Avian Bornaviruses Escape Recognition by the Innate Immune System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
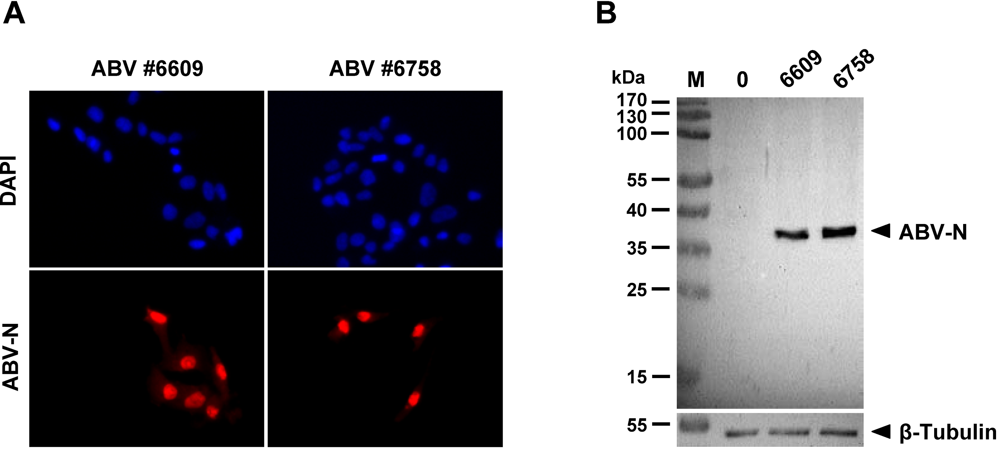
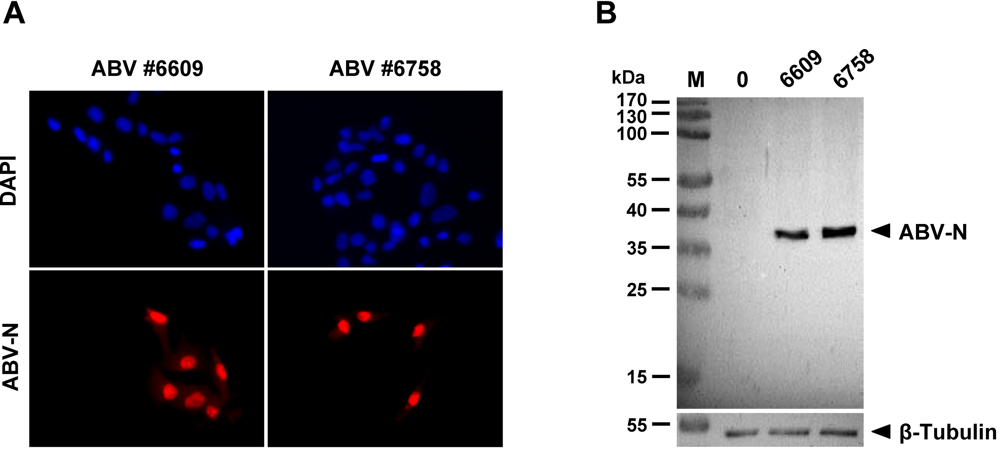
2.1. Sensitive monitoring of ABV infection using antiserum specific for the viral N protein
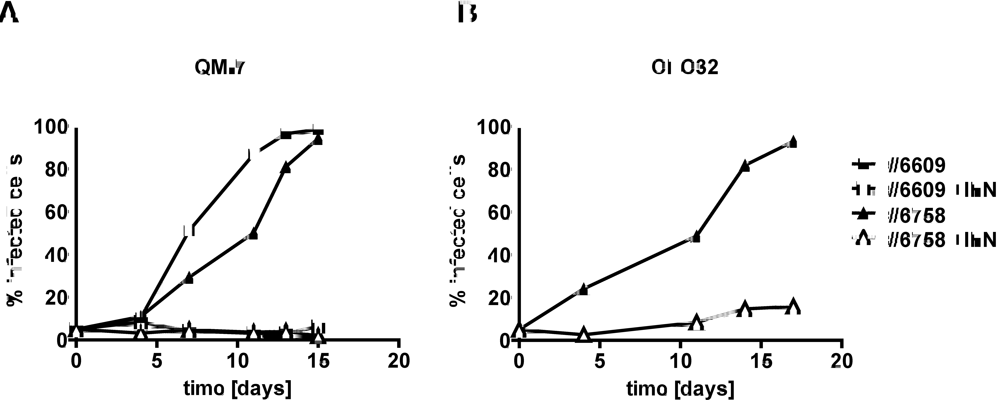
2.2. Type I IFN prevents efficient spread of ABV to uninfected quail cells

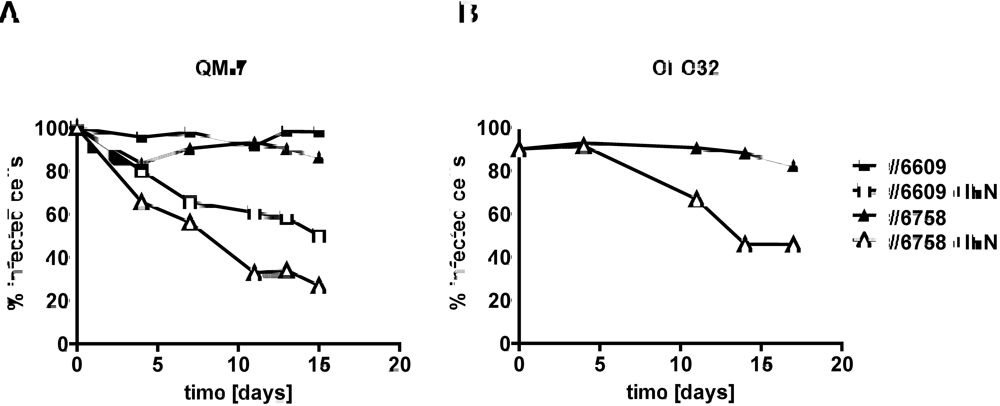
2.3. Type I IFN inhibits the activity of ABV in persistently infected quail cells

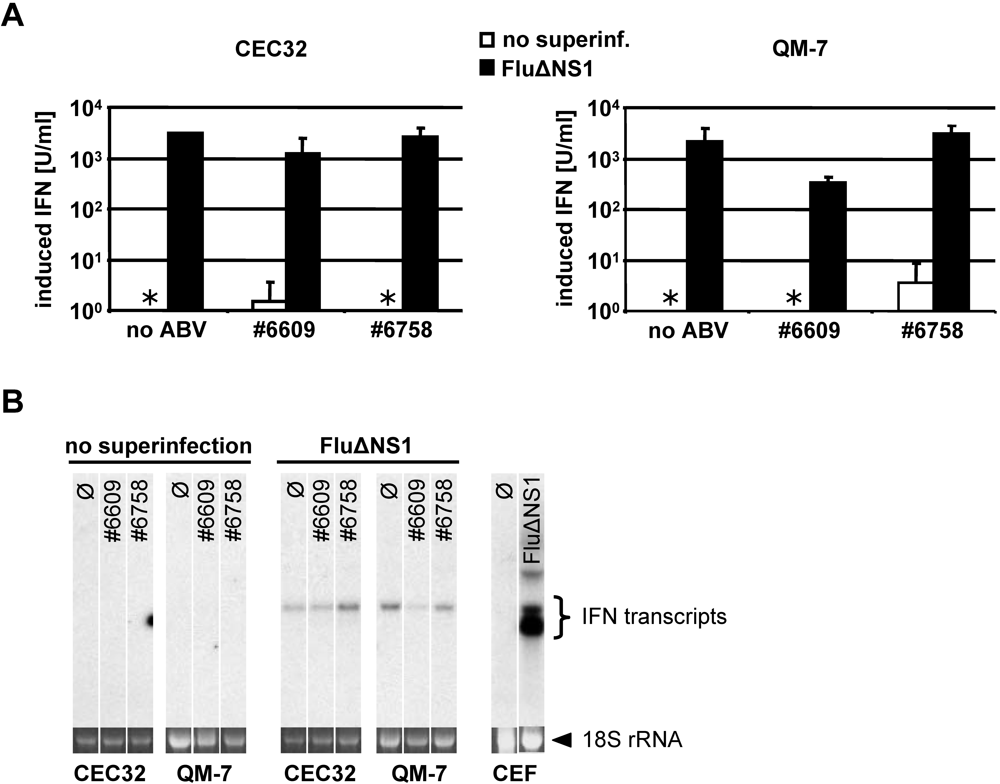
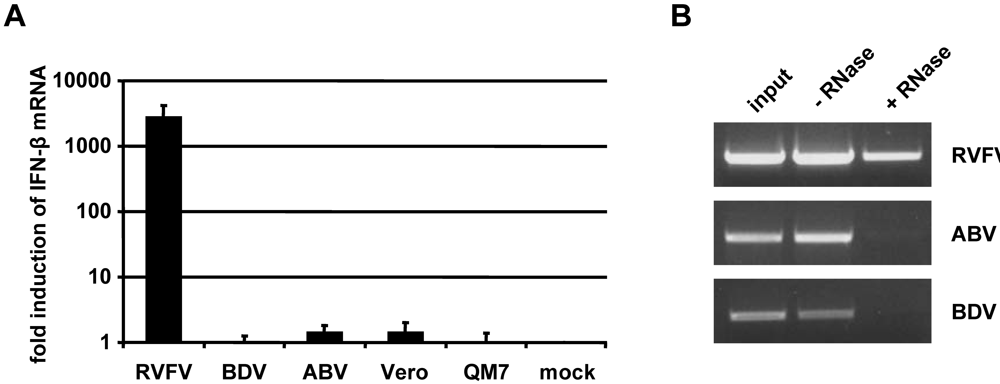
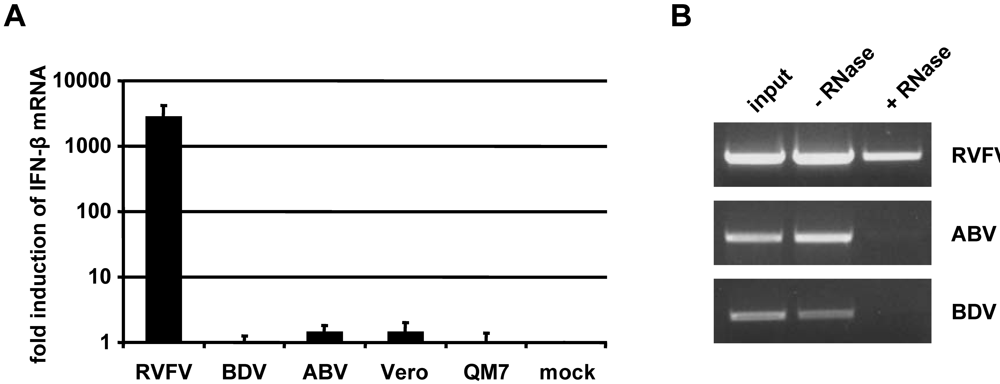
2.4. Inefficient activation of type I IFN genes in ABV-infected quail cells
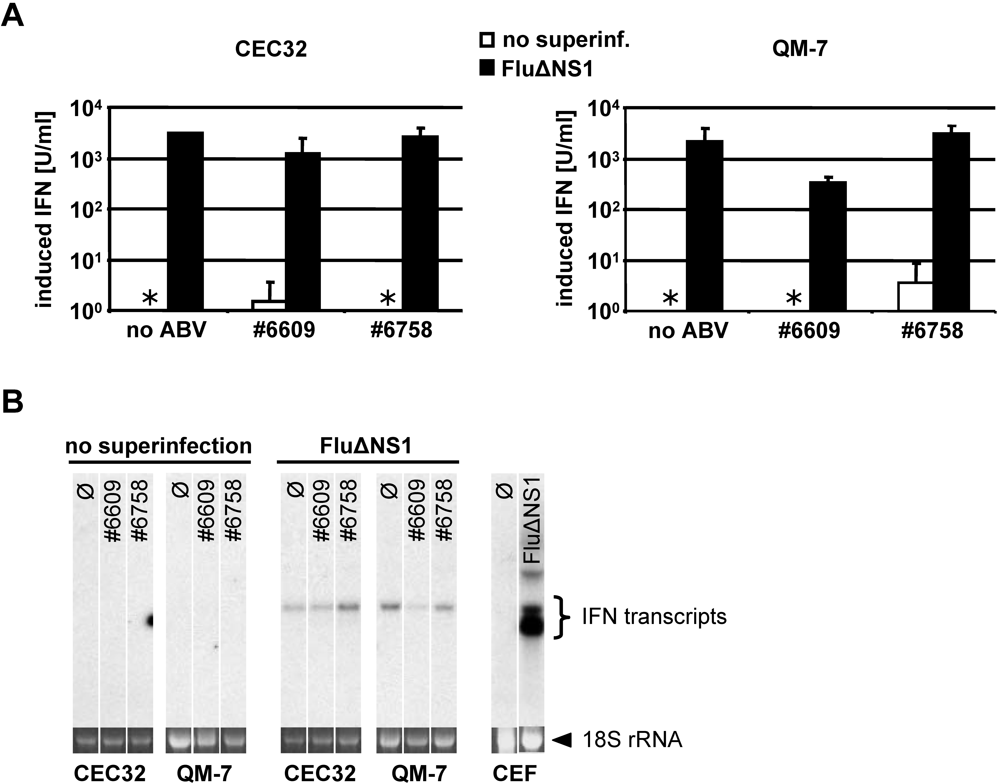
2.5. Persisting ABV infection does not prevent IFN induction by super-infecting influenza virus
2.6. No evidence for triphosphorylated 5’-termini of ABV genomic RNA
3. Experimental Section
3.1. Viruses
3.2. Production of antiserum against ABV-N
3.3. Cell lines and culture conditions
3.4. RNA isolation
3.5. Measuring IFN-β gene induction after RNA transfection
3.6. Digestion of RNA with 5’-monophosphate specific RNase
3.7. Detection of ABV-infected cells by immunofluorescence analysis
3.8. Detection of ABV-N in QM-7 cell lysates by Western blotting
3.9. Bioassay for type I IFN
3.10. IFN induction using SC35M-delNS1
3.11. Northern blot analysis
4. Conclusions
Acknowledgement
References
- Hallensleben, W.; Staeheli, P. Inhibition of Borna disease virus multiplication by interferon: cell line differences in susceptibility. Arch. Virol. 1999, 144, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G.; Weber, F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 2006, 344, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Gale Jr., M.; Foy, E.M. Evasion of intracellular host defence by hepatitis C virus . Nature 2005, 436, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Andersson, I.; Klingstrom, J.; Schumann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Muhlberger, E.; Mirazimi, A.; Weber, F. Processing of genome 5' termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction . PLoS One 2008, 3, e2032. [Google Scholar] [CrossRef] [PubMed]
- Staeheli, P.; Sentandreu, M.; Pagenstecher, A.; Hausmann, J. Alpha/beta interferon promotes transcription and inhibits replication of borna disease virus in persistently infected cells. J. Virol. 2001, 75, 8216–8223. [Google Scholar] [CrossRef] [PubMed]
- Unterstab, G.; Ludwig, S.; Anton, A.; Planz, O.; Dauber, B.; Krappmann, D.; Heins, G.; Ehrhardt, C.; Wolff, T. Viral targeting of the interferon-{beta}-inducing Traf family member-associated NF-{kappa}B activator (TANK)-binding kinase-1. Proc. Natl. Acad. Sci. USA 2005, 102, 13640–13645. [Google Scholar] [CrossRef]
- Schneider, U.; Martin, A.; Schwemmle, M.; Staeheli, P. Genome trimming by Borna disease viruses: viral replication control or escape from cellular surveillance? Cell Mol. Life Sci. 2007, 64, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.; Schwemmle, M.; Staeheli, P. Genome trimming: a unique strategy for replication control employed by Borna disease virus. Proc. Natl. Acad. Sci. USA 2005, 102, 3441–3446. [Google Scholar] [CrossRef]
- Kistler, A.L.; Gancz, A.; Clubb, S.; Skewes-Cox, P.; Fischer, K.; Sorber, K.; Chiu, C.Y.; Lublin, A.; Mechani, S.; Farnoushi, Y.; Greninger, A.; Wen, C.C.; Karlene, S.B.; Ganem, D.; DeRisi, J.L. Recovery of divergent avian bornaviruses from cases of proventricular dilatation disease: identification of a candidate etiologic agent. Virol. J. 2008, 5, 88. [Google Scholar] [CrossRef]
- Honkavuori, K.S.; Shivaprasad, H.L.; Williams, B.L.; Quan, P.L.; Hornig, M.; Street, C.; Palacios, G.; Hutchison, S.K.; Franca, M.; Egholm, M.; Briese, T.; Lipkin, W.I. Novel borna virus in psittacine birds with proventricular dilatation disease. Emerg. Infect. Dis. 2008, 14, 1883–1886. [Google Scholar] [CrossRef] [PubMed]
- Rinder, M.; Ackermann, A.; Kempf, H.; Kaspers, B.; Korbel, R.; Staeheli, P. Broad tissue and cell tropism of avian bornavirus in parrots with proventricular dilatation disease. J. Virol. 2009, 83, 5401–5407. [Google Scholar] [CrossRef] [PubMed]
- Gancz, A.Y.; Kistler, A.L.; Greninger, A.L.; Farnoushi, Y.; Mechani, S.; Perl, S.; Berkowitz, A.; Perez, N.; Clubb, S.; Derisi, J.L.; Ganem, D.; Lublin, A. Experimental induction of proventricular dilatation disease in cockatiels (Nymphicus hollandicus) inoculated with brain homogenates containing avian bornavirus 4. Virol. J. 2009, 6, 100. [Google Scholar] [CrossRef]
- Weissenböck, H.; Bakonyi, T.; Sekulin, K.; Ehrensperger, F.; Doneley, R.; Dürrwald, R.; Hoop, R.; Erdélyi, K.; Gál, J.; Kolodziejek, J.; Nowotny, N. Avian bornaviruses in psittacine birds from Europe and Australia with proventricular dilatation disease. Emerg. Infect. Dis. 2009, 15, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Weissenböck, H.; Sekulin, K.; Bakonyi, T.; Högler, S.; Nowotny, N. Novel avian bornavirus in a nonpsittacine species (Canary; Serinus canaria) with enteric ganglioneuritis and encephalitis. J. Virol. 2009, 83, 11367–11371. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, H.; Harlin, O.; Ohnemus, A.; Kaspers, B.; Staeheli, P. Synthesis of IFN-beta by virus-infected chicken embryo cells demonstrated with specific antisera and a new bioassay . J. Interferon Cytokine Res. 2004, 24, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Koerner, I.; Thiel, L.; Kothlow, S.; Kaspers, B.; Ruggli, N.; Summerfield, A.; Pavlovic, J.; Stech, J.; Staeheli, P. Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J. Gen. Virol. 2007, 88, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Schultz, U.; Rinderle, C.; Sekellick, M. J.; Marcus, P.I.; Staeheli, P. Recombinant chicken interferon from Escherichia coli and transfected COS cells is biologically active. Eur. J. Biochem. 1995, 229, 73–76. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Reuter, A.; Ackermann, A.; Kothlow, S.; Rinder, M.; Kaspers, B.; Staeheli, P. Avian Bornaviruses Escape Recognition by the Innate Immune System. Viruses 2010, 2, 927-938. https://doi.org/10.3390/v2040927
Reuter A, Ackermann A, Kothlow S, Rinder M, Kaspers B, Staeheli P. Avian Bornaviruses Escape Recognition by the Innate Immune System. Viruses. 2010; 2(4):927-938. https://doi.org/10.3390/v2040927
Chicago/Turabian StyleReuter, Antje, Andreas Ackermann, Sonja Kothlow, Monika Rinder, Bernd Kaspers, and Peter Staeheli. 2010. "Avian Bornaviruses Escape Recognition by the Innate Immune System" Viruses 2, no. 4: 927-938. https://doi.org/10.3390/v2040927
APA StyleReuter, A., Ackermann, A., Kothlow, S., Rinder, M., Kaspers, B., & Staeheli, P. (2010). Avian Bornaviruses Escape Recognition by the Innate Immune System. Viruses, 2(4), 927-938. https://doi.org/10.3390/v2040927