
In Vivo Emergence of Podovirus Resistance via tarS Mutation During Phage-Antibiotic Treatment of Experimental MSSA Endocarditis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Bacterial and Phage Strains and Growth Conditions
2.2. Phage Susceptibility Testing (PST)
2.3. Genomic DNA Extraction and Purification, PCR, Plasmid Construction, and Transformation
2.4. In Vitro Assays
2.5. Whole Genome Sequencing (WGS) and Comparative Genomics
2.6. RNA Purification, Sequencing, and Analysis
3. Results
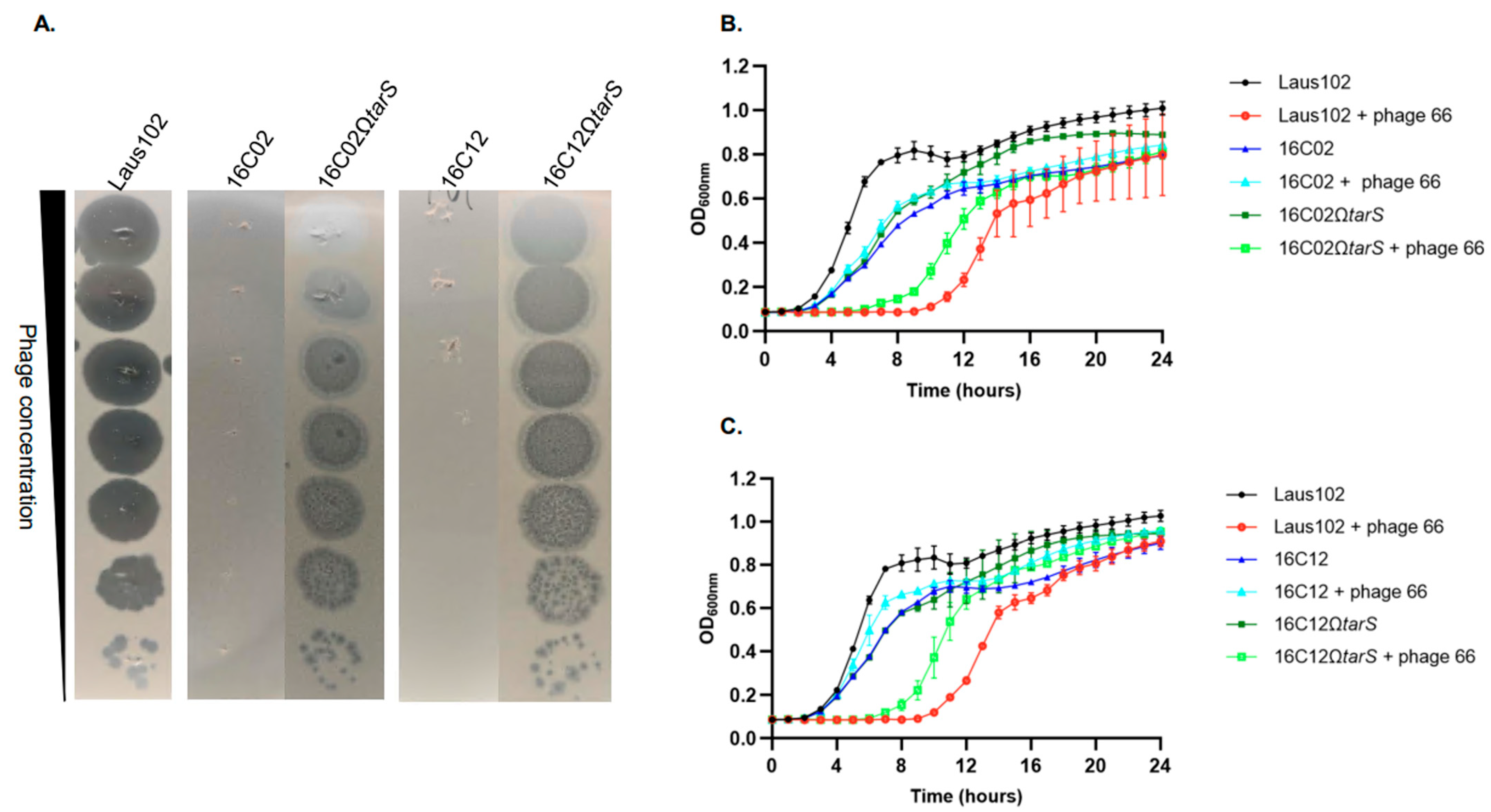
3.1. Resistance to the Podovirus, but Not to the Myovirus or Flucloxacillin, Was Selected in Rat Vegetations Treated with a Flucloxacillin/Two-Phage Cocktail Combination
3.2. Mutations in tarS as the Likely Genetic Basis for In Vivo Podovirus Resistance
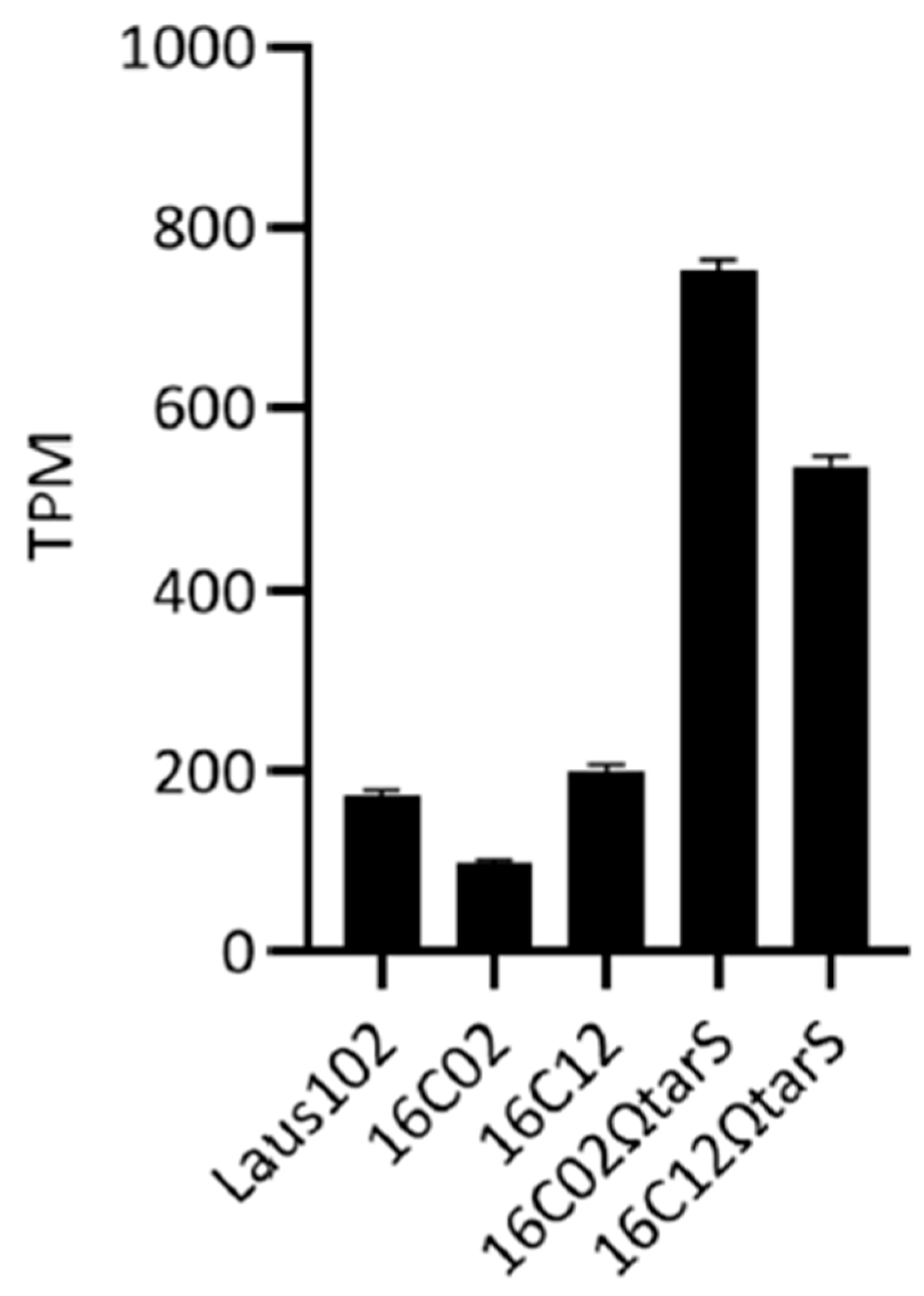
3.3. Expression Levels of tarS Did Not Account for Resistance
3.4. Complementation with Wild-Type tarS Restored Phage 66 Susceptibility
3.5. Resistance Correlated with Impaired Phage Adsorption
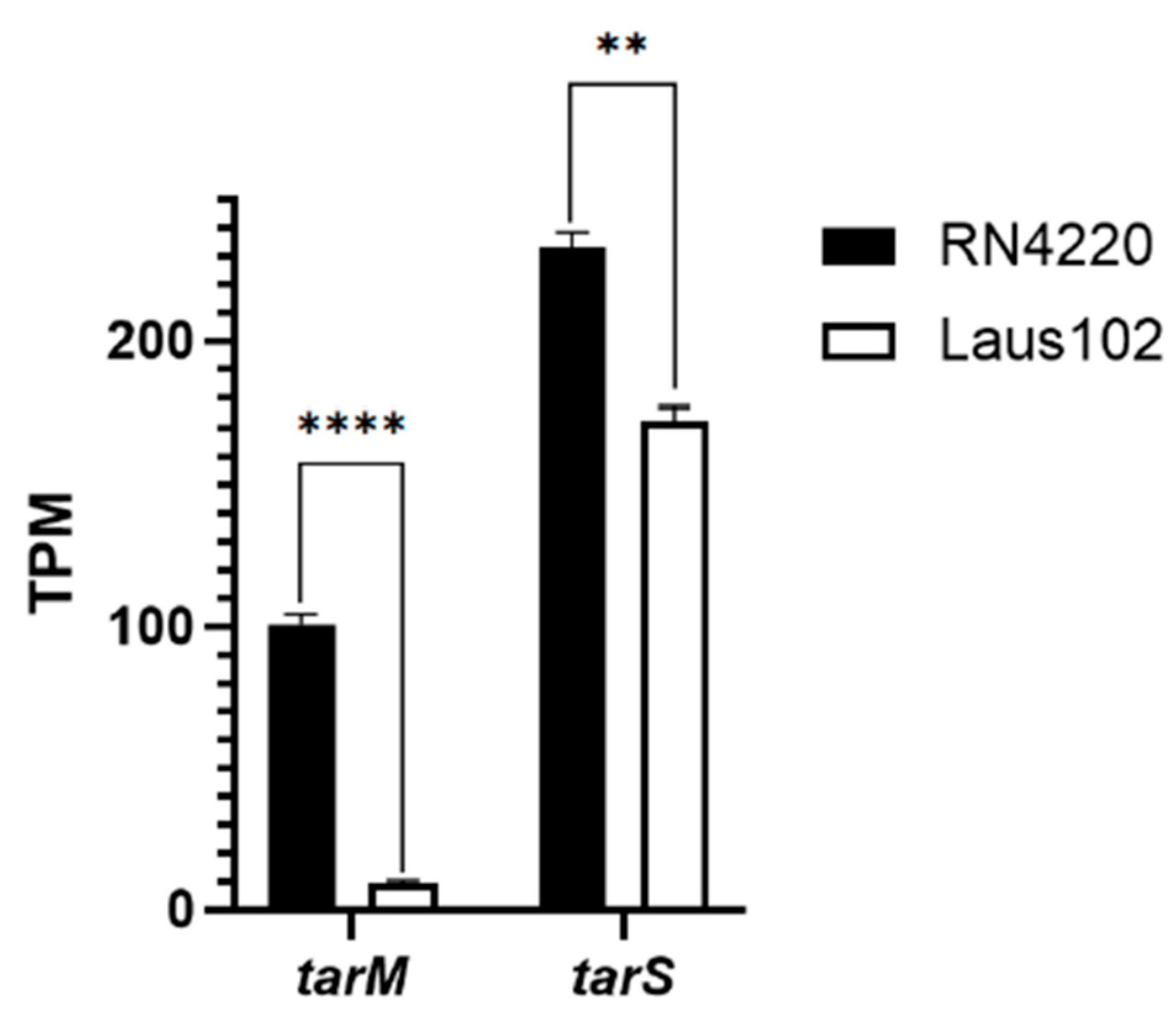
3.6. Analysis of tarM and tarP Sequences
3.7. Low tarM Expression Likely Explains Podovirus Innate Susceptibility of Laus102
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frieri, M.; Kumar, K.; Boutin, A. Antibiotic resistance. J. Infect. Public Health 2017, 10, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Pirnay, J.P.; Ferry, T.; Resch, G. Recent progress toward the implementation of phage therapy in Western medicine. FEMS Microbiol. Rev. 2022, 46, fuab040. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Azam, A.H.; Tanji, Y. Bacteriophage-host arm race: An update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy. Appl. Microbiol. Biotechnol. 2019, 103, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Corrigan, R.M.; Winstel, V.; Goerke, C.; Grundling, A.; Peschel, A. Wall teichoic Acid-dependent adsorption of staphylococcal siphovirus and myovirus. J. Bacteriol. 2011, 193, 4006–4009. [Google Scholar] [CrossRef] [PubMed]
- Azam, A.H.; Kadoi, K.; Miyanaga, K.; Usui, M.; Tamura, Y.; Cui, L.; Tanji, Y. Analysis host-recognition mechanism of staphylococcal kayvirus ΦSA039 reveals a novel strategy that protects Staphylococcus aureus against infection by Staphylococcus pseudintermedius Siphoviridae phages. Appl. Microbiol. Biotechnol. 2019, 103, 6809–6823. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, I.; Osada, K.; Azam, A.H.; Asakawa, H.; Miyanaga, K.; Tanji, Y. The Presence of Two Receptor-Binding Proteins Contributes to the Wide Host Range of Staphylococcal Twort-Like Phages. Appl. Environ. Microbiol. 2016, 82, 5763–5774. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Kohler, T.; Peschel, A. The wall teichoic acid and lipoteichoic acid polymers of Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Winstel, V.; Sanchez-Carballo, P.; Holst, O.; Xia, G.; Peschel, A. Biosynthesis of the unique wall teichoic acid of Staphylococcus aureus lineage ST395. mBio 2014, 5, e00869. [Google Scholar] [CrossRef] [PubMed]
- Wanner, S.; Schade, J.; Keinhorster, D.; Weller, N.; George, S.E.; Kull, L.; Bauer, J.; Grau, T.; Winstel, V.; Stoy, H.; et al. Wall teichoic acids mediate increased virulence in Staphylococcus aureus. Nat. Microbiol. 2017, 2, 16257. [Google Scholar] [CrossRef] [PubMed]
- Koc, C.; Gerlach, D.; Beck, S.; Peschel, A.; Xia, G.; Stehle, T. Structural and enzymatic analysis of TarM glycosyltransferase from Staphylococcus aureus reveals an oligomeric protein specific for the glycosylation of wall teichoic acid. J. Biol. Chem. 2015, 290, 9874–9885. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pfahler, N.M.; Volpel, S.L.; Stehle, T. Cell wall glycosylation in Staphylococcus aureus: Targeting the tar glycosyltransferases. Curr. Opin. Struct. Biol. 2021, 68, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gerlach, D.; Du, X.; Larsen, J.; Stegger, M.; Kuhner, P.; Peschel, A.; Xia, G.; Winstel, V. An accessory wall teichoic acid glycosyltransferase protects Staphylococcus aureus from the lytic activity of Podoviridae. Sci. Rep. 2015, 5, 17219. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Melo, L.; Vilas Boas, D.; Sillankorva, S.; Azeredo, J. Phage therapy as an alternative or complementary strategy to prevent and control biofilm-related infections. Curr. Opin. Microbiol. 2017, 39, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Save, J.; Que, Y.A.; Entenza, J.M.; Kolenda, C.; Laurent, F.; Resch, G. Bacteriophages Combined With Subtherapeutic Doses of Flucloxacillin Act Synergistically Against Staphylococcus aureus Experimental Infective Endocarditis. J. Am. Heart Assoc. 2022, 11, e023080. [Google Scholar] [CrossRef] [PubMed]
- Sakwinska, O.; Giddey, M.; Moreillon, M.; Morisset, D.; Waldvogel, A.; Moreillon, P. Staphylococcus aureus host range and human-bovine host shift. Appl. Environ. Microbiol. 2011, 77, 5908–5915. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.; Memmi, G.; Hernandez, D.; Bard, J.; Beaume, M.; Gill, S.; Francois, P.; Cheung, A.L. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol. 2011, 193, 2332–2335. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Glass, E.M.; Schneewind, O.; Missiakas, D. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. Methods Mol. Biol. 2008, 416, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Lofblom, J.; Kronqvist, N.; Uhlen, M.; Stahl, S.; Wernerus, H. Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. J. Appl. Microbiol. 2007, 102, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Beltrame, C.O.; Cortes, M.F.; Bandeira, P.T.; Figueiredo, A.M. Optimization of the RNeasy Mini Kit to obtain high-quality total RNA from sessile cells of Staphylococcus aureus. Braz. J. Med. Biol. Res. 2015, 48, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Sobhanifar, S.; Worrall, L.J.; King, D.T.; Wasney, G.A.; Baumann, L.; Gale, R.T.; Nosella, M.; Brown, E.D.; Withers, S.G.; Strynadka, N.C. Structure and Mechanism of Staphylococcus aureus TarS, the Wall Teichoic Acid beta-glycosyltransferase Involved in Methicillin Resistance. PLoS Pathog. 2016, 12, e1006067. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, A.; van Dalen, R.; Ali, S.; Gerlach, D.; van der Marel, G.A.; Fuchsberger, F.F.; Aerts, P.C.; de Haas, C.J.C.; Peschel, A.; Rademacher, C.; et al. Impact of Glycan Linkage to Staphylococcus aureus Wall Teichoic Acid on Langerin Recognition and Langerhans Cell Activation. ACS Infect. Dis. 2021, 7, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Winstel, V.; Xia, G.; Peschel, A. Pathways and roles of wall teichoic acid glycosylation in Staphylococcus aureus. Int. J. Med. Microbiol. 2014, 304, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.; Guo, Y.; De Castro, C.; Kim, S.H.; Schlatterer, K.; Xu, F.F.; Pereira, C.; Seeberger, P.H.; Ali, S.; Codee, J.; et al. Methicillin-resistant Staphylococcus aureus alters cell wall glycosylation to evade immunity. Nature 2018, 563, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Sobhanifar, S.; Worrall, L.J.; Gruninger, R.J.; Wasney, G.A.; Blaukopf, M.; Baumann, L.; Lameignere, E.; Solomonson, M.; Brown, E.D.; Withers, S.G.; et al. Structure and mechanism of Staphylococcus aureus TarM, the wall teichoic acid alpha-glycosyltransferase. Proc. Natl. Acad. Sci. USA 2015, 112, E576–E585. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Levin, B.R. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 1999, 2, 489–493. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherbuin, J.; Save, J.; Osswald, E.; Resch, G. In Vivo Emergence of Podovirus Resistance via tarS Mutation During Phage-Antibiotic Treatment of Experimental MSSA Endocarditis. Viruses 2025, 17, 1039. https://doi.org/10.3390/v17081039
Cherbuin J, Save J, Osswald E, Resch G. In Vivo Emergence of Podovirus Resistance via tarS Mutation During Phage-Antibiotic Treatment of Experimental MSSA Endocarditis. Viruses. 2025; 17(8):1039. https://doi.org/10.3390/v17081039
Chicago/Turabian StyleCherbuin, Jérémy, Jonathan Save, Emma Osswald, and Grégory Resch. 2025. "In Vivo Emergence of Podovirus Resistance via tarS Mutation During Phage-Antibiotic Treatment of Experimental MSSA Endocarditis" Viruses 17, no. 8: 1039. https://doi.org/10.3390/v17081039
APA StyleCherbuin, J., Save, J., Osswald, E., & Resch, G. (2025). In Vivo Emergence of Podovirus Resistance via tarS Mutation During Phage-Antibiotic Treatment of Experimental MSSA Endocarditis. Viruses, 17(8), 1039. https://doi.org/10.3390/v17081039