Dengue Virus and Autophagy

Abstract
:1. Introduction to Autophagy
2. Dengue Virus Infection Induces and Requires Autophagy
3. The Structure of Dengue Virus Replication Complexes
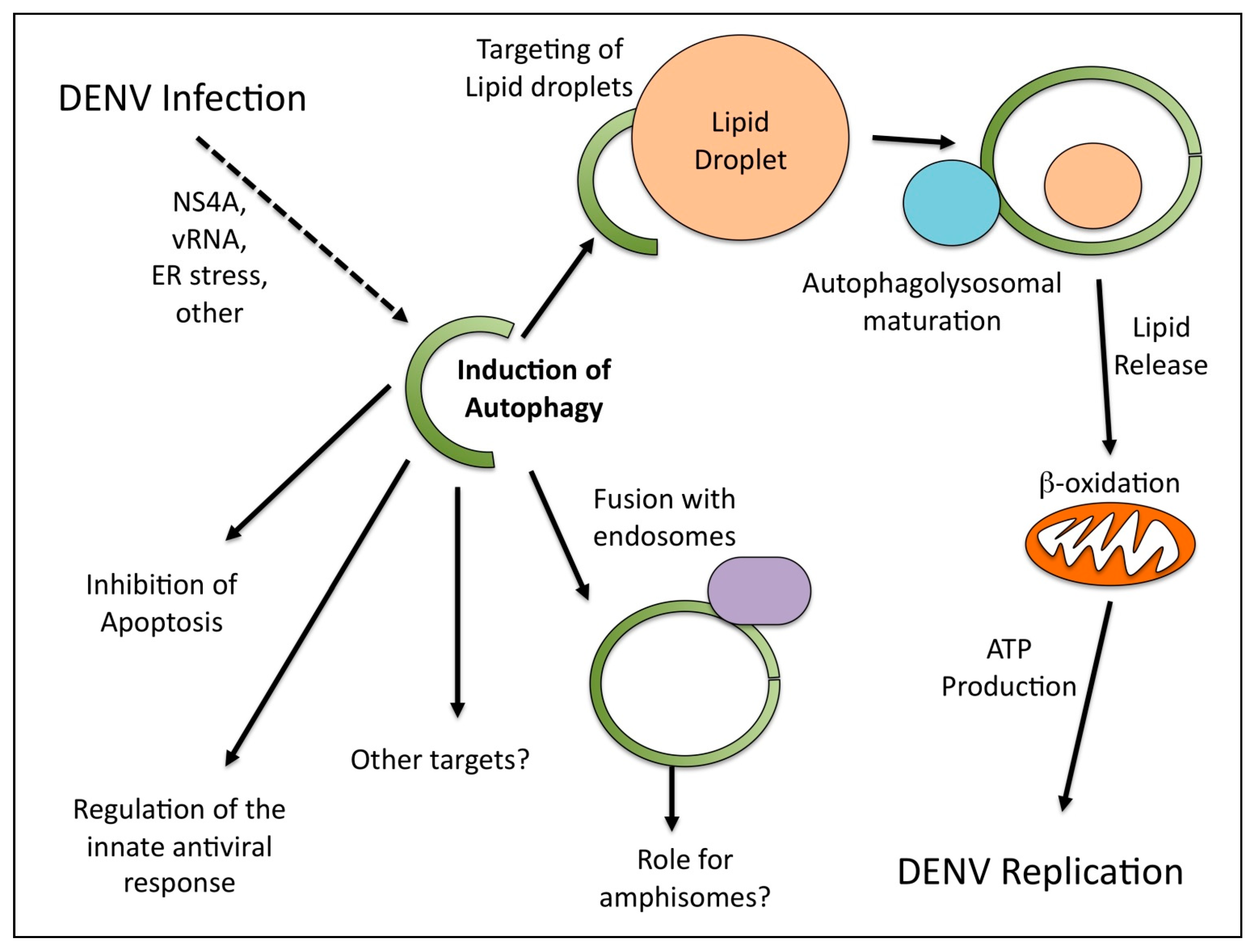
4. DENV Induces a Selective Autophagy that Stimulates Lipid Metabolism

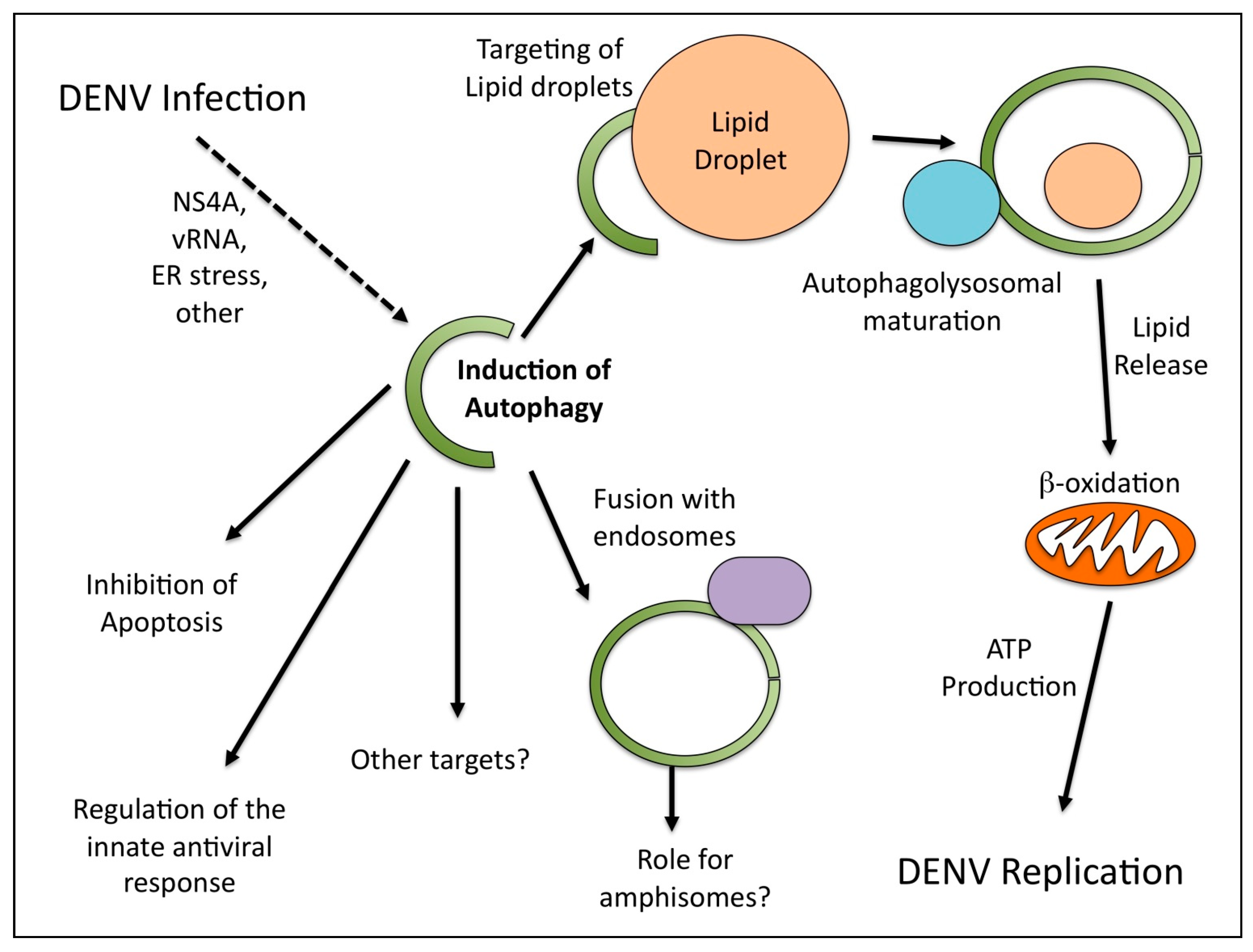{kind=link}
| Study | DENV Serotype | Cell Lines Used | Major Conclusions |
|---|---|---|---|
| Lee et al., 2008 [31] | DENV2 | Huh7 |
|
| Panyasrivanit et al., 2009 [33] | DENV2 | HepG2 |
|
| Khakpoor et al., 2009 [32] | DENV3 | HepG2 |
|
| Heaton et al., 2010a [30] | DENV2 | Huh7.5 |
|
| Heaton et al., 2010b [39] | DENV2 | Huh7.5, BHK, Huh7, HepG2 |
|
| Ke et al., 2011 [24] | DENV2 | Huh7 |
|
| McLean et al., 2011 [34] | DENV2 | MDCK, MEF, 293T, HeLa, Vero |
|
5. Conclusions and Future Directions
Acknowledgements
Conflict of Interest
References and Notes
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Kundu, M.; Thompson, C.B. Autophagy: Basic principles and relevance to disease. Annu. Rev. Pathol. 2008, 3, 427–455. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes. Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Codogno, P.; Cuervo, A.M.; Deretic, V.; Elazar, Z.; Fueyo-Margareto, J.; Gewirtz, D.A.; Kroemer, G.; Levine, B.; Mizushima, N.; et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010, 6, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell. Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The beclin 1-vps34 complex—At the crossroads of autophagy and beyond. Trends Cell. Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Kuma, A.; Mizushima, N. Physiological role of autophagy as an intracellular recycling system: With an emphasis on nutrient metabolism. Semin. Cell. Dev. Biol. 2010, 21, 683–690. [Google Scholar] [CrossRef]
- Kudchodkar, S.B.; Levine, B. Viruses and autophagy. Rev. Med. Virol. 2009, 19, 359–378. [Google Scholar] [CrossRef]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar] [CrossRef]
- Delgado, M.A.; Deretic, V. Toll-like receptors in control of immunological autophagy. Cell. Death. Differ. 2009, 16, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Multiple regulatory and effector roles of autophagy in immunity. Curr. Opin. Immunol. 2009, 21, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Milosevic, S.; Behrends, U.; Jaffee, E.M.; Pardoll, D.M.; Bornkamm, G.W.; Mautner, J. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 2003, 33, 1250–1259. [Google Scholar] [CrossRef]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Muller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy promotes mhc class ii presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 7922–7927. [Google Scholar] [CrossRef]
- Schmid, D.; Pypaert, M.; Munz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef]
- Kirkegaard, K. Subversion of the cellular autophagy pathway by viruses. Curr. Top. Microbiol. Immunol. 2009, 335, 323–333. [Google Scholar]
- Taylor, M.P.; Kirkegaard, K. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 2007, 81, 12543–12553. [Google Scholar] [CrossRef]
- Taylor, M.P.; Kirkegaard, K. Potential subversion of autophagosomal pathway by picornaviruses. Autophagy 2008, 4, 286–289. [Google Scholar] [CrossRef]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Cali, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses Hijack the LC3-I-positive edemosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500-508. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14046–14051. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis c virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 2011, 121, 37-56. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef]
- Esler, D. Dengue—Clinical and public health ramifications. Aust. Fam. Physician 2009, 38, 876-879. [Google Scholar]
- van der Schaar, H.M.; Rust, M.J.; Chen, C.; van der Ende-Metselaar, H.; Wilschut, J.; Zhuang, X.; Smit, J.M. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008, 4, e1000244. [Google Scholar] [CrossRef]
- Zaitseva, E.; Yang, S.T.; Melikov, K.; Pourmal, S.; Chernomordik, L.V. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog. 2010, 6, e1001131. [Google Scholar] [CrossRef]
- Clyde, K.; Kyle, J.L.; Harris, E. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J. Virol. 2006, 80, 11418–11431. [Google Scholar] [CrossRef]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 17345–17350. [Google Scholar] [CrossRef]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Khakpoor, A.; Panyasrivanit, M.; Wikan, N.; Smith, D.R. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J. Gen. Virol. 2009, 90, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 2009, 90, 448–456. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell. 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiology 2011, 19, 368–375. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Heaton, N.S.; Randall, G. Dengue Virus and Autophagy. Viruses 2011, 3, 1332-1341. https://doi.org/10.3390/v3081332
Heaton NS, Randall G. Dengue Virus and Autophagy. Viruses. 2011; 3(8):1332-1341. https://doi.org/10.3390/v3081332
Chicago/Turabian StyleHeaton, Nicholas S., and Glenn Randall. 2011. "Dengue Virus and Autophagy" Viruses 3, no. 8: 1332-1341. https://doi.org/10.3390/v3081332
APA StyleHeaton, N. S., & Randall, G. (2011). Dengue Virus and Autophagy. Viruses, 3(8), 1332-1341. https://doi.org/10.3390/v3081332