
Laboratory Diagnosis of Hendra and Nipah: Two Emerging Zoonotic Diseases with One Health Significance



Abstract
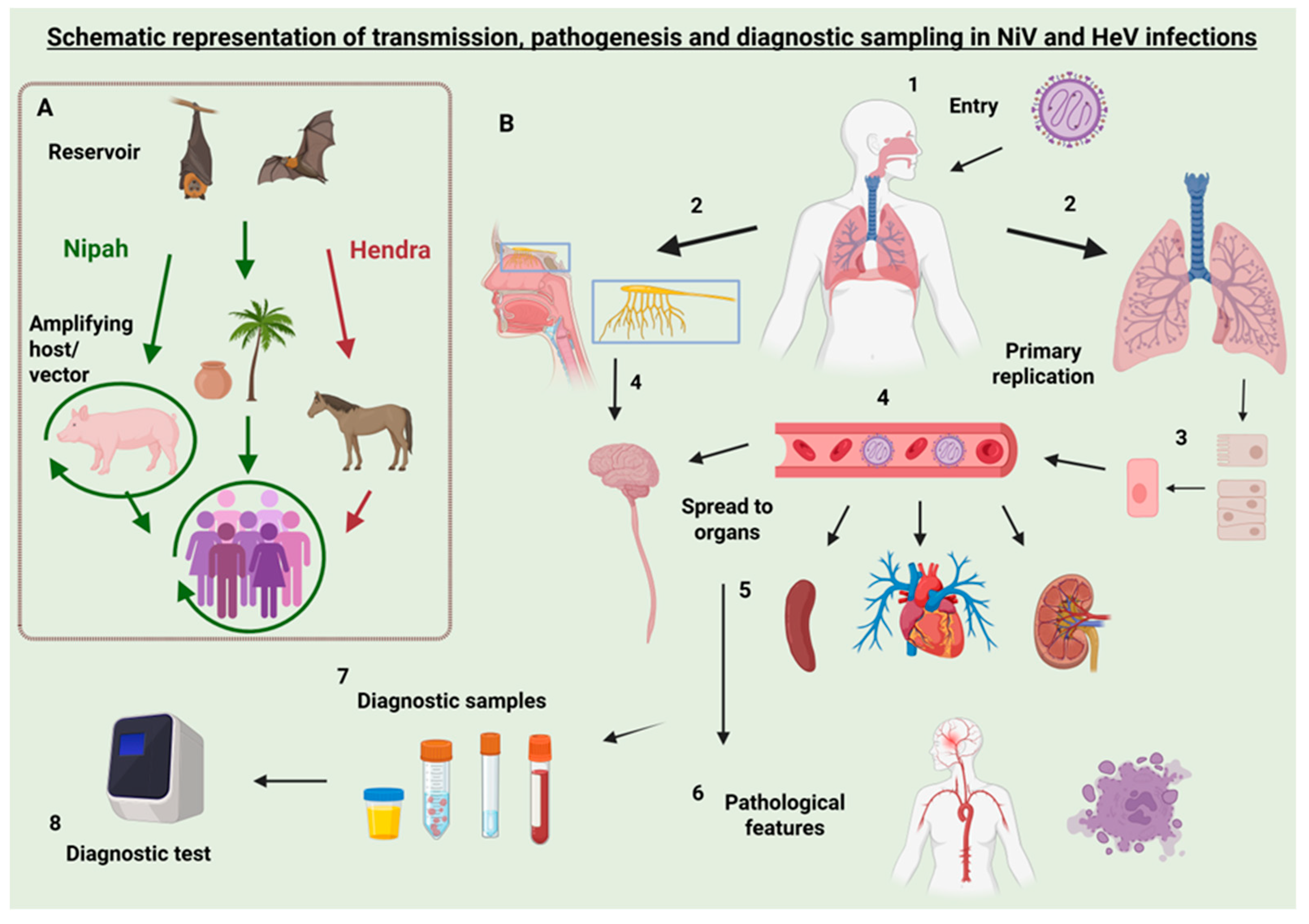
1. Introduction
2. Hendra Virus
2.1. Historical Background of HeV
2.2. Virus Structure and Classification
2.3. Hosts and Reservoirs
2.4. Transmission and Prevention
2.5. Clinical Signs
3. Nipah Virus
3.1. Historical Background of NiV
3.2. Hosts and Reservoirs
3.3. Geographic Distribution and Human Outbreaks
3.4. Molecular Virology
3.5. Clinical Signs, Pathology, and Animal Models
4. Diagnostic Methods
4.1. Molecular Tests
4.1.1. Nucleic Acid Amplification Tests
4.1.2. Sequencing
4.2. Serological Tests
4.2.1. Immunosorbent Assays
4.2.2. Neutralization Tests
4.2.3. Immunohistochemistry
4.2.4. Virus Isolation
4.2.5. Electron Microscopy
5. Diagnostic Considerations for Prevention and Control
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1

{kind=link}
| Year | Event | References |
|---|---|---|
| 1994 | Hendra virus first outbreak in Brisbane, Australia (horses and human deaths) | [3] |
| 1995 | HeV experimentally reproduced in horses; first human fatality | [4] |
| 1995 | HeV genome characterized; virus named equine morbilivirus | [4] |
| 1998 | NiV outbreak in Malaysia, linked to pigs | [34] |
| 1999 | NiV virus Singapore cases from imported pigs | [35] |
| 2000 | HeV isolated from Pteropus bats, confirming reservoir | [18] |
| 2001 | First NiV outbreak in Bangladesh and in India | [34,39] |
| 2010 | African green monkey NiV model established | [53] |
| 2012 | Equivac HeV vaccine released | [27] |
| 2014 | Philippines NiV outbreak linked to horse contact | [35] |
| 2024 | 15 NiV genetic clusters reported by Cortes-Azuero et al. | [35] |
| Indirect ELISA | Competitive ELISA | Sandwich ELISA | Capture ELISA | Solid-Phase Blocking ELISA | Luciferase Immunosorbent Assay (LISA) | Luminex Array Assay | |
|---|---|---|---|---|---|---|---|
| Assay target | Antibody (IgG, IgM) | Antibody or Antigen | Antigen | Antigen and Antibody | Antigen and Antibody (neutralizing antibody) | Antigens, Antibodies | Antigen and antibodies (multiple targets) |
| Mechanism | Antigen (recombinant NiV) coats plate, sample antibodies bind, detection by enzyme-conjugated secondary antibody | Antigen (or antibody) coats plate, antibody (or antigen) competes with a labeled reference for binding, signal is inversely proportional to concentration | Captures antigen between two antibodies (capture and detection), typically using mAbs | Uses antibody or antigen for initial capture step, uses second antibody for detection similar to sandwich ELISA | Antibody binds to plate, blocks non-specific binding, detected via secondary antibody | Uses luciferase-based detection for enhanced signal | Bead-based multiplex technology with fluorescent detection |
| Species used | Tested in most species; studies included humans and pigs | Pig, mini pig, non-human primate | Human | Human | Swine, equine, bat, human | Mouse, horse | Human, horse |
| Potential use | Checking antibodies for infection, checking vaccination response/status, serological surveillance, | Broad species testing, detection of low-concentration samples | Active infection diagnosis, viral load estimation | Acute infection detection | Surveillance, high-throughput testing | Large-scale screening, high-throughput testing | Multiplex detection of multiple targets, outbreak screening |
| Advantages | High sensitivity, Recombinant NiV/HeV virus avoids BSL-4 | High sensitivity and specificity | Can differentiate NiV and HeV, dose-dependent viral dose estimation | Good sensitivity and specificity, robust signal | High specificity, does not require species-specific reagents | Very high sensitivity, species-independent detection | Allows multiplex detection of multiple analytes |
| Disadvantages | Can achieve cross-reactions with secondary antibodies | Requires labeled competitor (monoclonal antibodies) | Lower sensitivity than RT-PCR, requires Mabs, requires high-affinity antibodies | More steps, antibodies determine results (may have reduced sensitivity) | Moderate sensitivity (confirm with second test), requires optimization for large-scale use | Requires specialized equipment, not readily available/tested in field conditions | Requires bead-based technology and flow cytometry, not suited to POCT/field use |
| Studies/References | [78,81,82] | [84] | [83] | [79] | [85] | [86] | [87,88] |
| Assay | Mechanism | Target(s) | Key Features | Use Case | Limitations/Notes | References |
|---|---|---|---|---|---|---|
| RT-qPCR | Reverse-transcription + quantitative PCR | N (main), L, M, P, F, G genes | Gold standard; WOAH-recommended; high sensitivity/specificity | Confirmatory diagnosis of acute infection | Requires thermocycler, RNA extraction | [66,67,68,69] |
| RT-RPA | Isothermal recombinase polymerase amplification | N gene | Field-suitable, inactivation step included, works for NiV-M and NiV-B | Rapid low-resource molecular testing | Sensitivity varies by assay (e.g., 100% to 62.5%) | [74] |
| RT-LAMP | Isothermal amplification (loop-mediated) | N gene | Highly sensitive; used for HeV preclinical detection | Surveillance or early diagnosis | Needs validation in field samples | [72,73] |
| RPA-CRISPR/Cas13a | One-pot isothermal and CRISPR detection | Synthetic cDNA of NiV | Visual detection via lateral flow/fluorescence; no cross-reactions | Lab-based rapid detection | Only tested on synthetic samples | [75] |
| PRNT/VNT | Live virus or pseudovirus-based neutralization | NiV and HeV neutralizing antibodies | Gold-standard serological confirmation | Confirming neutralizing Ab presence | Requires BSL-4 (live virus) or pseudovirus system | [67,89,90] |
| IHC | Antibody staining of virus antigen in tissue | NiV and HeV proteins | Used in formalin-fixed tissue; supports pathology studies | Post-mortem evaluation | Not first-line diagnostic | [89,90] |
| Virus Isolation | Virus grown in Vero E6 or other cell lines | Viable virus | Enables sequencing, further testing | Confirming infectious virus | Time-consuming, low sensitivity, BSL-4 required | [18,67,89,91] |
| Electron Microscopy | Visualizes virion ultrastructure | Virus particles | Historically used for genus-level ID | Primarily a research tool | Needs high virus concentration, BSL-4 | [89,92,93] |
Appendix A.2. Current and Investigational Control Strategies for Henipaviruses (NiV/HeV)
References
- Aditi; Shariff, M. Nipah virus infection: A review. Epidemiol. Infect. 2019, 147, e95. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wise, J.C.; Stewart, A.J. Hendra Virus: An Update on Diagnosis, Vaccination, and Biosecurity Protocols for Horses. Vet. Clin. N. Am. Equine Pract. 2023, 39, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Selvey, L.A.; Wells, R.M.; McCormack, J.G.; Ansford, A.J.; Murray, K.; Rogers, R.J.; Lavercombe, P.S.; Selleck, P.; Sheridan, J.W. Infection of humans and horses by a newly described morbillivirus. Med. J. Aust. 1995, 162, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Selleck, P.; Hooper, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Westbury, H.; Hiley, L.; Selvey, L.; Rodwell, B.; et al. A morbillivirus that caused fatal disease in horses and humans. Science 1995, 268, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Gould, A.R. Comparison of the deduced matrix and fusion protein sequences of equine morbillivirus with cognate genes of the Paramyxoviridae. Virus Res. 1996, 43, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hansson, E.; Langedijk, J.P.; Eaton, B.T.; Wang, L.F. The attachment protein of Hendra virus has high structural similarity but limited primary sequence homology compared with viruses in the genus Paramyxovirus. Virology 1998, 251, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hansson, E.; Shiell, B.; Michalski, W.; Eaton, B.T.; Wang, L.F. Sequence analysis of the Hendra virus nucleoprotein gene: Comparison with other members of the subfamily Paramyxovirinae. J. Gen. Virol. 1998, 79, 1775–1780. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Michalski, W.P.; Yu, M.; Pritchard, L.I.; Crameri, G.; Shiell, B.; Eaton, B.T. A novel P/V/C gene in a new member of the Paramyxoviridae family, which causes lethal infection in humans, horses, and other animals. J. Virol. 1998, 72, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Yu, M.; Hansson, E.; Pritchard, L.I.; Shiell, B.; Michalski, W.P.; Eaton, B.T. The exceptionally large genome of Hendra virus: Support for creation of a new genus within the family Paramyxoviridae. J. Virol. 2000, 74, 9972–9979. [Google Scholar] [CrossRef] [PubMed]
- Mayo, M.A. Virus taxonomy—Houston 2002. Arch. Virol. 2002, 147, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, A.D.; Zaki, S.R.; Goldsmith, C.S.; Wise, T.G.; Hengstberger, S.G. Ultrastructure of Hendra virus and Nipah virus within cultured cells and host animals. Microb. Infect. 2001, 3, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Harcourt, B.H.; Yu, M.; Tamin, A.; Rota, P.A.; Bellini, W.J.; Eaton, B.T. Molecular biology of Hendra and Nipah viruses. Microb. Infect. 2001, 3, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.A.; Stantchev, T.S.; Hickey, A.C.; Khetawat, D.; Bossart, K.N.; Krasnoperov, V.; Gill, P.; Feng, Y.R.; Wang, L.; Eaton, B.T.; et al. Identification of Hendra virus G glycoprotein residues that are critical for receptor binding. J. Virol. 2007, 81, 5893–5901. [Google Scholar] [CrossRef] [PubMed]
- Eaton, B.T.; Broder, C.C.; Wang, L.F. Hendra and Nipah viruses: Pathogenesis and therapeutics. Curr. Mol. Med. 2005, 5, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, J.C.; Kung, N.Y.; Selleck, P.W.; Field, H.E. Survival of hendra virus in the environment: Modelling the effect of temperature. EcoHealth 2015, 12, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, R.; Halpin, K.; Hyatt, A.D.; Daszak, P.; Mungall, B.A. Henipavirus susceptibility to environmental variables. Virus Res. 2008, 132, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Young, P.L.; Halpin, K.; Selleck, P.W.; Field, H.; Gravel, J.L.; Kelly, M.A.; Mackenzie, J.S. Serologic evidence for the presence in Pteropus bats of a paramyxovirus related to equine morbillivirus. Emerg. Infect. Dis. 1996, 2, 239–240. [Google Scholar] [CrossRef] [PubMed]
- Halpin, K.; Young, P.L.; Field, H.E.; Mackenzie, J.S. Isolation of Hendra virus from pteropid bats: A natural reservoir of Hendra virus. J. Gen. Virol. 2000, 81, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Broos, A.; de Jong, C.; Zeddeman, A.; Smith, C.; Smith, G.; Moore, F.; Barr, J.; Crameri, G.; Marsh, G.; et al. Identifying Hendra Virus Diversity in Pteropid Bats. PLoS ONE 2011, 6, e25275. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.M.; Hooper, P.T.; Selleck, P.W.; Gleeson, L.J.; Daniels, P.W.; Westbury, H.A.; Murray, P.K. Transmission studies of Hendra virus (equine morbillivirus) in fruit bats, horses and cats. Aust. Vet. J. 1998, 76, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.M.; Hooper, P.T.; Selleck, P.W.; Westbury, H.A.; Slocombe, R.F. Experimental hendra virus infection in pregnant guinea-pigs and fruit Bats (Pteropus poliocephalus). J. Comp. Pathol. 2000, 122, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Giles, J.R.; Eby, P.; Parry, H.; Peel, A.J.; Plowright, R.K.; Westcott, D.A.; McCallum, H. Environmental drivers of spatiotemporal foraging intensity in fruit bats and implications for Hendra virus ecology. Sci. Rep. 2018, 8, 9555. [Google Scholar] [CrossRef] [PubMed]
- Field, H.E.; Barratt, P.C.; Hughes, R.J.; Shield, J.; Sullivan, N.D. A fatal case of Hendra virus infection in a horse in north Queensland: Clinical and epidemiological features. Aust. Vet. J. 2000, 78, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Playford, E.G.; McCall, B.; Smith, G.; Slinko, V.; Allen, G.; Smith, I.; Moore, F.; Taylor, C.; Kung, Y.H.; Field, H. Human Hendra virus encephalitis associated with equine outbreak, Australia, 2008. Emerg. Infect. Dis. 2010, 16, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, P.D.; Gabor, M.; Poe, I.; Neale, K.; Chaffey, K.; Finlaison, D.S.; Gu, X.; Hick, P.M.; Read, A.J.; Wright, T.; et al. Hendra Virus Infection in Dog, Australia, 2013. Emerg. Infect. Dis. 2015, 21, 2182–2185. [Google Scholar] [CrossRef] [PubMed]
- Westbury, H.A.; Hooper, P.T.; Selleck, P.W.; Murray, P.K. Equine morbillivirus pneumonia: Susceptibility of laboratory animals to the virus. Aust. Vet. J. 1995, 72, 278–279. [Google Scholar] [CrossRef] [PubMed]
- Bossart, K.N.; Crameri, G.; Dimitrov, A.S.; Mungall, B.A.; Feng, Y.R.; Patch, J.R.; Choudhary, A.; Wang, L.F.; Eaton, B.T.; Broder, C.C. Receptor binding, fusion inhibition, and induction of cross-reactive neutralizing antibodies by a soluble G glycoprotein of Hendra virus. J. Virol. 2005, 79, 6690–6702. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.D.; Allworth, A.M.; Paterson, D.L.; Snow, T.M.; Boots, R.; Gleeson, L.J.; Gould, A.R.; Hyatt, A.D.; Bradfield, J. Fatal encephalitis due to novel paramyxovirus transmitted from horses. Lancet 1997, 349, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Field, H.; Schaaf, K.; Kung, N.; Simon, C.; Waltisbuhl, D.; Hobert, H.; Moore, F.; Middleton, D.; Crook, A.; Smith, G.; et al. Hendra virus outbreak with novel clinical features, Australia. Emerg. Infect. Dis. 2010, 16, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.J.; Douglas, I.C.; Baldock, F.C.; Glanville, R.J.; Seppanen, K.T.; Gleeson, L.J.; Selleck, P.N.; Dunn, K.J. Investigation of a second focus of equine morbillivirus infection in coastal Queensland. Aust. Vet. J. 1996, 74, 243–244. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.Y.; Fraser, N.S.; Henning, J.; Halpin, K.; Gibson, J.S.; Betzien, L.; Stewart, A.J. Hendra virus: Epidemiology dynamics in relation to climate change, diagnostic tests and control measures. One Health 2021, 12, 100207. [Google Scholar] [CrossRef] [PubMed]
- Marsh, G.A.; Haining, J.; Hancock, T.J.; Robinson, R.; Foord, A.J.; Barr, J.A.; Riddell, S.; Heine, H.G.; White, J.R.; Crameri, G.; et al. Experimental infection of horses with Hendra virus/Australia/horse/2008/Redlands. Emerg. Infect. Dis. 2011, 17, 2232–2238. [Google Scholar] [CrossRef] [PubMed]
- Field, H.; de Jong, C.; Melville, D.; Smith, C.; Smith, I.; Broos, A.; Kung, Y.H.; McLaughlin, A.; Zeddeman, A. Hendra virus infection dynamics in Australian fruit bats. PLoS ONE 2011, 6, e28678. [Google Scholar] [CrossRef] [PubMed]
- Ang, B.S.P.; Lim, T.C.C.; Wang, L. Nipah Virus Infection. J. Clin. Microbiol. 2018, 56, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Azuero, O.; Lefrancq, N.; Nikolay, B.; McKee, C.; Cappelle, J.; Hul, V.; Ou, T.P.; Hoem, T.; Lemey, P.; Rahman, M.Z.; et al. The Genetic Diversity of Nipah Virus Across Spatial Scales. J. Infect. Dis. 2024, 230, e1235–e1244. [Google Scholar] [CrossRef] [PubMed]
- Faus-Cotino, J.; Reina, G.; Pueyo, J. Nipah Virus: A Multidimensional Update. Viruses 2024, 16, 179. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Dhama, K.; Chakraborty, S.; Tiwari, R.; Natesan, S.; Khandia, R.; Munjal, A.; Vora, K.S.; Latheef, S.K.; Karthik, K.; et al. Nipah virus: Epidemiology, pathology, immunobiology and advances in diagnosis, vaccine designing and control strategies—A comprehensive review. Vet. Q. 2019, 39, 26–55. [Google Scholar] [CrossRef] [PubMed]
- Bruno, L.; Nappo, M.A.; Ferrari, L.; Di Lecce, R.; Guarnieri, C.; Cantoni, A.M.; Corradi, A. Nipah Virus Disease: Epidemiological, Clinical, Diagnostic and Legislative Aspects of This Unpredictable Emerging Zoonosis. Animals 2022, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, S.; Shete, A.; Sarkale, P.; Kumar, A.; Mote, C.; Yadav, P. Genomic characterization, transcriptome analysis, and pathogenicity of the Nipah virus (Indian isolate). Virulence 2023, 14, 2224642. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, M.; Sudeep, A.B.; Mathapati, B.; Balasubramanian, R.; Ullas, P.T.; Mohandas, S.; Patil, D.R.; Shete, A.M.; Gopale, S.; Sawant, P.; et al. Serosurvey for Nipah virus in bat population of southern part of India. Comp. Immunol. Microbiol. Infect. Dis. 2022, 85, 101800. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lu, D.; Yang, M.; Chai, S.; Du, H.; Jiang, H. Nipah virus: Epidemiology, pathogenesis, treatment, and prevention. Front. Med. 2024, 18, 969–987. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.W.; Young, T.A.; Zhang, J.; Liu, S.; Leser, G.P.; Komives, E.A.; Lamb, R.A.; Zhou, Z.H.; Salafsky, J.; Jardetzky, T.S. Monomeric ephrinB2 binding induces allosteric changes in Nipah virus G that precede its full activation. Nat. Commun. 2017, 8, 781. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cross, R.W.; Doyle, M.P.; Kose, N.; Mousa, J.J.; Annand, E.J.; Borisevich, V.; Agans, K.N.; Sutton, R.; Nargi, R.; et al. Potent Henipavirus Neutralization by Antibodies Recognizing Diverse Sites on Hendra and Nipah Virus Receptor Binding Protein. Cell 2020, 183, 1536–1550.e1517. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.E.; Dutch, R.E. A Conserved Region in the F2 Subunit of Paramyxovirus Fusion Proteins Is Involved In Fusion Regulation. J. Virol. 2007, 81, 8303–8314. [Google Scholar] [CrossRef] [PubMed]
- Moll, M.; Kaufmann, A.; Maisner, A. Influence of N-Glycans on Processing and Biological Activity of the Nipah Virus Fusion Protein. J. Virol. 2004, 78, 7274–7278. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.; Lowe, L.; Tamin, A.; Yu, Z.; Bankamp, B.; Bowden, N.; Rollin, P.; Comer, J.; Ksiazek, T.; Hossain, M.J.; et al. Genetic Characterization of Nipah Virus, Bangladesh, 2004. Emerg. Infect. Dis. J. 2005, 11, 1594. [Google Scholar] [CrossRef] [PubMed]
- Clayton, B.; Middleton, D.; Bergfeld, J.; Haining, J.; Arkinstall, R.; Wang, L.; Marsh, G. Transmission Routes for Nipah Virus from Malaysia and Bangladesh. Emerg. Infect. Dis. J. 2012, 18, 1983. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.N.; Agans, K.N.; Sivasubramani, S.K.; Geisbert, J.B.; Borisevich, V.; Mire, C.E.; Lawrence, W.S.; Fenton, K.A.; Geisbert, T.W. A Lethal Aerosol Exposure Model of Nipah Virus Strain Bangladesh in African Green Monkeys. J. Infect. Dis. 2020, 221 (Suppl. 4), S431–S435. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.Z.; Islam, M.M.; Hossain, M.E.; Rahman, M.M.; Islam, A.; Siddika, A.; Hossain, M.S.S.; Sultana, S.; Islam, A.; Rahman, M.; et al. Genetic diversity of Nipah virus in Bangladesh. Int. J. Infect. Dis. 2021, 102, 144–151. [Google Scholar] [CrossRef] [PubMed]
- DeBuysscher, B.L.; de Wit, E.; Munster, V.J.; Scott, D.; Feldmann, H.; Prescott, J. Comparison of the Pathogenicity of Nipah Virus Isolates from Bangladesh and Malaysia in the Syrian Hamster. PLoS Negl. Trop. Dis. 2013, 7, e2024. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.T.; Grosjean, I.; Brisson, C.; Blanquier, B.; Fevre-Montange, M.; Bernard, A.; Loth, P.; Georges-Courbot, M.C.; Chevallier, M.; Akaoka, H.; et al. A golden hamster model for human acute Nipah virus infection. Am. J. Pathol. 2003, 163, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Clayton, B.A.; Middleton, D.; Arkinstall, R.; Frazer, L.; Wang, L.-F.; Marsh, G.A. The Nature of Exposure Drives Transmission of Nipah Viruses from Malaysia and Bangladesh in Ferrets. PLoS Negl. Trop. Dis. 2016, 10, e0004775. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Hickey, A.C.; Smith, M.A.; Chan, Y.P.; Wang, L.F.; Mattapallil, J.J.; Geisbert, J.B.; Bossart, K.N.; Broder, C.C. Development of an acute and highly pathogenic nonhuman primate model of Nipah virus infection. PLoS ONE 2010, 5, e10690. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, J.B.; Borisevich, V.; Prasad, A.N.; Agans, K.N.; Foster, S.L.; Deer, D.J.; Cross, R.W.; Mire, C.E.; Geisbert, T.W.; Fenton, K.A. An Intranasal Exposure Model of Lethal Nipah Virus Infection in African Green Monkeys. J. Infect. Dis. 2020, 221 (Suppl. 4), S414–S418. [Google Scholar] [CrossRef] [PubMed]
- Griffin, B.D.; Leung, A.; Chan, M.; Warner, B.M.; Ranadheera, C.; Tierney, K.; Audet, J.; Frost, K.L.; Safronetz, D.; Embury-Hyatt, C.; et al. Establishment of an RNA polymerase II-driven reverse genetics system for Nipah virus strains from Malaysia and Bangladesh. Sci. Rep. 2019, 9, 11171. [Google Scholar] [CrossRef] [PubMed]
- Kasloff, S.B.; Leung, A.; Pickering, B.S.; Smith, G.; Moffat, E.; Collignon, B.; Embury-Hyatt, C.; Kobasa, D.; Weingartl, H.M. Pathogenicity of Nipah henipavirus Bangladesh in a swine host. Sci. Rep. 2019, 9, 5230. [Google Scholar] [CrossRef] [PubMed]
- Clayton, B.A.; Marsh, G.A. Nipah Viruses from Malaysia and Bangladesh: Two of a Kind? Future Virol. 2014, 9, 935–946. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hossain, M.J.; Sultana, S.; Homaira, N.; Khan, S.U.; Rahman, M.; Gurley, E.S.; Rollin, P.E.; Lo, M.K.; Comer, J.A.; et al. Date Palm Sap Linked to Nipah Virus Outbreak in Bangladesh, 2008. Vector-Borne Zoonotic Dis. 2012, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.T.; Shieh, W.-J.; Kumar, S.; Norain, K.; Abdullah, W.; Guarner, J.; Goldsmith, C.S.; Chua, K.B.; Lam, S.K.; Tan, C.T.; et al. Nipah Virus Infection: Pathology and Pathogenesis of an Emerging Paramyxoviral Zoonosis. Am. J. Pathol. 2002, 161, 2153–2167. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.T.; Tan, C.T. Clinical and Pathological Manifestations of Human Henipavirus Infection. In Henipavirus: Ecology, Molecular Virology, and Pathogenesis; Lee, B., Rota, P.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 95–104. [Google Scholar]
- Elvert, M.; Sauerhering, L.; Maisner, A. Cytokine Induction in Nipah Virus–Infected Primary Human and Porcine Bronchial Epithelial Cells. J. Infect. Dis. 2019, 221 (Suppl. 4), S395–S400. [Google Scholar] [CrossRef] [PubMed]
- Maisner, A.; Neufeld, J.; Weingartl, H. Organ- and endotheliotropism of Nipah virus infections in vivo and in vitro. Thromb. Haemost. 2009, 102, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Devnath, P.; Wajed, S.; Chandra Das, R.; Kar, S.; Islam, I.; Masud, H.M.A.A. The pathogenesis of Nipah virus: A review. Microb. Pathog. 2022, 170, 105693. [Google Scholar] [CrossRef] [PubMed]
- Escaffre, O.; Borisevich, V.; Rockx, B. Pathogenesis of Hendra and Nipah virus infection in humans. J. Infect. Dev. Ctries. 2013, 7, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.T.; Goh, K.J.; Wong, K.T.; Sarji, S.A.; Chua, K.B.; Chew, N.K.; Murugasu, P.; Loh, Y.L.; Chong, H.T.; Tan, K.S.; et al. Relapsed and late-onset Nipah encephalitis. Ann. Neurol. 2002, 51, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Huang, P.; Li, B.; Cao, Z.; Huang, Z.; Zhang, Z.; Liu, M.; Li, H.; Niu, L.; Zhang, T.; et al. A Single-Copy Sensitive and Field-Deployable One-Pot RT-RPA CRISPR/Cas12a Assay for the Specific Visual Detection of the Nipah Virus. Transbound. Emerg. Dis. 2024, 2024, 4118007. [Google Scholar] [CrossRef] [PubMed]
- Garbuglia, A.R.; Lapa, D.; Pauciullo, S.; Raoul, H.; Pannetier, D. Nipah Virus: An Overview of the Current Status of Diagnostics and Their Role in Preparedness in Endemic Countries. Viruses 2023, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.L.; Halpin, K.; Warrilow, D.; Smith, G.A. Development of a fluorogenic RT-PCR assay (TaqMan) for the detection of Hendra virus. J. Virol. Methods 2001, 98, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Feldman, K.S.; Foord, A.; Heine, H.G.; Smith, I.L.; Boyd, V.; Marsh, G.A.; Wood, J.L.; Cunningham, A.A.; Wang, L.F. Design and evaluation of consensus PCR assays for henipaviruses. J. Virol. Methods 2009, 161, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cai, L.; Ye, D.; Chen, J.; Ai, X.; Tang, X.; Deng, A.; Gao, Z.; Xiang, M.; Yu, M.; et al. A Stable and Dependable Visual Technique for On-Site Nipah Virus Nucleic Acids Detection. Sci. Rep. 2025, 15, 7037. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Marsh, G.A.; Olsson, M.; McMillan, D.; Macdonald, J. Rapid, sensitive, and specific, low-resource molecular detection of Hendra virus. One Health 2023, 16, 100504. [Google Scholar] [CrossRef] [PubMed]
- Foord, A.J.; Middleton, D.; Heine, H.G. Hendra virus detection using Loop-Mediated Isothermal Amplification. J. Virol. Methods 2012, 181, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Z.; Guan, W.; Chen, Q.; Liu, D. Rapid and Specific Detection of All Known Nipah virus Strains’ Sequences With Reverse Transcription-Loop-Mediated Isothermal Amplification. Front. Microbiol. 2019, 10, 418. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Olsson, M.; Marsh, G.A.; Macdonald, J.; McMillan, D. Evaluation of three rapid low-resource molecular tests for Nipah virus. Front. Microbiol. 2022, 13, 1101914. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Zuo, L.; He, D.; Fang, Z.; Berthet, N.; Yu, C.; Wong, G. Rapid detection of Nipah virus using the one-pot RPA-CRISPR/Cas13a assay. Virus Res. 2023, 332, 199130. [Google Scholar] [CrossRef] [PubMed]
- Bonsall, D.; Golubchik, T.; Cesare, M.d.; Limbada, M.; Kosloff, B.; MacIntyre-Cockett, G.; Hall, M.; Wymant, C.; Ansari, M.A.; Abeler-Dörner, L.; et al. A Comprehensive Genomics Solution for HIV Surveillance and Clinical Monitoring in Low-Income Settings. J. Clin. Microbiol. 2020, 58, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V. Enzyme-Linked Immunosorbent Assays. Curr. Protoc. Immunol. 2015, 110, 2.1.1–2.1.23. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, D.D.; Venkatesh, G.; Tosh, C.; Patel, P.; Mashoria, A.; Gupta, V.; Gupta, S. Development and Evaluation of Recombinant Nucleocapsid Protein Based Diagnostic ELISA for Detection of Nipah Virus Infection in Pigs. J. Immunoassay Immunochem. 2016, 37, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Shete, A.M.; Jain, R.; Mohandas, S.; Pardeshi, P.; Yadav, P.D.; Gupta, N.; Mourya, D. Development of Nipah virus-specific IgM & IgG ELISA for screening human serum samples. Indian J. Med. Res. 2022, 156, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Ramasundram, V. Kinetics of IgM and IgG Seroconversion in Nipah Virus Infection. Neurol. J. Southeast Asia 2000, 5, 23–28. [Google Scholar]
- Yu, F.; Khairullah, N.S.; Inoue, S.; Balasubramaniam, V.; Berendam, S.J.; Teh, L.K.; Ibrahim, N.S.W.; Rahman, S.A.; Hassan, S.S.; Hasebe, F.; et al. Serodiagnosis Using Recombinant Nipah Virus Nucleocapsid Protein Expressed in Escherichia coli. J. Clin. Microbiol. 2006, 44, 3134–3138. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Diederich, S.; Smith, G.; Reiche, S.; Pinho dos Reis, V.; Stroh, E.; Groschup, M.H.; Weingartl, H.M.; Balkema-Buschmann, A. Indirect ELISA based on Hendra and Nipah virus proteins for the detection of henipavirus specific antibodies in pigs. PLoS ONE 2018, 13, e0194385. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhu, W.; Truong, T.; Pickering, B.; Babiuk, S.; Kobasa, D.; Banadyga, L. Detection of Nipah and Hendra Viruses Using Recombinant Human Ephrin B2 Capture Virus in Immunoassays. Viruses 2022, 14, 1657. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Pickering, B.; Smith, G.; Pinette, M.; Truong, T.; Babiuk, S.; Kobasa, D.; Banadyga, L.; Yang, M. Development and laboratory evaluation of a competitive ELISA for serodiagnosis of Nipah and Hendra virus infection using recombinant Nipah glycoproteins and a monoclonal antibody. Front. Vet. Sci. 2023, 10, 1120367. [Google Scholar] [CrossRef] [PubMed]
- Kashiwazaki, Y.; Na, Y.N.; Tanimura, N.; Imada, T. A solid-phase blocking ELISA for detection of antibodies to Nipah virus. J. Virol. Methods 2004, 121, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, Y.; Huang, X.; Zhao, Y.; Wan, C. A Luciferase Immunosorbent Assay Based on Attachment Glycoprotein for the Rapid and Easy Detection of Nipah Virus IgG Antibodies. Microorganisms 2024, 12, 983. [Google Scholar] [CrossRef] [PubMed]
- Foord, A.J.; White, J.R.; Colling, A.; Heine, H.G. Microsphere suspension array assays for detection and differentiation of Hendra and Nipah viruses. Biomed. Res. Int. 2013, 2013, 289295. [Google Scholar] [CrossRef] [PubMed]
- McNabb, L.; Barr, J.; Crameri, G.; Juzva, S.; Riddell, S.; Colling, A.; Boyd, V.; Broder, C.; Wang, L.F.; Lunt, R. Henipavirus microsphere immuno-assays for detection of antibodies against Hendra virus. J. Virol. Methods 2014, 200, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Daniels, P.; Ksiazek, T.; Eaton, B.T. Laboratory diagnosis of Nipah and Hendra virus infections. Microb. Infect. 2001, 3, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Bailey, D. Advances in diagnostics, vaccines and therapeutics for Nipah virus. Microb. Infect. 2019, 21, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Soman Pillai, V.; Krishna, G.; Valiya Veettil, M. Nipah Virus: Past Outbreaks and Future Containment. Viruses 2020, 12, 465. [Google Scholar] [CrossRef] [PubMed]
- Popov, V.L.; Tesh, R.B.; Weaver, S.C.; Vasilakis, N. Electron Microscopy in Discovery of Novel and Emerging Viruses from the Collection of the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA). Viruses 2019, 11, 477. [Google Scholar] [CrossRef] [PubMed]
- Curry, A.; Appleton, H.; Dowsett, B. Application of transmission electron microscopy to the clinical study of viral and bacterial infections: Present and future. Micron 2006, 37, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.; Prajapat, V.; Nayak, D. Advancements in Nipah virus treatment: Analysis of current progress in vaccines, antivirals, and therapeutics. Immunology 2024, 171, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Pedrera, M.; McLean, R.K.; Medfai, L.; Thakur, N.; Todd, S.; Marsh, G.; Bailey, D.; Donofrio, G.; Muramatsu, H.; Pardi, N.; et al. Evaluation of the immunogenicity of an mRNA vectored Nipah virus vaccine candidate in pigs. Front. Immunol. 2024, 15, 1384417. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; Li, R.; Wang, S.; Yang, L.; Wang, S.; Yin, Y.; Zai, X.; Zhang, J.; Xu, J. Nipah virus attachment glycoprotein ectodomain delivered by type 5 adenovirus vector elicits broad immune response against NiV and HeV. Front. Cell. Infect. Microbiol. 2023, 13, 1180344. [Google Scholar] [CrossRef] [PubMed]
- Halpin, K.; Graham, K.; Durr, P.A. Sero-Monitoring of Horses Demonstrates the Equivac® HeV Hendra Virus Vaccine to Be Highly Effective in Inducing Neutralising Antibody Titres. Vaccines 2021, 9, 731. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Bobb, K.; Borisevich, V.; Geisbert, J.B.; Agans, K.N.; Cross, R.W.; Prasad, A.N.; Fenton, K.A.; Yu, H.; Fouts, T.R.; et al. A single dose investigational subunit vaccine for human use against Nipah virus and Hendra virus. NPJ Vaccines 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van den Hurk, S.; Yondo, A.; Velayudhan, B.T. Laboratory Diagnosis of Hendra and Nipah: Two Emerging Zoonotic Diseases with One Health Significance. Viruses 2025, 17, 1003. https://doi.org/10.3390/v17071003
van den Hurk S, Yondo A, Velayudhan BT. Laboratory Diagnosis of Hendra and Nipah: Two Emerging Zoonotic Diseases with One Health Significance. Viruses. 2025; 17(7):1003. https://doi.org/10.3390/v17071003
Chicago/Turabian Stylevan den Hurk, Shaun, Aurelle Yondo, and Binu T. Velayudhan. 2025. "Laboratory Diagnosis of Hendra and Nipah: Two Emerging Zoonotic Diseases with One Health Significance" Viruses 17, no. 7: 1003. https://doi.org/10.3390/v17071003
APA Stylevan den Hurk, S., Yondo, A., & Velayudhan, B. T. (2025). Laboratory Diagnosis of Hendra and Nipah: Two Emerging Zoonotic Diseases with One Health Significance. Viruses, 17(7), 1003. https://doi.org/10.3390/v17071003