
Two Years of SARS-CoV-2 Omicron Genomic Evolution in Brazil (2022–2024): Subvariant Tracking and Assessment of Regional Sequencing Efforts

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Data Retrieval
2.2. Data Processing
2.3. Pango Lineages and Phylogenetics Analysis
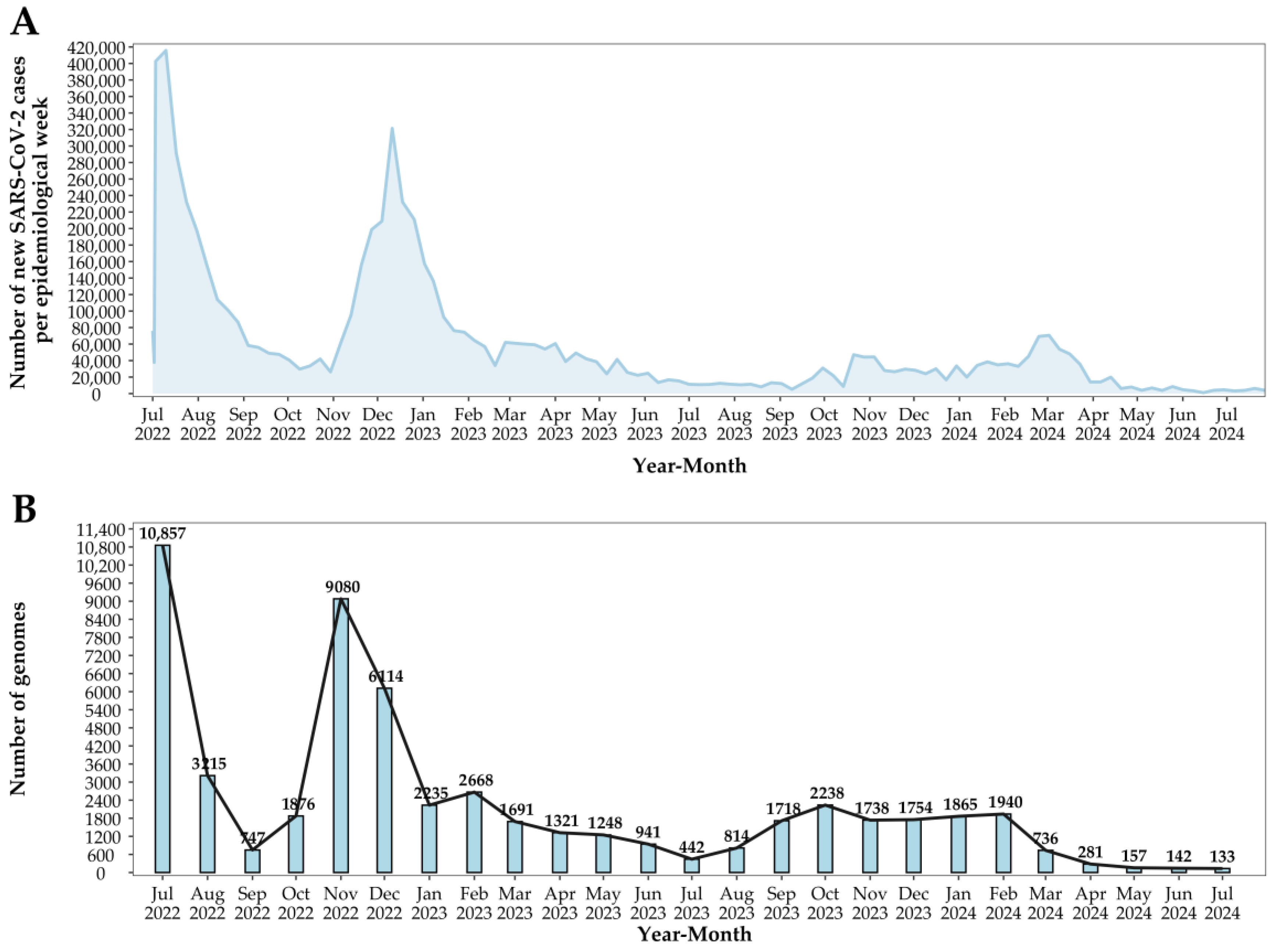
3. Results
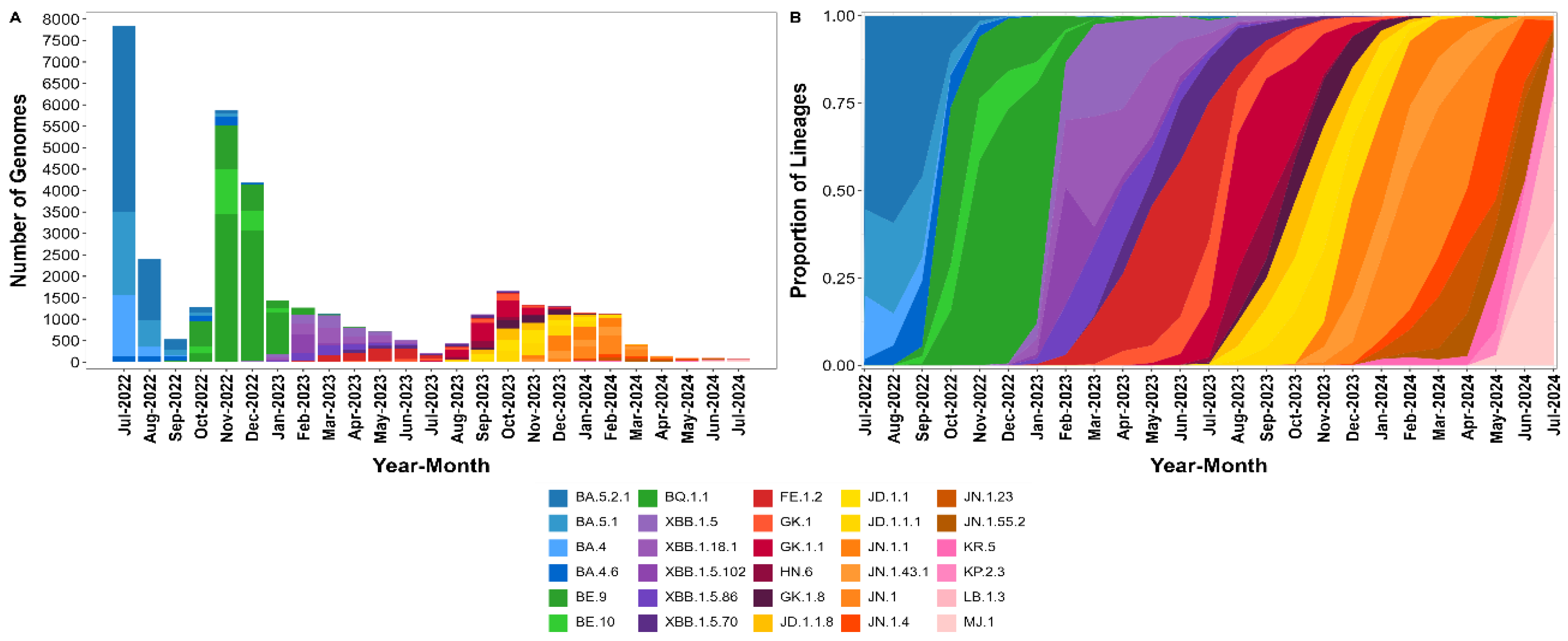
3.1. Pango Lineages
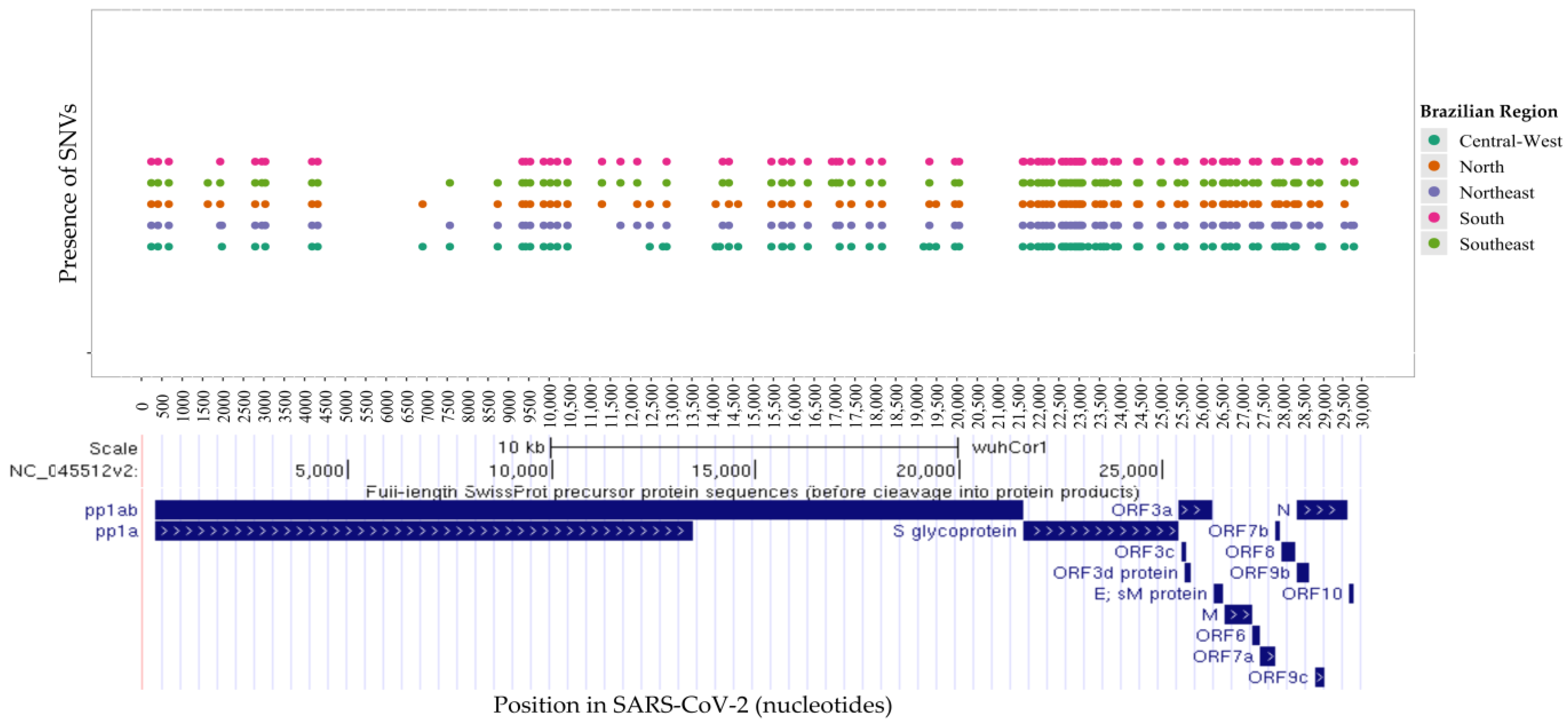
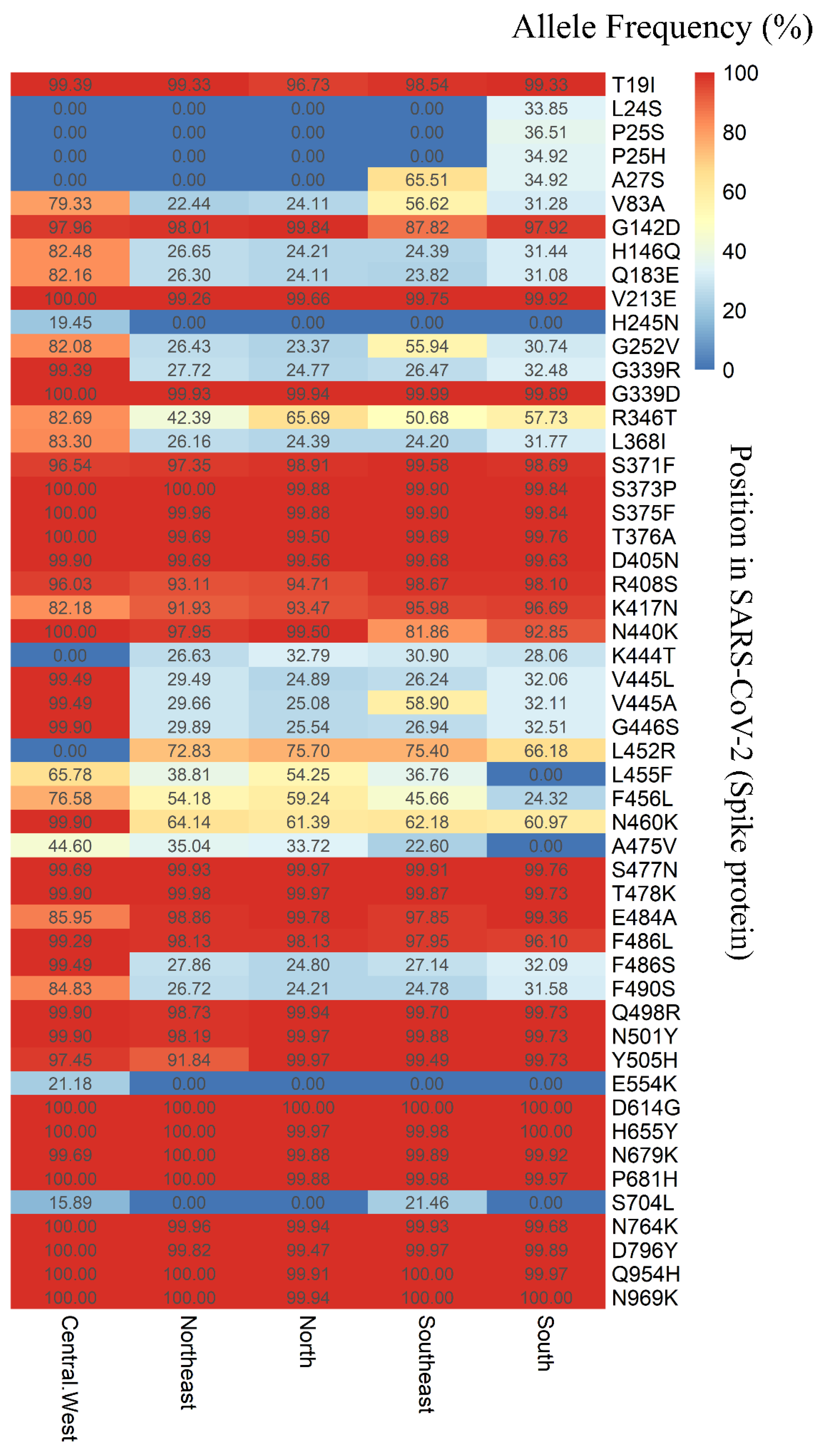
3.2. An Analysis of SARS-CoV-2 Mutations Across Brazilian Regions
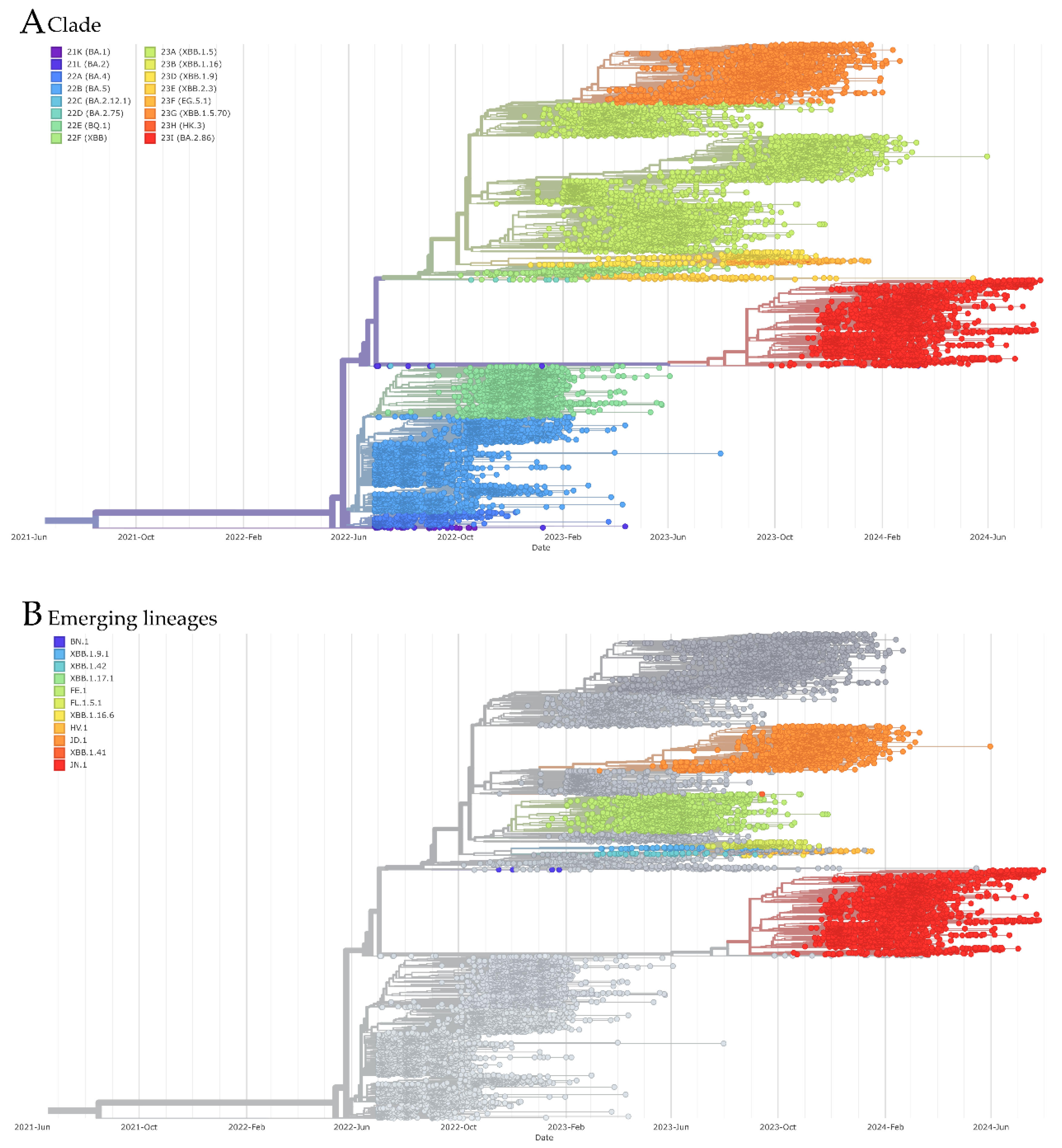
3.3. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Penrice-Randal, R.; Hiscox, J.A.; Barclay, W.S. SARS-CoV-2 One Year on: Evidence for Ongoing Viral Adaptation. J. Gen. Virol. 2021, 102, 001584. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Statement on the Fifteenth Meeting of the IHR (2005) Emergency Committee on the COVID-19 Pandemic; WHO Director General’s Speeches 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Woo, P.C.Y.; de Groot, R.J.; Haagmans, B.; Lau, S.K.P.; Neuman, B.W.; Perlman, S.; Sola, I.; van der Hoek, L.; Wong, A.C.P.; Yeh, S.H. ICTV Virus Taxonomy Profile: Coronaviridae 2023. J. Gen. Virol. 2023, 104, 001843. [Google Scholar] [CrossRef] [PubMed]
- Moeller, N.H.; Shi, K.; Demir, Ö.; Belica, C.; Banerjee, S.; Yin, L.; Durfee, C.; Amaro, R.E.; Aihara, H. Structure and Dynamics of SARS-CoV-2 Proofreading Exoribonuclease ExoN. Proc. Natl. Acad. Sci. USA 2022, 119, e2106379119. [Google Scholar] [CrossRef]
- Liu, C.; Shi, W.; Becker, S.T.; Schatz, D.G.; Liu, B.; Yang, Y. Structural Basis of Mismatch Recognition by a SARS-CoV-2 Proofreading Enzyme. Science 2021, 373, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Ghafari, M.; Beer, M.; Lythgoe, K.; Simmonds, P.; Stilianakis, N.I.; Katzourakis, A. The Evolution of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 361–379. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G Spike Mutation Increases Entry Efficiency with Enhanced ACE2-Binding Affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef]
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Zé-Zé, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation Rate of SARS-CoV-2 and Emergence of Mutators during Experimental Evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- Abavisani, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Mollapour Siasakht, M.; Farhadi, A.; Kodori, M.; Mahmanzar, M.; Meshkat, Z. Mutations in SARS-CoV-2 Structural Proteins: A Global Analysis. Virol. J. 2022, 19, 220. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol Biol 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Landman, S.R.; Reilly, C.S.; Mansky, L.M. HIV-1 and HIV-2 Exhibit Similar Mutation Frequencies and Spectra in the Absence of G-to-A Hypermutation. Retrovirology 2015, 12, 60. [Google Scholar] [CrossRef]
- Ribeiro, R.M.; Li, H.; Wang, S.; Stoddard, M.B.; Learn, G.H.; Korber, B.T.; Bhattacharya, T.; Guedj, J.; Parrish, E.H.; Hahn, B.H.; et al. Quantifying the Diversification of Hepatitis C Virus (HCV) during Primary Infection: Estimates of the In Vivo Mutation Rate. PLoS Pathog. 2012, 8, e1002881. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.N.; Russell, C.A. The Evolution of Seasonal Influenza Viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Pauly, M.D.; Procario, M.C.; Lauring, A.S. A Novel Twelve Class Fluctuation Test Reveals Higher than Expected Mutation Rates for Influenza A Viruses. eLife 2017, 6, e26437. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, E.; Sato, K. Comparison of the Mutation Rates of Human Influenza A and B Viruses. J. Virol. 2006, 80, 3675–3678. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Barmania, F.; Mellet, J.; Peta, K.; Strydom, A.; Viljoen, I.M.; James, W.; Gordon, S.; Pepper, M.S. SARS-CoV-2 Variants, Vaccines, and Host Immunity. Front. Immunol. 2022, 12, 809244. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D. da S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Paton, R.S.; Overton, C.E.; Ward, T. The Rapid Replacement of the SARS-CoV-2 Delta Variant by Omicron (B.1.1.529) in England. Sci. Transl. Med. 2022, 14, eabo5395. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Van Nam, L.; Dien, T.C.; Bang, L.V.N.; Thach, P.N.; Van Duyet, L. Genetic features of SARS-CoV-2 Alpha, Delta, and Omicron variants and their association with the clinical severity of COVID-19 in Vietnam. IJID Reg. 2024, 11, 100348. [Google Scholar] [CrossRef]
- Giovanetti, M.; Slavov, S.N.; Fonseca, V.; Wilkinson, E.; Tegally, H.; Patané, J.S.L.; Viala, V.L.; San, E.J.; Rodrigues, E.S.; Santos, E.V.; et al. Genomic Epidemiology of the SARS-CoV-2 Epidemic in Brazil. Nat. Microbiol. 2022, 7, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Menezes, D.; Fonseca, P.L.C.; de Araújo, J.L.F.; Souza, R.P. SARS-CoV-2 Genomic Surveillance in Brazil: A Systematic Review with Scientometric Analysis. Viruses 2022, 14, 2715. [Google Scholar] [CrossRef]
- Lamarca, A.P.; Souza, U.J.B.; Moreira, F.R.R.; Almeida, L.G.P.; Menezes, M.T.; Souza, A.B.; Ferreira, A.C.S.; Gerber, A.L.; Lima, A.B.; Guimarães, A.P.C.; et al. The Omicron Lineages BA.1 and BA.2 (Betacoronavirus SARS-CoV-2) Have Repeatedly Entered Brazil through a Single Dispersal Hub. Viruses 2023, 15, 888. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data–from Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- de Souza, U.J.B.; dos Santos, R.N.; Campos, F.S.; Lourenço, K.L.; da Fonseca, F.G.; Spilki, F.R. High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021. Viruses 2021, 13, 1806. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Brito, A.F.; Semenova, E.; Dudas, G.; Hassler, G.W.; Kalinich, C.C.; Kraemer, M.U.G.; Ho, J.; Tegally, H.; Githinji, G.; Agoti, C.N.; et al. Global Disparities in SARS-CoV-2 Genomic Surveillance. Nat. Commun. 2022, 13, 7003. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The Emergence, Genomic Diversity and Global Spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Candido, D.S.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and Epidemic Spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef]
- Moreira, F.R.R.; Bonfim, D.M.; Zauli, D.A.G.; Silva, J.P.; Lima, A.B.; Malta, F.S.V.; Ferreira, A.C.S.; Pardini, V.C.; Magalhães, W.C.S.; Queiroz, D.C.; et al. Epidemic Spread of SARS-CoV-2 Lineage B.1.1.7 in Brazil. Viruses 2021, 13, 984. [Google Scholar] [CrossRef] [PubMed]
- de Souza, U.J.B.; dos Santos, R.N.; de Melo, F.L.; Belmok, A.; Galvão, J.D.; de Rezende, T.C.V.; Cardoso, F.D.P.; Carvalho, R.F.; da Silva Oliveira, M.; Ribeiro Junior, J.C.; et al. Genomic Epidemiology of SARS-CoV-2 in Tocantins State and the Diffusion of P.1.7 and AY.99.2 Lineages in Brazil. Viruses 2022, 14, 659. [Google Scholar] [CrossRef]
- Arantes, I.; Gomes, M.; Ito, K.; Sarafim, S.; Gräf, T.; Miyajima, F.; Khouri, R.; de Carvalho, F.C.; de Almeida, W.A.F.; Siqueira, M.M.; et al. Spatiotemporal Dynamics and Epidemiological Impact of SARS-CoV-2 XBB Lineage Dissemination in Brazil in 2023. Microbiol. Spectr. 2024, 12, e03831-23. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, B.C.; y Castro, T.R.; Tessele, L.F.; Casarin, B.C.; Seerig, A.P.; de Almeida Vieira, A.; Santos, V.T.; Schwarzbold, A.V.; Trindade, P.A. Genomic Surveillance and Vaccine Response to the Dominant SARS-CoV-2 XBB Lineage in Rio Grande Do Sul. Sci. Rep. 2024, 14, 16831. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Staropoli, I.; Michel, V.; Lemoine, F.; Donati, F.; Prot, M.; Porrot, F.; Guivel-Benhassine, F.; Jeyarajah, B.; Brisebarre, A.; et al. Distinct Evolution of SARS-CoV-2 Omicron XBB and BA.2.86/JN.1 Lineages Combining Increased Fitness and Antibody Evasion. Nat. Commun. 2024, 15, 2254. [Google Scholar] [CrossRef]
- Looi, M.-K. Covid-19: WHO Adds JN.1 as New Variant of Interest. BMJ 2023, 383, p2975. [Google Scholar] [CrossRef] [PubMed]
- Araf, Y.; Akter, F.; Tang, Y.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron Variant of SARS-CoV-2: Genomics, Transmissibility, and Responses to Current COVID-19 Vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza, U.J.B.d.; Spilki, F.R.; Tanuri, A.; Roehe, P.M.; Campos, F.S. Two Years of SARS-CoV-2 Omicron Genomic Evolution in Brazil (2022–2024): Subvariant Tracking and Assessment of Regional Sequencing Efforts. Viruses 2025, 17, 64. https://doi.org/10.3390/v17010064
Souza UJBd, Spilki FR, Tanuri A, Roehe PM, Campos FS. Two Years of SARS-CoV-2 Omicron Genomic Evolution in Brazil (2022–2024): Subvariant Tracking and Assessment of Regional Sequencing Efforts. Viruses. 2025; 17(1):64. https://doi.org/10.3390/v17010064
Chicago/Turabian StyleSouza, Ueric José Borges de, Fernando Rosado Spilki, Amilcar Tanuri, Paulo Michel Roehe, and Fabrício Souza Campos. 2025. "Two Years of SARS-CoV-2 Omicron Genomic Evolution in Brazil (2022–2024): Subvariant Tracking and Assessment of Regional Sequencing Efforts" Viruses 17, no. 1: 64. https://doi.org/10.3390/v17010064
APA StyleSouza, U. J. B. d., Spilki, F. R., Tanuri, A., Roehe, P. M., & Campos, F. S. (2025). Two Years of SARS-CoV-2 Omicron Genomic Evolution in Brazil (2022–2024): Subvariant Tracking and Assessment of Regional Sequencing Efforts. Viruses, 17(1), 64. https://doi.org/10.3390/v17010064