi, M.J.; Etulain, J.; Rivadeneyra, L.; Pozner, R.G.; Gomez, R.M.; Schattner, M. Expression and functionality of type i interferon receptor in the megakaryocytic lineage. J. Thromb. Haemost. 2011, 9, 2477–2485. [Google Scholar] [CrossRef]
- Loria, G.D.; Romagnoli, P.A.; Moseley, N.B.; Rucavado, A.; Altman, J.D. Platelets support a protective immune response to lcmv by preventing splenic necrosis. Blood 2012. [Google Scholar]
- Yun, N.E.; Poussard, A.L.; Seregin, A.V.; Walker, A.G.; Smith, J.K.; Aronson, J.F.; Smith, J.N.; Soong, L.; Paessler, S. Functional interferon system is required for clearance of lassa virus. J. Virol. 2012, 86, 3389–3392. [Google Scholar] [CrossRef]
- Kolokoltsova, O.A.; Yun, N.E.; Poussard, A.L.; Smith, J.K.; Smith, J.N.; Salazar, M.; Walker, A.; Tseng, C.T.; Aronson, J.F.; Paessler, S. Mice lacking alpha/beta and gamma interferon receptors are susceptible to junin virus infection. J. Virol. 2010, 84, 13063–13067. [Google Scholar]
- Bradfute, S.B.; Stuthman, K.S.; Shurtleff, A.C.; Bavari, S. A stat-1 knockout mouse model for machupo virus pathogenesis. Virol. J. 2011, 8, 300. [Google Scholar] [CrossRef]
- Flatz, L.; Rieger, T.; Merkler, D.; Bergthaler, A.; Regen, T.; Schedensack, M.; Bestmann, L.; Verschoor, A.; Kreutzfeldt, M.; Bruck, W.; et al. T cell-dependence of lassa fever pathogenesis. PLoS Pathog. 2010, 6, e1000836. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schattner, M.; Rivadeneyra, L.; Pozner, R.G.; Gómez, R.M. Pathogenic Mechanisms Involved in the Hematological Alterations of Arenavirus-induced Hemorrhagic Fevers. Viruses 2013, 5, 340-351. https://doi.org/10.3390/v5010340
Schattner M, Rivadeneyra L, Pozner RG, Gómez RM. Pathogenic Mechanisms Involved in the Hematological Alterations of Arenavirus-induced Hemorrhagic Fevers. Viruses. 2013; 5(1):340-351. https://doi.org/10.3390/v5010340
Chicago/Turabian StyleSchattner, Mirta, Leonardo Rivadeneyra, Roberto G. Pozner, and Ricardo M. Gómez. 2013. "Pathogenic Mechanisms Involved in the Hematological Alterations of Arenavirus-induced Hemorrhagic Fevers" Viruses 5, no. 1: 340-351. https://doi.org/10.3390/v5010340
APA StyleSchattner, M., Rivadeneyra, L., Pozner, R. G., & Gómez, R. M. (2013). Pathogenic Mechanisms Involved in the Hematological Alterations of Arenavirus-induced Hemorrhagic Fevers. Viruses, 5(1), 340-351. https://doi.org/10.3390/v5010340