GCRV-II Triggers B and T Lymphocyte Apoptosis via Mitochondrial ROS Pathway

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Isolation and Culture
2.3. Viral Infection
2.4. Apoptosis Detection
2.5. Mitochondrial Membrane Potential Measurement
2.6. Determination of Reactive Oxygen Species (ROS) and Mitochondrial ROS
2.7. Inhibitor Treatment
2.8. Transmission Electron Microscopy
2.9. RNA-seq
2.10. Semi-Quantitative and Quantitative Real-Time PCR
2.11. SDS-PAGE and Western Blot
2.12. Statistical Analysis
3. Results
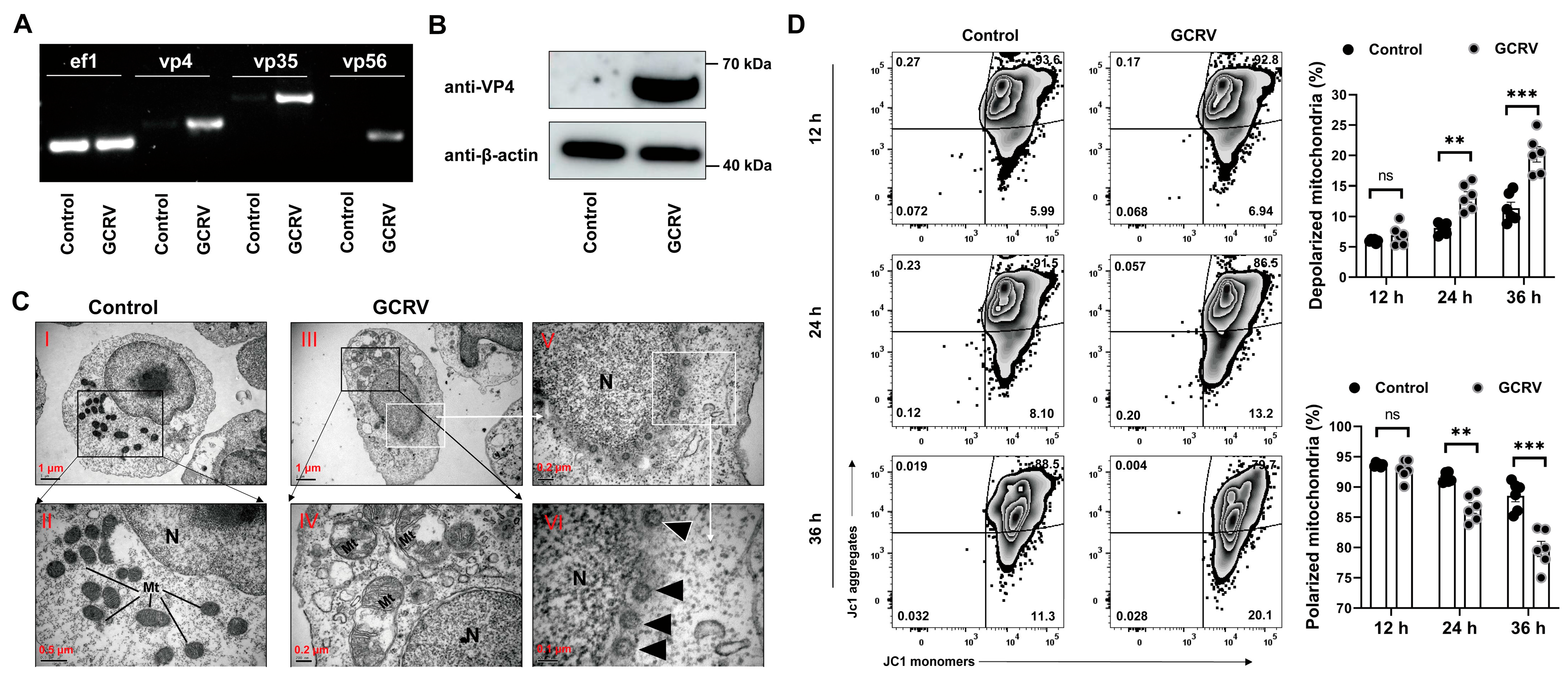
3.1. GCRV-II Infects PBLs and Induces Mitochondrial Dysfunction
3.2. GCRV-II Triggers Mitochondria-Dependent Apoptosis Pathway in PBLs
3.3. GCRV-II Induces Cell Apoptosis and Necrosis Through Caspase-3 Activation
3.4. GCRV-II Induces ROS Production and Apoptosis in IgM+ B and CD4+T Lymphocytes
3.5. Inhibition of mtROS Production Attenuates GCRV-II-Induced Apoptosis in IgM+ B and CD4+ T Lymphocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PBLs | Peripheral blood leukocytes |
| GCRV-II | Type II grass carp reovirus |
| ER | Endoplasmic reticulum |
| Mo/Mφs | Monocytes/macrophages |
| ROS | Reactive oxygen species |
| MMP | Mitochondrial permeabilization |
| mtROS | Mitochondrial reactive oxygen species |
| MOI | Multiplicity of infection |
| TEM | Transmission electron microscopy |
| GSEA | Gene set enrichment analysis |
References
- Li, P.W.; Zhang, J.; Chang, M.X. Structure, function and immune evasion strategies of aquareoviruses, with focus on grass carp reovirus. Rev. Aquac. 2023, 16, 410–432. [Google Scholar] [CrossRef]
- Jiang, R.; Zhang, J.; Liao, Z.W.; Zhu, W.T.; Su, H.; Zhang, Y.A.; Su, J.G. Temperature-regulated type II grass carp reovirus establishes latent infection in Ctenopharyngodon idella brain. Virol. Sin. 2023, 38, 440–447. [Google Scholar] [CrossRef]
- Zhu, W.T.; Qiao, M.H.; Hu, M.D.; Huo, X.C.; Zhang, Y.A.; Su, J.G. Type II grass carp reovirus rapidly invades grass carp (Ctenopharyngodon idella) via nostril–olfactory system–brain axis, gill, and skin on head. Viruses 2023, 15, 1614. [Google Scholar] [CrossRef]
- Yang, L.; Su, J.G. Type II grass carp reovirus infects leukocytes but not erythrocytes and thrombocytes in grass carp (Ctenopharyngodon idella). Viruses 2021, 13, 870. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, A.Q.; Zhang, C.; Zhang, Y.A.; Tu, J.G. Hsp90 regulates GCRV-II proliferation by interacting with VP35 as its receptor and chaperone. J. Virol. 2022, 96, 19. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.L.; Li, Y.Q.; Lin, G.; Zhang, J.J.; Jiang, N.; Liu, W.Z.; Meng, Y.; Zhou, Y.; Fan, Y.D. Non-pathogenic grass carp reovirus infection leads to both apoptosis and autophagy in a grass carp cell line. Fish Shellfish Immunol. 2022, 127, 681–689. [Google Scholar] [CrossRef]
- Jia, R.; Cao, L.P.; Du, J.L.; Liu, Y.J.; Wang, J.H.; Jeney, G.; Yin, G.J. Grass carp reovirus induces apoptosis and oxidative stress in grass carp (Ctenopharyngodon idellus) kidney cell line. Virus Res. 2014, 185, 77–81. [Google Scholar] [CrossRef]
- Liang, B.; Su, J.G. Inducible nitric oxide synthase (iNOS) mediates vascular endothelial cell apoptosis in grass carp reovirus (GCRV)-induced hemorrhage. Int. J. Mol. Sci. 2019, 20, 6335. [Google Scholar] [CrossRef]
- Zhao, K.W.; Zhang, Y.S.; Yin, Z.J.; Tan, L.B.; Juario, M.; Zhang, H.Y.; Liu, Y.L.; Xu, P.X.; Zhang, Q.; Zhao, G.A.; et al. GCRV-II major outer capsid protein VP4 promotes cell apoptosis by VDAC2-mediated calcium pathway facilitation. Int. J. Biol. Macromol. 2025, 285, 138273. [Google Scholar] [CrossRef]
- Xia, N.; Zhang, Y.Q.; Zhu, W.T.; Su, J.G. GCRV-II invades monocytes/macrophages and induces macrophage polarization and apoptosis in tissues to facilitate viral replication and dissemination. J. Virol. 2024, 98, 3. [Google Scholar] [CrossRef]
- Barry, G.; Fragkoudis, R.; Ferguson, M.C.; Lulla, A.; Merits, A.; Kohl, A.; Fazakerley, J.K. Semliki forest virus-induced endoplasmic reticulum stress accelerates apoptotic death of mammalian cells. J. Virol. 2010, 84, 7369–7377. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gorshkov, K.; Lee, E.M.; Xu, M.; Cheng, Y.S.; Sun, N.; Soheilian, F.; de Val, N.; Ming, G.L.; Song, H.J.; et al. Zika virus-induced neuronal apoptosis via increased mitochondrial fragmentation. Front. Microbiol. 2020, 11, 598203. [Google Scholar] [CrossRef] [PubMed]
- Reshi, M.L.; Su, Y.-C.; Hong, J.-R. RNA viruses: ROS-mediated cell death. Int. J. Cell Biol. 2014, 2014, 1–16. [Google Scholar] [CrossRef]
- Mayank, A.K.; Sharma, S.; Nailwal, H.; Lal, S.K. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef]
- Ampomah, P.B.; Lim, L.H.K. Influenza A virus-induced apoptosis and virus propagation. Apoptosis 2020, 25, 1–11. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Zhou, X.C.; Jiang, W.B.; Liu, Z.S.; Liu, S.; Liang, X.Z. Virus infection and death receptor-mediated apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef]
- Finlay, B.B.; Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008, 4, e1000018. [Google Scholar]
- Incrocci, R.; Hussain, S.; Stone, A.; Bieging, K.; Alt, L.A.C.; Fay, M.J.; Swanson-Mungerson, M. Epstein-barr virus latent membrane protein 2A (LMP2A)-mediated changes in Fas expression and Fas-dependent apoptosis: Role of Lyn/Syk activation. Cell Immunol. 2015, 297, 108–119. [Google Scholar] [CrossRef]
- Takahashi, M.; Higuchi, M.; Makokha, G.N.; Matsuki, H.; Yoshita, M.; Tanaka, Y.; Fujii, M. HTLV-1 Tax oncoprotein stimulates ROS production and apoptosis in T cells by interacting with USP10. Blood 2013, 122, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Schank, M.; Zhao, J.; Wang, L.; Nguyen, L.N.T.; Zhang, Y.; Wu, X.Y.; Zhang, J.Y.; Jiang, Y.; Ning, S.B.; El Gazzar, M.; et al. ROS-induced mitochondrial dysfunction in CD4 T cells from ART-controlled people living with HIV. Viruses 2023, 15, 1061. [Google Scholar] [CrossRef]
- Stapleford, K.; Verburg, S.G.; Lelievre, R.M.; Westerveld, M.J.; Inkol, J.M.; Sun, Y.L.; Workenhe, S.T. Viral-mediated activation and inhibition of programmed cell death. PLoS Pathog. 2022, 18, e1010718. [Google Scholar]
- El-Amine, R.; Germini, D.; Zalcharova, V.V.; Tsfasman, T.; Sheval, E.V.; Louzada, R.A.N.; Dupuy, C.; Bilhou-Nabera, C.; Hamade, A.; Najjar, F.; et al. HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 2018, 15, 97–108. [Google Scholar] [CrossRef]
- Gimenez, S.; Hamrouni, E.; André, S.; Picard, M.; Soundaramourty, C.; Lozano, C.; Vincent, T.; Tran, T.-A.; Kundura, L.; Estaquier, J.; et al. Monocytic reactive oxygen species-induced T cell apoptosis impairs cellular immune response to SARS-CoV-2 mRNA vaccine. J. Allergy Clin. Immunol. 2025, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kundura, L.; Gimenez, S.; Cezar, R.; André, S.; Younas, M.; Lin, Y.L.; Portalès, P.; Lozano, C.; Boulle, C.; Reynes, J.; et al. Angiotensin II induces reactive oxygen species, DNA damage, and T-cell apoptosis in severe COVID-19. J. Allergy Clin. Immun. 2022, 150, 594–603. [Google Scholar] [CrossRef]
- Rahmani, Z.; Huh, K.W.; Lasher, R.; Siddiqui, A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef]
- Piccoli, C.; Scrima, R.; Quarato, G.; D′Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Moradpour, D.; Capitanio, N. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 2007, 46, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Lecoeur, H.; Borgne-Sanchez, A.; Chaloin, O.; El-Khoury, R.; Brabant, M.; Langonné, A.; Porceddu, M.; Brière, J.J.; Buron, N.; Rebouillat, D.; et al. HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome oxidase. Cell Death Dis. 2012, 3, e282. [Google Scholar] [CrossRef]
- Boonnate, P.; Kariya, R.; Okada, S. Shikonin induces ROS-dependent apoptosis via mitochondria depolarization and ER stress in adult T cell leukemia/lymphoma. Antioxidants 2023, 12, 864. [Google Scholar] [CrossRef]
- Meuren, L.M.; Prestes, E.B.; Papa, M.P.; de Carvalho, L.R.P.; Mustafa, Y.M.; da Costa, L.S.; Da Poian, A.T.; Bozza, M.T.; Arruda, L.B. Infection of endothelial cells by dengue virus induces ROS production by different sources affecting virus replication, cellular activation, death and vascular permeability. Front. Immunol. 2022, 13, 810376. [Google Scholar] [CrossRef]
- Zhang, Y.A.; Salinas, I.; Li, J.; Parra, D.; Bjork, S.; Xu, Z.; LaPatra, S.E.; Bartholomew, J.; Sunyer, J.O. IgT, a primitive immunoglobulin class specialized in mucosal immunity. Nat. Immunol. 2010, 11, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.W.; Zhang, X.Y.; Wu, C.S.; Zhang, Y.A.; Zhou, Y.; Zhang, X.J. Membrane IgM+ plasma cells in grass carp (Ctenopharyngodon idella): Insights into the conserved evolution of IgM+ plasma cells in vertebrates. Dev. Comp. Immunol. 2020, 106, 103613. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.Z.; Liu, X.; Wu, C.S.; Ma, Z.Y.; Wang, Y.; Zhang, Y.A.; Zhang, X.J. Molecular and functional analyses of the primordial costimulatory molecule CD80/86 and its receptors CD28 and CD152 (CTLA-4) in a teleost fish. Front. Immunol. 2022, 13, 885005. [Google Scholar] [CrossRef] [PubMed]
- Han, X.Q.; Pan, Y.R.; Zhong, Y.Q.; Tian, T.T.; Liu, X.; Zhang, X.J.; Zhang, Y.A. Identification and functional analyses of CD4-1+ cells in grass carp (Ctenopharyngodon idella). Fish Shellfish Immunol. 2024, 150, 109649. [Google Scholar] [CrossRef]
- Liu, J.X.; Ren, J.Q.; Wang, D.; Wang, Z.; Ma, X.Y.; Zhou, H. Edwardsiella piscicida promotes mitophagy to escape autophagy-mediated antibacterial defense in teleost monocytes/macrophages. Aquaculture 2025, 596, 741–784. [Google Scholar] [CrossRef]
- Lv, M.Y.; Wang, Y.W.; Yu, J.Z.; Kong, Y.Y.; Zhou, H.; Zhang, A.Y.; Wang, X.Y. Grass carp IL-2 promotes neutrophil extracellular traps formation via inducing ROS production and autophagy in vitro. Fish Shellfish Immunol. 2024, 144, 109261. [Google Scholar] [CrossRef]
- Li, J.; Barreda, D.R.; Zhang, Y.A.; Boshra, H.; Gelman, A.E.; Lapatra, S.; Tort, L.; Sunyer, J.O. B lymphocytes from early vertebrates have potent phagocytic and microbicidal abilities. Nat. Immunol. 2006, 7, 1116–1124. [Google Scholar] [CrossRef]
- Wu, C.S.; Dai, Y.S.; Yuan, G.L.; Su, J.G.; Liu, X.L. Immunomodulatory effects and induction of apoptosis by different molecular weight chitosan oligosaccharides in head kidney macrophages from blunt snout bream (Megalobrama amblycephala). Front. Immunol. 2019, 10, 869. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Cui, Y.; Huang, Y.F.; Liu, H.; Li, L.; Li, M.; Ruan, K.C.; Zhou, Q.; Wang, C. Tom70 mediates sendai virus-induced apoptosis on mitochondria. J. Virol. 2015, 89, 3804–3818. [Google Scholar] [CrossRef]
- Rao, Y.L.; Wan, Q.Y.; Su, H.; Xiao, X.; Liao, Z.W.; Ji, J.F.; Yang, C.R.; Lin, L.; Su, J.G. ROS-induced HSP70 promotes cytoplasmic translocation of high-mobility group box 1b and stimulates antiviral autophagy in grass carp kidney cells. J. Biol. Chem. 2018, 293, 17387–17401. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.S.; Li, C.; Bai, C.M.; Wang, C.M. Ostreid Herpesvirus-1 Infects Specific Hemocytes in Ark Clam. Scapharca Broughtonii Viruses 2018, 10, 529. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Liu, W.; Seah, J.N.; Lam, C.S.; Xiang, J.H.; Korzh, V.; Kwang, J. White spot syndrome virus (WSSV) infects specific hemocytes of the shrimp. Dis. Aquat. Organ. 2002, 52, 249–259. [Google Scholar] [CrossRef]
- Raja, R.; Sen, G.C. The antiviral action of the RIG-I induced pathway of apoptosis (RIPA) is enhanced by its ability to degrade Otulin, which deubiquitinates IRF3. Cell Death Differ. 2022, 29, 504–513. [Google Scholar] [CrossRef]
- Zhu, H.C.; Hou, P.L.; Chu, F.Y.; Li, X.Y.; Zhang, W.J.; Sun, X.N.; Liu, Y.; Zhao, G.M.; Gao, Y.W.; He, D.C.; et al. PBLD promotes IRF3 mediated the type I interferon (IFN-I) response and apoptosis to inhibit viral replication. Cell Death Dis. 2024, 15, 727. [Google Scholar] [CrossRef]
- Simpson, D.S.; Pang, J.; Weir, A.; Kong, I.Y.; Fritsch, M.; Rashidi, M.; Cooney, J.P.; Davidson, K.C.; Speir, M.; Djajawi, T.M.; et al. Interferon-γ primes macrophages for pathogen ligand-induced killing via a caspase-8 and mitochondrial cell death pathway. Immunity 2022, 55, 423–441.e9. [Google Scholar] [CrossRef]
- Nichols, J.E.; Niles, J.A.; Roberts, N.J. Human lymphocyte apoptosis after exposure to influenza A virus. J. Virol. 2001, 75, 5921–5929. [Google Scholar] [CrossRef]
- Roberts, N.J. The enigma of lymphocyte apoptosis in the response to influenza virus infection. Viruses 2023, 15, 759. [Google Scholar] [CrossRef]
- Nichols, J.E.; Niles, J.A.; Fleming, E.H.; Roberts, N.J. The role of cell surface expression of influenza virus neuraminidase in induction of human lymphocyte apoptosis. Virology 2019, 534, 80–86. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence 5′-3′ | Tm (°C) | Product Size (bp) |
|---|---|---|---|
| Ef1-F | CGCCAGTGTTGCCTTCGT | 58 | 99 |
| Ef1-R | CGCTCAATCTTCCATCCCTT | ||
| Bax-F | CATCTATGAGCGGGTTCGTC | 57 | 94 |
| Bax-R | TTTATGGCTGGGGTCACACA | ||
| Cytc-F | TGCATACTTAAAGTCAGCCACATC | 58 | 97 |
| Cytc-R | TAAAGACATCCGAATCCAAACAAC | ||
| Apaf1-F | AAGTTCTGGAGCCTGGACAC | 56 | 106 |
| Apaf1-R | AACTCAAGACCCCACAGCAC | ||
| Caspase8-F | GGTAATCTGGTTGAAATCCGTG | 56 | 98 |
| Caspase8-R | CCTTGGCAGGCTTGAATGA | ||
| Caspase9-F | CCTGGAGCAGTTCATGGTGT | 57 | 212 |
| Caspase9-R | ATGGCGTCCATCTGGTCATC | ||
| Caspase3-F | CTGATGGGGCATCTGGACTG | 58 | 147 |
| Caspase3-R | GTTGGTTCATGCCTGTCGTG | ||
| Vp4-F | CGAAAACCTACCAGTGGATAATG | 60 | 135 |
| Vp4-R | CCAGCTAGTACGCCGACGAC | ||
| Vp35-F | CATGCCAGTCATATTTGATC | 51 | 195 |
| Vp35-R | TGGGAGGTTGTGGTAGAA | ||
| Vp56-F | AGCAGGCTATTCATCACCAGT | 57 | 107 |
| Vp56-R | GCTCTAACACTCACCGTCTTTTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Dong, W.-J.; Wu, C.-S.; Tian, T.-T.; Zhang, X.-J.; Zhang, Y.-A. GCRV-II Triggers B and T Lymphocyte Apoptosis via Mitochondrial ROS Pathway. Viruses 2025, 17, 930. https://doi.org/10.3390/v17070930
Wang J, Dong W-J, Wu C-S, Tian T-T, Zhang X-J, Zhang Y-A. GCRV-II Triggers B and T Lymphocyte Apoptosis via Mitochondrial ROS Pathway. Viruses. 2025; 17(7):930. https://doi.org/10.3390/v17070930
Chicago/Turabian StyleWang, Jie, Wen-Jing Dong, Chang-Song Wu, Tian-Tian Tian, Xu-Jie Zhang, and Yong-An Zhang. 2025. "GCRV-II Triggers B and T Lymphocyte Apoptosis via Mitochondrial ROS Pathway" Viruses 17, no. 7: 930. https://doi.org/10.3390/v17070930
APA StyleWang, J., Dong, W.-J., Wu, C.-S., Tian, T.-T., Zhang, X.-J., & Zhang, Y.-A. (2025). GCRV-II Triggers B and T Lymphocyte Apoptosis via Mitochondrial ROS Pathway. Viruses, 17(7), 930. https://doi.org/10.3390/v17070930