Molecular Mechanisms of HIV Immune Evasion of the Innate Immune Response in Myeloid Cells

{kind=link}
{kind=link}
Abstract
:1. Introduction
Intrinsic Anti-Viral Factors Limit Infection of Myeloid Cells by HIV and SIV
2. Viral Factors Counteract Intrinsic Antiviral Factors; the Role of Vpx in SIV Pathogenesis
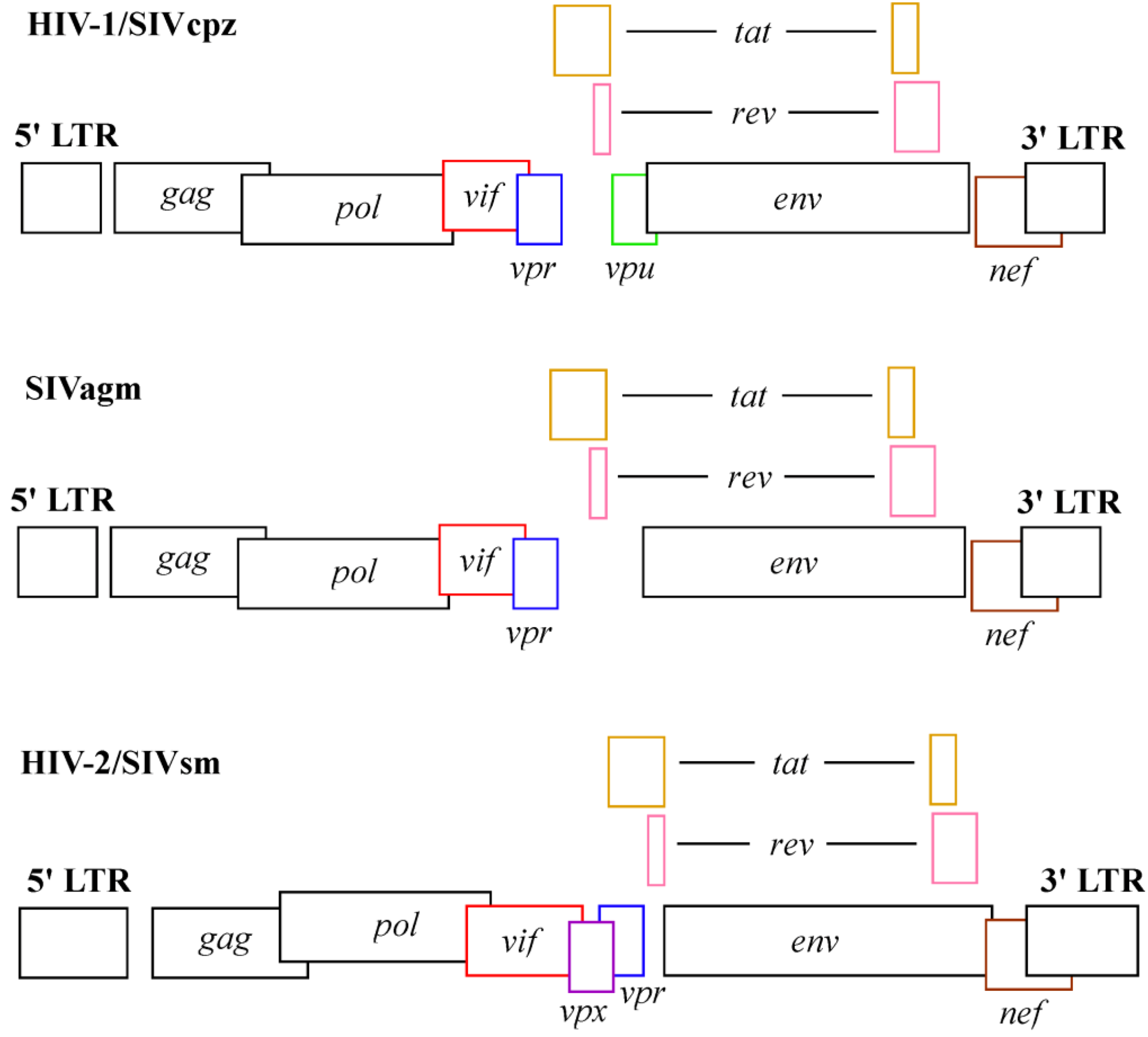
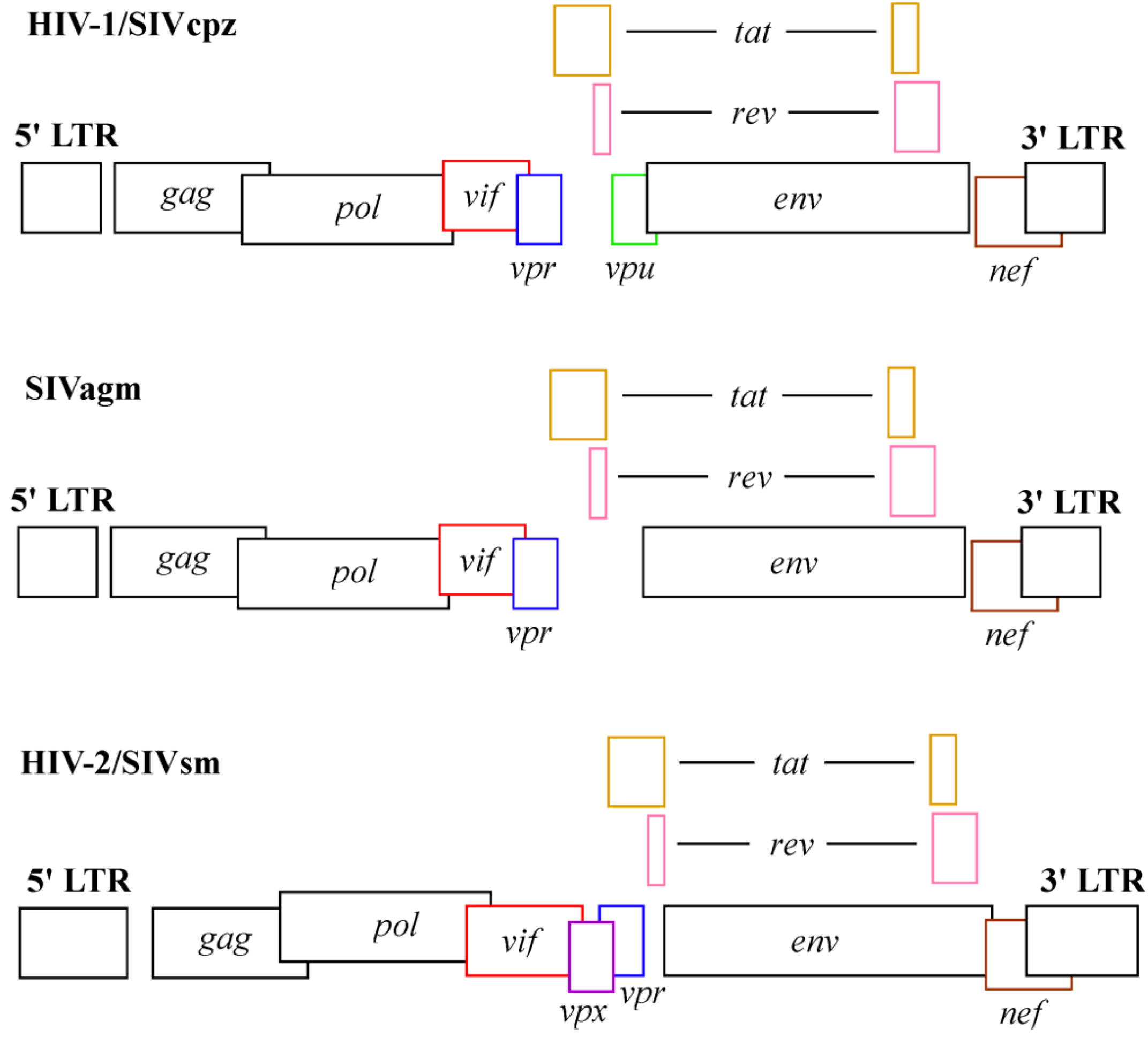
2.1. In vivo Studies of Vpx Function
2.2. In vitro Studies of Vpx Function
2.2.1. Vpx Targets the Host Factor SAMHD1
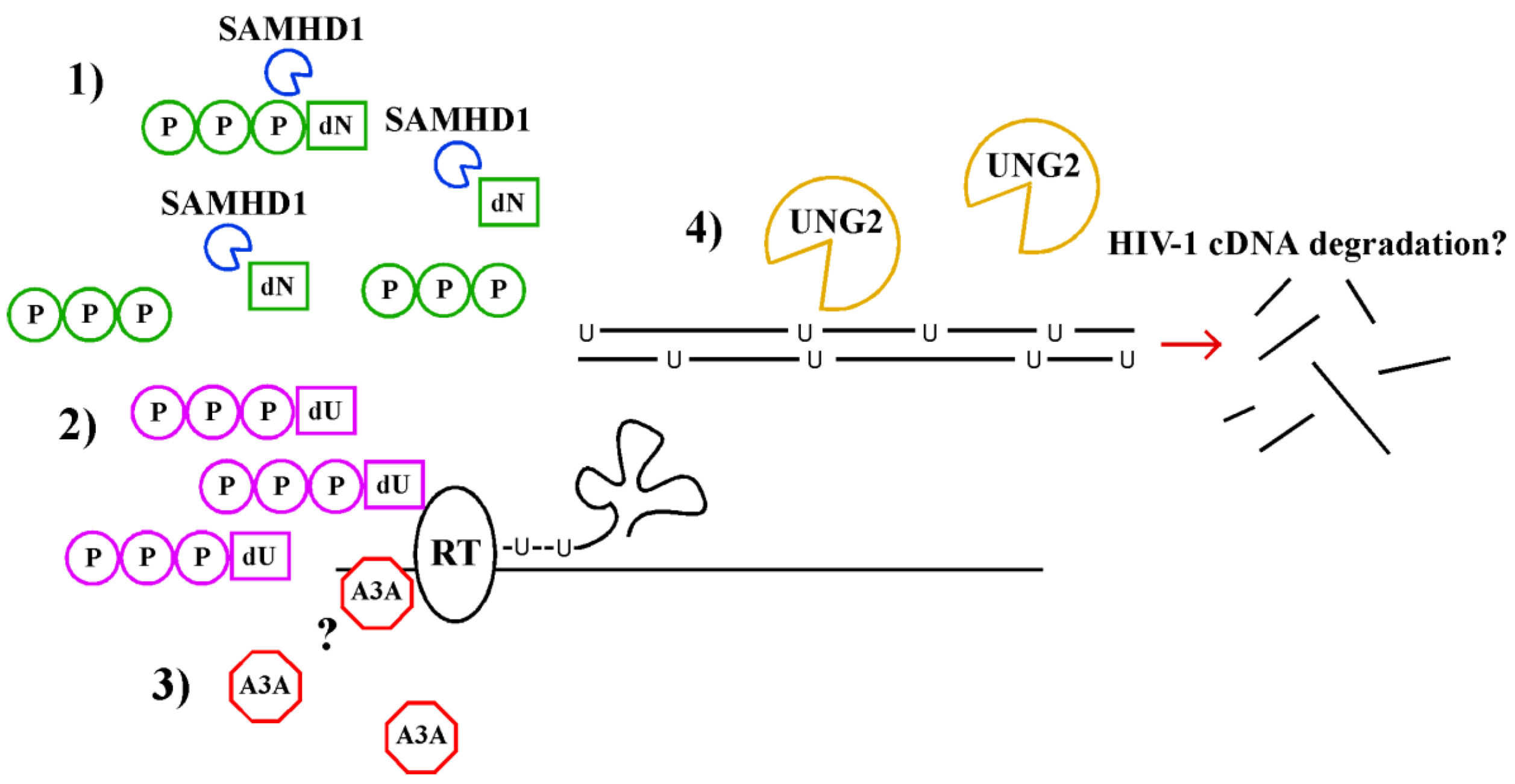
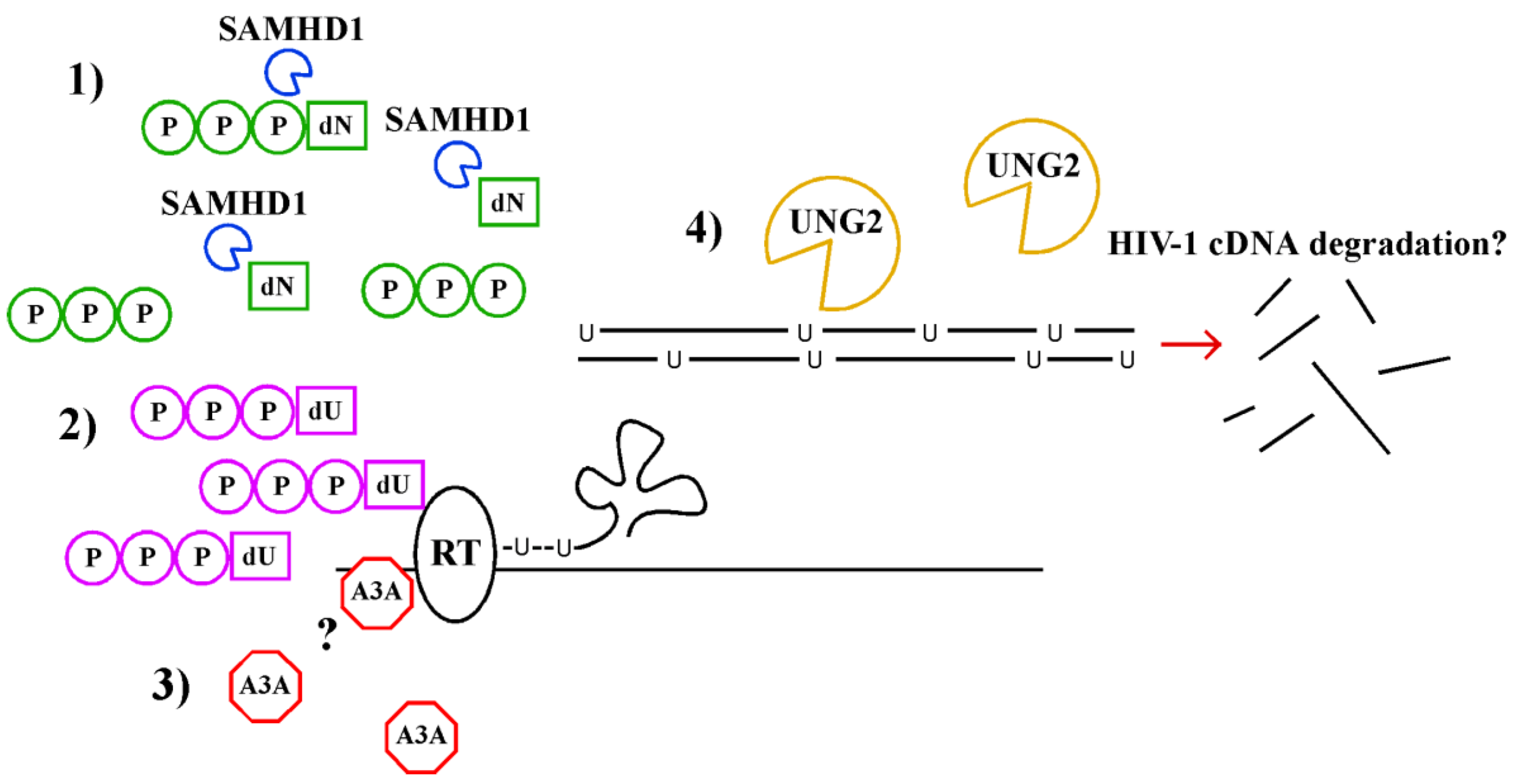
2.2.2. Vpx Counteracts SAMHD1 via the Ubiquitin Ligase Complex DDB1 and CUL4A
5. Conclusion
Acknowledgements
Conflict of Interest
References and Notes
- Cameron, P.U.; Forsum, U.; Teppler, H.; Granelli-Piperno, A.; Steinman, R.M. During hiv-1 infection most blood dendritic cells are not productively infected and can induce allogeneic cd4+ t cells clonal expansion. Clin. Exp. Immunol. 1992, 88, 226–236. [Google Scholar]
- Kaushik, R.; Zhu, X.; Stranska, R.; Wu, Y.; Stevenson, M. A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell Host Microbe 2009, 6, 68–80. [Google Scholar] [CrossRef]
- Gupta, P.; Collins, K.B.; Ratner, D.; Watkins, S.; Naus, G.J.; Landers, D.V.; Patterson, B.K. Memory cd4(+) t cells are the earliest detectable human immunodeficiency virus type 1 (hiv-1)-infected cells in the female genital mucosal tissue during hiv-1 transmission in an organ culture system. J. Virol. 2002, 76, 9868–9876. [Google Scholar]
- Zhang, Z.; Schuler, T.; Zupancic, M.; Wietgrefe, S.; Staskus, K.A.; Reimann, K.A.; Reinhart, T.A.; Rogan, M.; Cavert, W.; Miller, C.J.; et al. Sexual transmission and propagation of siv and hiv in resting and activated cd4+ t cells. Science 1999, 286, 1353–1357. [Google Scholar] [CrossRef]
- Hladik, F.; Sakchalathorn, P.; Ballweber, L.; Lentz, G.; Fialkow, M.; Eschenbach, D.; McElrath, M.J. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity 2007, 26, 257–270. [Google Scholar] [CrossRef]
- Hu, J.; Gardner, M.B.; Miller, C.J. Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J. Virol. 2000, 74, 6087–6095. [Google Scholar] [CrossRef]
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute hiv-1 infection. New Engl. J. Med. 2011, 364, 1943–1954. [Google Scholar] [CrossRef]
- Reinhart, T.A.; Rogan, M.J.; Huddleston, D.; Rausch, D.M.; Eiden, L.E.; Haase, A.T. Simian immunodeficiency virus burden in tissues and cellular compartments during clinical latency and aids. J. Infect. Dis. 1997, 176, 1198–1208. [Google Scholar]
- Schacker, T.; Little, S.; Connick, E.; Gebhard, K.; Zhang, Z.Q.; Krieger, J.; Pryor, J.; Havlir, D.; Wong, J.K.; Schooley, R.T.; et al. Productive infection of t cells in lymphoid tissues during primary and early human immunodeficiency virus infection. J. Infect. Dis. 2001, 183, 555–562. [Google Scholar] [CrossRef]
- Koenig, S.; Gendelman, H.E.; Orenstein, J.M.; Dal Canto, M.C.; Pezeshkpour, G.H.; Yungbluth, M.; Janotta, F.; Aksamit, A.; Martin, M.A.; Fauci, A.S. Detection of aids virus in macrophages in brain tissue from aids patients with encephalopathy. Science 1986, 233, 1089–1093. [Google Scholar]
- Desrosiers, R.C.; Hansen-Moosa, A.; Mori, K.; Bouvier, D.P.; King, N.W.; Daniel, M.D.; Ringler, D.J. Macrophage-tropic variants of siv are associated with specific aids-related lesions but are not essential for the development of aids. Am. J. Pathol. 1991, 139, 29–35. [Google Scholar]
- Berger, G.; Durand, S.; Fargier, G.; Nguyen, X.N.; Cordeil, S.; Bouaziz, S.; Muriaux, D.; Darlix, J.L.; Cimarelli, A. Apobec3a is a specific inhibitor of the early phases of hiv-1 infection in myeloid cells. PLoS Pathog. 2011, 7, e1002221. [Google Scholar] [CrossRef]
- Laguette, N.; Rahm, N.; Sobhian, B.; Chable-Bessia, C.; Munch, J.; Snoeck, J.; Sauter, D.; Switzer, W.M.; Heneine, W.; Kirchhoff, F.; et al. Evolutionary and functional analyses of the interaction between the myeloid restriction factor samhd1 and the lentiviral vpx protein. Cell Host Microbe 2012, 11, 205–217. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Middel, J.; Cornelissen, I.L.; Nottet, H.S.; KewalRamani, V.N.; Littman, D.R.; Figdor, C.G.; van Kooyk, Y. Dc-sign, a dendritic cell-specific hiv-1-binding protein that enhances trans-infection of t cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Saifuddin, M.; Hart, M.L.; Gewurz, H.; Zhang, Y.; Spear, G.T. Interaction of mannose-binding lectin with primary isolates of human immunodeficiency virus type 1. J. Gen. Virol. 2000, 81, 949–955. [Google Scholar]
- Bobardt, M.D.; Saphire, A.C.; Hung, H.C.; Yu, X.; Van der Schueren, B.; Zhang, Z.; David, G.; Gallay, P.A. Syndecan captures, protects, and transmits hiv to t lymphocytes. Immunity 2003, 18, 27–39. [Google Scholar] [CrossRef]
- Gummuluru, S.; Rogel, M.; Stamatatos, L.; Emerman, M. Binding of human immunodeficiency virus type 1 to immature dendritic cells can occur independently of dc-sign and mannose binding c-type lectin receptors via a cholesterol-dependent pathway. J. Virol. 2003, 77, 12865–12874. [Google Scholar] [CrossRef]
- Izquierdo-Useros, N.; Lorizate, M.; Contreras, F.X.; Rodriguez-Plata, M.T.; Glass, B.; Erkizia, I.; Prado, J.G.; Casas, J.; Fabrias, G.; Krausslich, H.G.; et al. Sialyllactose in viral membrane gangliosides is a novel molecular recognition pattern for mature dendritic cell capture of hiv-1. PLoS Biol. 2012, 10, e1001315. [Google Scholar] [CrossRef]
- Puryear, W.B.; Yu, X.; Ramirez, N.P.; Reinhard, B.M.; Gummuluru, S. Hiv-1 incorporation of host-cell-derived glycosphingolipid gm3 allows for capture by mature dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 7475–7480. [Google Scholar]
- Magerus-Chatinet, A.; Yu, H.; Garcia, S.; Ducloux, E.; Terris, B.; Bomsel, M. Galactosyl ceramide expressed on dendritic cells can mediate hiv-1 transfer from monocyte derived dendritic cells to autologous t cells. Virology 2007, 362, 67–74. [Google Scholar] [CrossRef]
- de Silva, S.; Planelles, V.; Wu, L. Differential effects of vpr on single-cycle and spreading hiv-1 infections in cd4+ t-cells and dendritic cells. PLoS One 2012, 7, e35385. [Google Scholar]
- Wu, L.; KewalRamani, V.N. Dendritic-cell interactions with hiv: Infection and viral dissemination. Nat. Rev. Immunol. 2006, 6, 859–868. [Google Scholar] [CrossRef]
- Cameron, P.U.; Freudenthal, P.S.; Barker, J.M.; Gezelter, S.; Inaba, K.; Steinman, R.M. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to cd4+ t cells. Science 1992, 257, 383–387. [Google Scholar]
- Altfeld, M.; Fadda, L.; Frleta, D.; Bhardwaj, N. Dcs and nk cells: Critical effectors in the immune response to hiv-1. Nat. Rev. Immunol. 2011, 11, 176–186. [Google Scholar] [CrossRef]
- Coleman, C.M.; Wu, L. Hiv interactions with monocytes and dendritic cells: Viral latency and reservoirs. Retrovirology 2009, 6, 51. [Google Scholar] [CrossRef]
- Sharova, N.; Swingler, C.; Sharkey, M.; Stevenson, M. Macrophages archive hiv-1 virions for dissemination in trans. EMBO J. 2005, 24, 2481–2489. [Google Scholar] [CrossRef]
- Lim, E.S.; Fregoso, O.I.; McCoy, C.O.; Matsen, F.A.; Malik, H.S.; Emerman, M. The ability of primate lentiviruses to degrade the monocyte restriction factor samhd1 preceded the birth of the viral accessory protein vpx. Cell Host Microbe 2012, 11, 194–204. [Google Scholar] [CrossRef]
- Tristem, M.; Marshall, C.; Karpas, A.; Hill, F. Evolution of the primate lentiviruses: Evidence from vpx and vpr. EMBO J. 1992, 11, 3405–3412. [Google Scholar]
- Ayinde, D.; Maudet, C.; Transy, C.; Margottin-Goguet, F. Limelight on two hiv/siv accessory proteins in macrophage infection: Is vpx overshadowing vpr? Retrovirology 2010, 7, 35. [Google Scholar]
- Le Rouzic, E.; Benichou, S. The vpr protein from hiv-1: Distinct roles along the viral life cycle. Retrovirology 2005, 2, 11. [Google Scholar] [CrossRef]
- Malim, M.H.; Emerman, M. Hiv-1 accessory proteins—Ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef]
- Peeters, M.; Courgnaud, V. Overview of primate lentiviruses and their evolution in non-human primates in Africa. In HIV Sequence Compendium; HIV sequence compendium; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: Los Alamos, NM, USA, 2002; pp. 2–23. [Google Scholar]
- Belshan, M.; Kimata, J.T.; Brown, C.; Cheng, X.; McCulley, A.; Larsen, A.; Thippeshappa, R.; Hodara, V.; Giavedoni, L.; Hirsch, V.; et al. Vpx is critical for sivmne infection of pigtail macaques. Retrovirology 2012, 9, 32. [Google Scholar] [CrossRef]
- Hirsch, V.M.; Sharkey, M.E.; Brown, C.R.; Brichacek, B.; Goldstein, S.; Wakefield, J.; Byrum, R.; Elkins, W.R.; Hahn, B.H.; Lifson, J.D.; et al. Vpx is required for dissemination and pathogenesis of siv(sm) pbj: Evidence of macrophage-dependent viral amplification. Nat. Med. 1998, 4, 1401–1408. [Google Scholar]
- Gibbs, J.S.; Lackner, A.A.; Lang, S.M.; Simon, M.A.; Sehgal, P.K.; Daniel, M.D.; Desrosiers, R.C. Progression to aids in the absence of a gene for vpr or vpx. J. Virol. 1995, 69, 2378–2383. [Google Scholar]
- Mandal, D.; Prasad, V.R. Analysis of 2-ltr circle junctions of viral DNA in infected cells. Meth. Mol. Biol. 2009, 485, 73–85. [Google Scholar] [CrossRef]
- Sharova, N.; Wu, Y.; Zhu, X.; Stranska, R.; Kaushik, R.; Sharkey, M.; Stevenson, M. Primate lentiviral vpx commandeers ddb1 to counteract a macrophage restriction. PLoS Pathog. 2008, 4, e1000057. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of hiv-1 infection of macrophages mediated by the samhd1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. Samhd1 is the dendritic- and myeloid-cell-specific hiv-1 restriction factor counteracted by vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Lim, E.S.; Emerman, M. Hiv: Going for the watchman. Nature 2011, 474, 587–588. [Google Scholar] [CrossRef]
- Planelles, V. Samhd1 joins the red queen’s court. Cell Host Microbe 2012, 11, 103–105. [Google Scholar] [CrossRef]
- Baldauf, H.M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. Samhd1 restricts hiv-1 infection in resting cd4(+) t cells. Nat. Med. 2012, 18, 1682–1689. [Google Scholar] [CrossRef]
- Descours, B.; Cribier, A.; Chable-Bessia, C.; Ayinde, D.; Rice, G.; Crow, Y.; Yatim, A.; Schwartz, O.; Laguette, N.; Benkirane, M. Samhd1 restricts hiv-1 reverse transcription in quiescent cd4+ t-cells. Retrovirology 2012, 9, 87. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The hd domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998, 23, 469–472. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. Hiv-1 restriction factor samhd1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. Samhd1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar]
- Jermy, A. Viral infection: Samhd1 cuts the power to hiv-1. Nat. Rev. Microbiol. 2012, 10, 237. [Google Scholar] [CrossRef]
- Schaller, T.; Goujon, C.; Malim, M.H. Aids/hiv. Hiv interplay with samhd1. Science 2012, 335, 1313–1314. [Google Scholar] [CrossRef]
- Manel, N.; Hogstad, B.; Wang, Y.; Levy, D.E.; Unutmaz, D.; Littman, D.R. A cryptic sensor for hiv-1 activates antiviral innate immunity in dendritic cells. Nature 2010, 467, 214–217. [Google Scholar] [CrossRef]
- Xu, W.; Santini, P.A.; Sullivan, J.S.; He, B.; Shan, M.; Ball, S.C.; Dyer, W.B.; Ketas, T.J.; Chadburn, A.; Cohen-Gould, L.; et al. Hiv-1 evades virus-specific igg2 and iga responses by targeting systemic and intestinal b cells via long-range intercellular conduits. Nat. Immunol. 2009, 10, 1008–1017. [Google Scholar] [CrossRef]
- Ogawa, K.; Shibata, R.; Kiyomasu, T.; Higuchi, I.; Kishida, Y.; Ishimoto, A.; Adachi, A. Mutational analysis of the human immunodeficiency virus vpr open reading frame. J. Virol. 1989, 63, 4110–4114. [Google Scholar]
- Balliet, J.W.; Kolson, D.L.; Eiger, G.; Kim, F.M.; McGann, K.A.; Srinivasan, A.; Collman, R. Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: Mutational analysis of a primary hiv-1 isolate. Virology 1994, 200, 623–631. [Google Scholar] [CrossRef]
- Rey, F.; BouHamdan, M.; Navarro, J.M.; Agostini, I.; Willetts, K.; Bouyac, M.; Tamalet, C.; Spire, B.; Vigne, R.; Sire, J. A role for human immunodeficiency virus type 1 vpr during infection of peripheral blood mononuclear cells. J. Gen. Virol. 1998, 79, 1083–1087. [Google Scholar]
- Heinzinger, N.K.; Bukinsky, M.I.; Haggerty, S.A.; Ragland, A.M.; Kewalramani, V.; Lee, M.A.; Gendelman, H.E.; Ratner, L.; Stevenson, M.; Emerman, M. The vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 7311–7315. [Google Scholar]
- Lang, S.M.; Weeger, M.; Stahl-Hennig, C.; Coulibaly, C.; Hunsmann, G.; Muller, J.; Muller-Hermelink, H.; Fuchs, D.; Wachter, H.; Daniel, M.M.; et al. Importance of vpr for infection of rhesus monkeys with simian immunodeficiency virus. J. Virol. 1993, 67, 902–912. [Google Scholar]
- Hoch, J.; Lang, S.M.; Weeger, M.; Stahl-Hennig, C.; Coulibaly, C.; Dittmer, U.; Hunsmann, G.; Fuchs, D.; Muller, J.; Sopper, S.; et al. Vpr deletion mutant of simian immunodeficiency virus induces aids in rhesus monkeys. J. Virol. 1995, 69, 4807–4813. [Google Scholar]
- Somasundaran, M.; Sharkey, M.; Brichacek, B.; Luzuriaga, K.; Emerman, M.; Sullivan, J.L.; Stevenson, M. Evidence for a cytopathogenicity determinant in hiv-1 vpr. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9503–9508. [Google Scholar]
- Schrofelbauer, B.; Hakata, Y.; Landau, N.R. Hiv-1 vpr function is mediated by interaction with the damage-specific DNA-binding protein ddb1. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4130–4135. [Google Scholar]
- Kennedy, E.M.; Daddacha, W.; Slater, R.; Gavegnano, C.; Fromentin, E.; Schinazi, R.F.; Kim, B. Abundant non-canonical dutp found in primary human macrophages drives its frequent incorporation by hiv-1 reverse transcriptase. J. Biol. Chem. 2011, 286, 25047–25055. [Google Scholar]
- Sire, J.; Querat, G.; Esnault, C.; Priet, S. Uracil within DNA: An actor of antiviral immunity. Retrovirology 2008, 5, 45. [Google Scholar] [CrossRef]
- Norman, J.M.; Mashiba, M.; McNamara, L.A.; Onafuwa-Nuga, A.; Chiari-Fort, E.; Shen, W.; Collins, K.L. The antiviral factor apobec3g enhances the recognition of hiv-infected primary t cells by natural killer cells. Nat. Immunol. 2011, 12, 975–983. [Google Scholar] [CrossRef]
- Croxford, J.L.; Gasser, S. Damage control: How hiv survives the editor apobec3g. Nat. Immunol. 2011, 12, 925–927. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Chen, R.; Le Rouzic, E.; Kearney, J.A.; Mansky, L.M.; Benichou, S. Vpr-mediated incorporation of ung2 into hiv-1 particles is required to modulate the virus mutation rate and for replication in macrophages. J. Biol. Chem. 2004, 279, 28419–28425. [Google Scholar]
- Zimmerman, E.S.; Sherman, M.P.; Blackett, J.L.; Neidleman, J.A.; Kreis, C.; Mundt, P.; Williams, S.A.; Warmerdam, M.; Kahn, J.; Hecht, F.M.; Grant, R.M.; de Noronha, C.M.; Weyrich, A.S.; Greene, W.C.; Planelles, V. Human immunodeficiency virus type 1 vpr induces DNA replication stress in vitro and in vivo. J. Virol. 2006, 80, 10407–10418. [Google Scholar]
- Lavin, M.F.; Kozlov, S. Atm activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. Atr: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef]
- Priet, S.; Gros, N.; Navarro, J.M.; Boretto, J.; Canard, B.; Querat, G.; Sire, J. Hiv-1-associated uracil DNA glycosylase activity controls dutp misincorporation in viral DNA and is essential to the hiv-1 life cycle. Mol. Cell 2005, 17, 479–490. [Google Scholar] [CrossRef]
- Gu, Y.; Sundquist, W.I. Good to cu. Nature 2003, 424, 21–22. [Google Scholar] [CrossRef]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of apobec3g-edited nascent hiv-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar]
- Yan, N.; O'Day, E.; Wheeler, L.A.; Engelman, A.; Lieberman, J. Hiv DNA is heavily uracilated, which protects it from autointegration. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9244–9249. [Google Scholar]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits hiv-1 infection and is suppressed by the viral vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Bishop, K.N.; Verma, M.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. Apobec3g inhibits elongation of hiv-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef]
- Koning, F.A.; Newman, E.N.; Kim, E.Y.; Kunstman, K.J.; Wolinsky, S.M.; Malim, M.H. Defining apobec3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009, 83, 9474–9485. [Google Scholar]
- Malim, M.H. Apobec proteins and intrinsic resistance to hiv-1 infection. Phil. Trans. Roy. Soc. Lond. B Biol. Sci. 2009, 364, 675–687. [Google Scholar] [CrossRef]
- Peng, G.; Greenwell-Wild, T.; Nares, S.; Jin, W.; Lei, K.J.; Rangel, Z.G.; Munson, P.J.; Wahl, S.M. Myeloid differentiation and susceptibility to hiv-1 are linked to apobec3 expression. Blood 2007, 110, 393–400. [Google Scholar] [CrossRef]
- Koning, F.A.; Goujon, C.; Bauby, H.; Malim, M.H. Target cell-mediated editing of hiv-1 cdna by apobec3 proteins in human macrophages. J. Virol. 2011, 85, 13448–13452. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Greene, W.C. The apobec3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Ann. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mashiba, M.; Collins, K.L. Molecular Mechanisms of HIV Immune Evasion of the Innate Immune Response in Myeloid Cells. Viruses 2013, 5, 1-14. https://doi.org/10.3390/v5010001
Mashiba M, Collins KL. Molecular Mechanisms of HIV Immune Evasion of the Innate Immune Response in Myeloid Cells. Viruses. 2013; 5(1):1-14. https://doi.org/10.3390/v5010001
Chicago/Turabian StyleMashiba, Mike, and Kathleen L. Collins. 2013. "Molecular Mechanisms of HIV Immune Evasion of the Innate Immune Response in Myeloid Cells" Viruses 5, no. 1: 1-14. https://doi.org/10.3390/v5010001
APA StyleMashiba, M., & Collins, K. L. (2013). Molecular Mechanisms of HIV Immune Evasion of the Innate Immune Response in Myeloid Cells. Viruses, 5(1), 1-14. https://doi.org/10.3390/v5010001