AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Nucleocapsid Protein Variants



Abstract
1. Introduction
2. Materials and Methods
3. Results
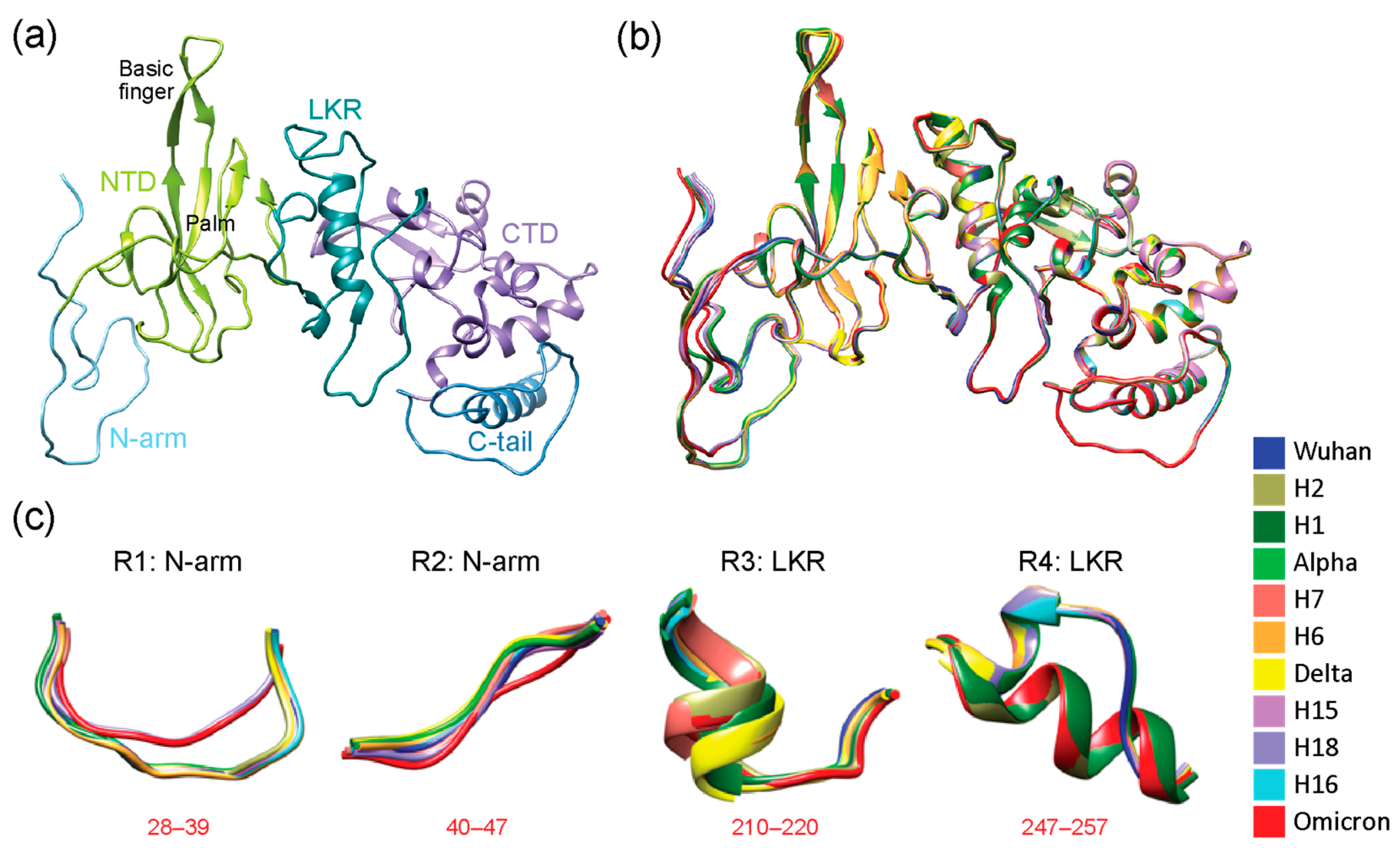
3.1. Full-Length Analysis of the N-Protein Using Backbone Root Mean Square Deviations (RMSD)
3.2. Regional Analysis with TM Scores Using US-Align
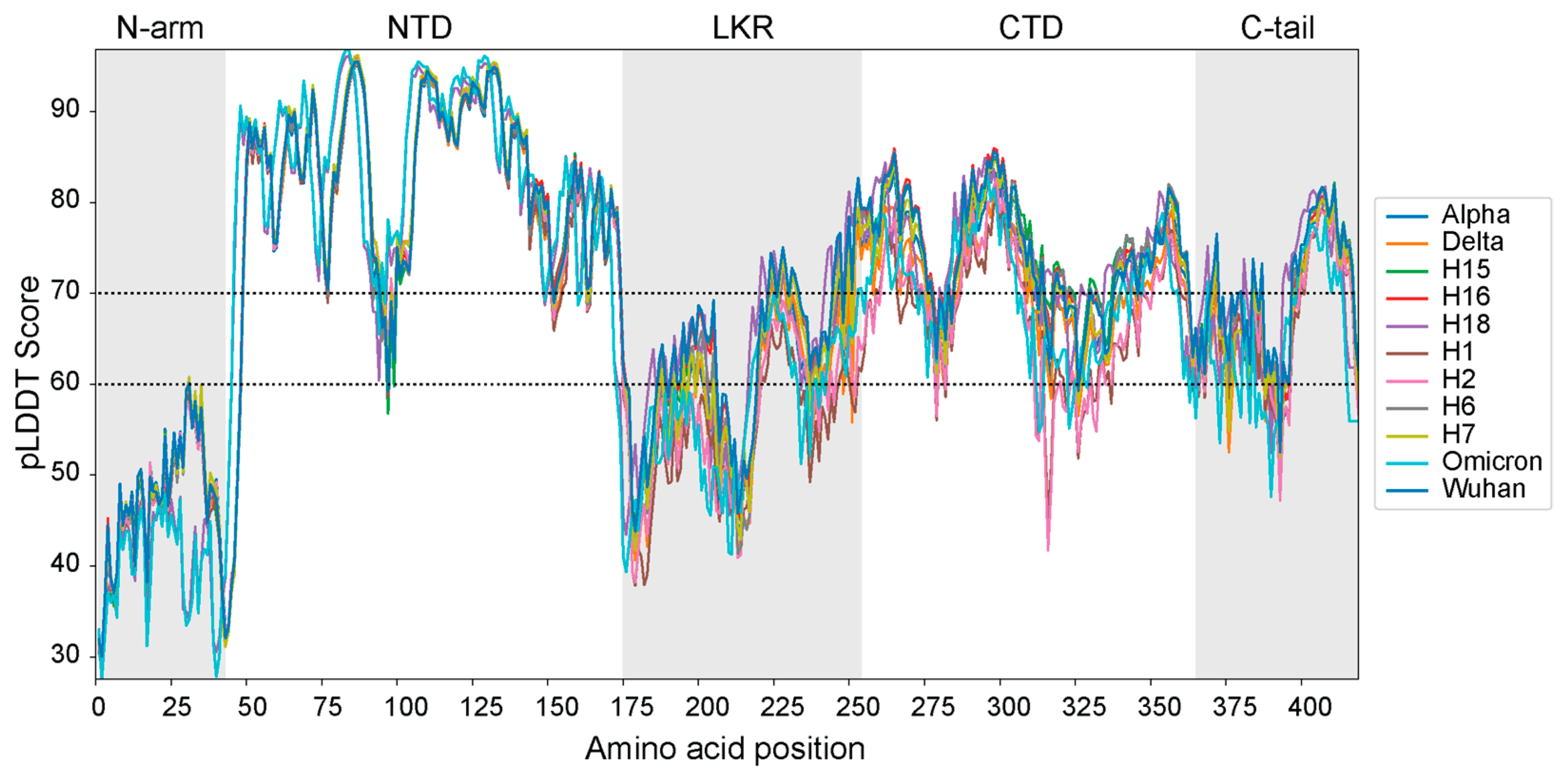
3.3. Protein Disorder and pLDDT Scores
3.4. Protein Disorder and Binding Capacity Across the Pandemic
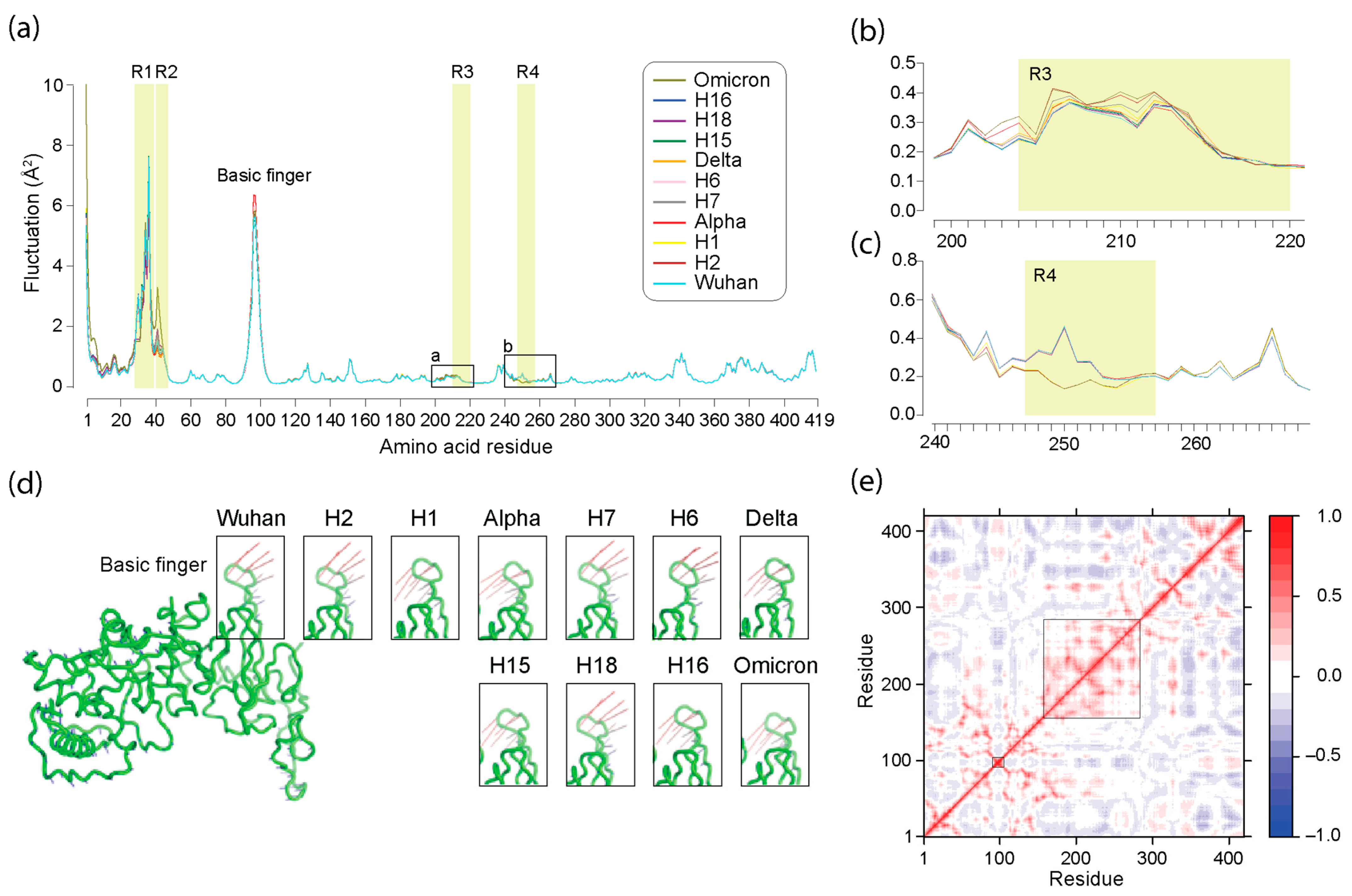
3.5. Normal Mode Analysis of Haplotypes and VOCs
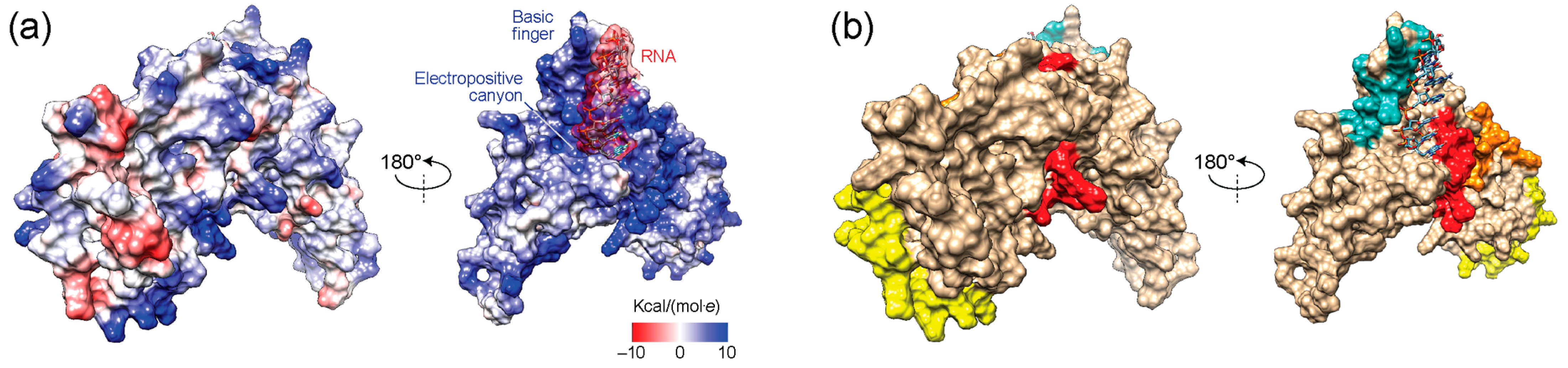
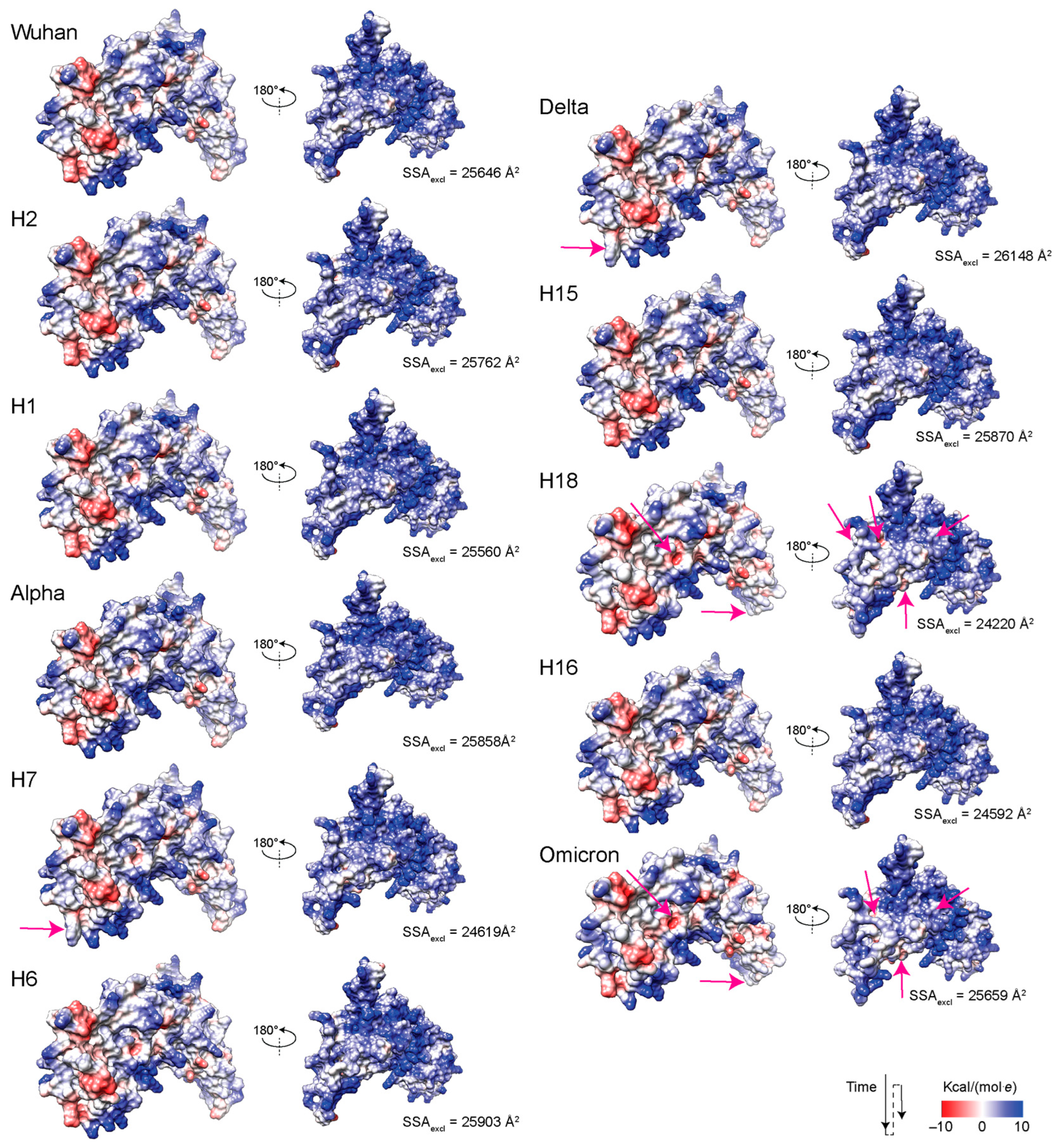
3.6. Exploring Electrostatic Potential Surface Fingerprints
3.7. Benchmarking Alphafold2 Reference Structures against Experimental Cryo-EM Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. COVID-19 Dashboard, COVID-19 Deaths. Available online: https://data.who.int/dashboards/covid19/cases (accessed on 1 July 2024).
- StatsAmerica. States in Profile, Population Estimate for 2023, Ranked List. Available online: https://www.statsamerica.org/sip/rank_list.aspx?rank_label=pop1&ct=S18 (accessed on 1 July 2024).
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects. Available online: https://population.un.org/wpp/Download/Standard/Population/ (accessed on 1 July 2024).
- Ramadan, N.; Shaib, H. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): A Review. Germs 2019, 9, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hodgens, A.; Gupta, V. Severe Acute Respiratory Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Summary of Probable SARS Cases with Onset of Illness from 1 November 2002 to 31 July 2003. Available online: https://www.who.int/publications/m/item/summary-of-probable-sars-cases-with-onset-of-illness-from-1-november-2002-to-31-july-2003 (accessed on 1 July 2024).
- MERS-CoV Worldwide Overview. Available online: https://www.ecdc.europa.eu/en/middle-east-respiratory-syndrome-coronavirus-mers-cov-situation-update (accessed on 1 July 2024).
- Pormohammad, A.; Ghorbani, S.; Khatami, A.; Farzi, R.; Baradaran, B.; Turner, D.L.; Turner, R.J.; Bahr, N.C.; Idrovo, J. Comparison of Confirmed COVID-19 with SARS and MERS Cases-Clinical Characteristics, Laboratory Findings, Radiographic Signs and Outcomes: A Systematic Review and Meta-analysis. Rev. Med. Virol. 2020, 30, e2112. [Google Scholar] [CrossRef] [PubMed]
- Pustake, M.; Tambolkar, I.; Giri, P.; Gandhi, C. SARS, MERS and CoVID-19: An Overview and Comparison of Clinical, Laboratory and Radiological Features. J. Fam. Med. Prim. Care 2022, 11, 10–17. [Google Scholar] [CrossRef]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, K.; Kagaya, K.; Zarebski, A.; Chowell, G. Estimating the Asymptomatic Proportion of Coronavirus Disease 2019 (COVID-19) Cases on Board the Diamond Princess Cruise Ship, Yokohama, Japan, 2020. Eurosurveillance 2020, 25, 2000180. [Google Scholar] [CrossRef]
- Liu, P.L. COVID-19 Information on Social Media and Preventive Behaviors: Managing the Pandemic through Personal Responsibility. Soc. Sci. Med. 2021, 277, 113928. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, T.; Ali, M.A.; Caetano-Anollés, K.; Caetano-Anollés, G. Seasonal Effects Decouple SARS-CoV-2 Haplotypes Worldwide. F1000Research 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Caetano-Anollés, G. AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Spike Protein Variants. Biology 2024, 13, 134. [Google Scholar] [CrossRef]
- Wu, W.; Cheng, Y.; Zhou, H.; Sun, C.; Zhang, S. The SARS-CoV-2 Nucleocapsid Protein: Its Role in the Viral Life Cycle, Structure and Functions, and Use as a Potential Target in the Development of Vaccines and Diagnostics. Virol. J. 2023, 20, 6. [Google Scholar] [CrossRef]
- Tomaszewski, T.; DeVries, R.S.; Dong, M.; Bhatia, G.; Norsworthy, M.D.; Zheng, X.; Caetano-Anollés, G. New Pathways of Mutational Change in SARS-CoV-2 Proteomes Involve Regions of Intrinsic Disorder Important for Virus Replication and Release. Evol. Bioinform. Online 2020, 16, 1176934320965149. [Google Scholar] [CrossRef]
- Peng, Y.; Du, N.; Lei, Y.; Dorje, S.; Qi, J.; Luo, T.; Gao, G.F.; Song, H. Structures of the SARS-CoV-2 Nucleocapsid and Their Perspectives for Drug Design. EMBO J. 2020, 39, e105938. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; West, A.M.V.; Silletti, S.; Corbett, K.D. Architecture and Self-assembly of the SARS-CoV-2 Nucleocapsid Protein. Protein Sci. 2020, 29, 1890–1901. [Google Scholar] [CrossRef]
- Chang, C.-K.; Hsu, Y.-L.; Chang, Y.-H.; Chao, F.-A.; Wu, M.-C.; Huang, Y.-S.; Hu, C.-K.; Huang, T.-H. Multiple Nucleic Acid Binding Sites and Intrinsic Disorder of Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein: Implications for Ribonucleocapsid Protein Packaging. J. Virol. 2009, 83, 2255–2264. [Google Scholar] [CrossRef]
- Hilser, V.J.; Thompson, E.B. Intrinsic Disorder as a Mechanism to Optimize Allosteric Coupling in Proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 8311–8315. [Google Scholar] [CrossRef]
- Chang, C.; Hou, M.-H.; Chang, C.-F.; Hsiao, C.-D.; Huang, T. The SARS Coronavirus Nucleocapsid Protein—Forms and Functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates, J.R.; Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 Nucleocapsid Phosphoprotein Forms Mutually Exclusive Condensates with RNA and the Membrane-Associated M Protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Shin, O.S. SARS-CoV-2 Nucleocapsid Protein Targets RIG-I-Like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells 2021, 10, 530. [Google Scholar] [CrossRef]
- de Haan, C.A.M.; Rottier, P.J.M. Molecular Interactions in the Assembly of Coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar] [CrossRef]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Structure and Replication Characterized by in Situ Cryo-Electron Tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martínez-Sobrido, L.; García-Sastre, A.; Weber, F.; Kochs, G. The Intracellular Sites of Early Replication and Budding of SARS-Coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Agnihotry, S.; Pathak, R.K.; Singh, D.B.; Tiwari, A.; Hussain, I. Chapter 11—Protein Structure Prediction. In Bioinformatics; Singh, D.B., Pathak, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 177–188. ISBN 978-0-323-89775-4. [Google Scholar]
- Yuan, X.; Shao, Y.; Bystroff, C. Ab Initio Protein Structure Prediction Using Pathway Models. Comp. Funct. Genom. 2003, 4, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XIV. Proteins: Struct. Funct. Bioinform. 2021, 89, 1607–1617. [Google Scholar] [CrossRef]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XV. Proteins: Struct. Funct. Bioinform. 2023, 91, 1539–1549. [Google Scholar] [CrossRef]
- Ford, C.T.; Jacob Machado, D.; Janies, D.A. Predictions of the SARS-CoV-2 Omicron Variant (B.1.1.529) Spike Protein Receptor-Binding Domain Structure and Neutralizing Antibody Interactions. Front. Virol. 2022, 2, 830202. [Google Scholar] [CrossRef]
- Kilim, O.; Mentes, A.; Pál, B.; Csabai, I.; Gellért, Á. SARS-CoV-2 Receptor-Binding Domain Deep Mutational AlphaFold2 Structures. Sci. Data 2023, 10, 134. [Google Scholar] [CrossRef]
- Yang, Q.; Jian, X.; Syed, A.A.S.; Fahira, A.; Zheng, C.; Zhu, Z.; Wang, K.; Zhang, J.; Wen, Y.; Li, Z.; et al. Structural Comparison and Drug Screening of Spike Proteins of Ten SARS-CoV-2 Variants. Research 2022, 2022, 9781758. [Google Scholar] [CrossRef]
- Tomaszewski, T.; Gurtler, V.; Caetano-Anollés, K.; Caetano-Anollés, G. Chapter 8—The Emergence of SARS-CoV-2 Variants of Concern in Australia by Haplotype Coalescence Reveals a Continental Link to COVID-19 Seasonality. In Methods in Microbiology; Pavia, C.S., Gurtler, V., Eds.; COVID-19: Biomedical Perspectives; Academic Press: Cambridge, MA, USA, 2022; Volume 50, pp. 233–268. [Google Scholar]
- Burra, P.; Soto-Díaz, K.; Chalen, I.; Gonzalez-Ricon, R.J.; Istanto, D.; Caetano-Anollés, G. Temperature and Latitude Correlate with SARS-CoV-2 Epidemiological Variables but Not with Genomic Change Worldwide. Evol. Bioinform. Online 2021, 17, 1176934321989695. [Google Scholar] [CrossRef]
- Hernandez, N.; Caetano-Anollés, G. Worldwide Correlations Support COVID-19 Seasonal Behavior and Impact of Global Change. Evol. Bioinform. 2023, 19, 11769343231169377. [Google Scholar] [CrossRef] [PubMed]
- Muradyan, N.; Arakelov, V.; Sargsyan, A.; Paronyan, A.; Arakelov, G.; Nazaryan, K. Impact of Mutations on the Stability of SARS-CoV-2 Nucleocapsid Protein Structure. Sci. Rep. 2024, 14, 5870. [Google Scholar] [CrossRef]
- Zhao, H.; Nguyen, A.; Wu, D.; Li, Y.; Hassan, S.A.; Chen, J.; Shroff, H.; Piszczek, G.; Schuck, P. Plasticity in Structure and Assembly of SARS-CoV-2 Nucleocapsid Protein. PNAS Nexus 2022, 1, pgac049. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Islam, M.R.; Alam, A.S.M.R.U.; Islam, I.; Hoque, M.N.; Akter, S.; Rahaman, M.M.; Sultana, M.; Hossain, M.A. Evolutionary Dynamics of SARS-CoV-2 Nucleocapsid Protein and Its Consequences. J. Med. Virol. 2021, 93, 2177–2195. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A Local Superposition-Free Score for Comparing Protein Structures and Models Using Distance Difference Tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Zhang, C.; Shine, M.; Pyle, A.M.; Zhang, Y. US-Align: Universal Structure Alignments of Proteins, Nucleic Acids, and Macromolecular Complexes. Nat. Methods 2022, 19, 1109–1115. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R Package for the Comparative Analysis of Protein Structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2024. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- The pandas development team. Pandas v.2.2.2 2024. p. 2024. [CrossRef]
- Zemla, A. LGA: A Method for Finding 3D Similarities in Protein Structures. Nucleic Acids Res. 2003, 31, 3370–3374. [Google Scholar] [CrossRef]
- Zemla, A.; Zhou, C.E.; Slezak, T.; Kuczmarski, T.; Rama, D.; Torres, C.; Sawicka, D.; Barsky, D. AS2TS System for Protein Structure Modeling and Analysis. Nucleic Acids Res. 2005, 33, W111–W115. [Google Scholar] [CrossRef] [PubMed]
- LGA. Protein Structure Comparison Facility. Available online: http://proteinmodel.org/AS2TS/LGA/lga.html (accessed on 2 July 2024).
- Chothia, C.; Lesk, A.M. The Relation between the Divergence of Sequence and Structure in Proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [CrossRef]
- Reva, B.A.; Finkelstein, A.V.; Skolnick, J. What Is the Probability of a Chance Prediction of a Protein Structure with an Rmsd of 6 å? Fold. Des. 1998, 3, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Olsen, K.S.; Gentry, K.M.; Sambade, M.; Beck, W.; Garness, J.; Entwistle, S.; Willis, C.; Vensko, S.; Woods, A.; et al. Landscape and Selection of Vaccine Epitopes in SARS-CoV-2. Genome Med. 2021, 13, 101. [Google Scholar] [CrossRef]
- TM-Score: Quantitative Assessment of Similarity between Protein Structures. Available online: https://zhanggroup.org/TM-score/ (accessed on 1 July 2024).
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A Structural Biology Community Assessment of AlphaFold2 Applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Bruley, A.; Mornon, J.-P.; Duprat, E.; Callebaut, I. Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond. Biomolecules 2022, 12, 1467. [Google Scholar] [CrossRef]
- Wilson, C.J.; Choy, W.-Y.; Karttunen, M. AlphaFold2: A Role for Disordered Protein/Region Prediction? Int. J. Mol. Sci. 2022, 23, 4591. [Google Scholar] [CrossRef]
- Alderson, T.R.; Pritišanac, I.; Kolarić, Đ.; Moses, A.M.; Forman-Kay, J.D. Systematic Identification of Conditionally Folded Intrinsically Disordered Regions by AlphaFold2. Proc. Natl. Acad. Sci. USA 2023, 120, e2304302120. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-Dependent Prediction of Protein Disorder as a Function of Redox State and Protein Binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Case, D.A. Normal Mode Analysis of Protein Dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Bauer, J.A.; Pavlović, J.; Bauerová-Hlinková, V. Normal Mode Analysis as a Routine Part of a Structural Investigation. Molecules 2019, 24, 3293. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Skjaerven, L.; Yao, X.-Q. The Bio3D Packages for Structural Bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Wako, H.; Endo, S. Normal Mode Analysis as a Method to Derive Protein Dynamics Information from the Protein Data Bank. Biophys. Rev. 2017, 9, 877–893. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, D.C.; Chalupska, D.; Silhan, J.; Koutna, E.; Nencka, R.; Veverka, V.; Boura, E. Structural Basis of RNA Recognition by the SARS-CoV-2 Nucleocapsid Phosphoprotein. PLoS Pathog. 2020, 16, e1009100. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, V.; Lehnert, U.; Echols, N.; Milburn, D.; Engelman, D.; Gerstein, M. Normal Modes for Predicting Protein Motions: A Comprehensive Database Assessment and Associated Web Tool. Protein Sci. A Publ. Protein Soc. 2005, 14, 633. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 Nucleocapsid Protein Is Dynamic, Disordered, and Phase Separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank. RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 2 July 2024).
- Bai, Z.; Cao, Y.; Liu, W.; Li, J. The SARS-CoV-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses 2021, 13, 1115. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.-Y.; Lee, K.-R.; Tarn, W.-Y. Phosphorylation of the Arginine/Serine Dipeptide-Rich Motif of the Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein Modulates Its Multimerization, Translation Inhibitory Activity and Cellular Localization. FEBS J. 2008, 275, 4152–4163. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Chang, C.; Ikeya, T.; Güntert, P.; Chang, Y.; Hsu, Y.; Huang, T.; Kainosho, M. Solution Structure of the C-Terminal Dimerization Domain of SARS Coronavirus Nucleocapsid Protein Solved by the SAIL-NMR Method. J. Mol. Biol. 2008, 380, 608–622. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chang, C.; Chang, Y.-W.; Sue, S.-C.; Bai, H.-I.; Riang, L.; Hsiao, C.-D.; Huang, T. Structure of the SARS Coronavirus Nucleocapsid Protein RNA-Binding Dimerization Domain Suggests a Mechanism for Helical Packaging of Viral RNA. J. Mol. Biol. 2007, 368, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Chen, C.-M.M.; Chiang, M.; Hsu, Y.; Huang, T. Transient Oligomerization of the SARS-CoV N Protein--Implication for Virus Ribonucleoprotein Packaging. PLoS ONE 2013, 8, e65045. [Google Scholar] [CrossRef]
- Surjit, M.; Kumar, R.; Mishra, R.N.; Reddy, M.K.; Chow, V.T.K.; Lal, S.K. The Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein Is Phosphorylated and Localizes in the Cytoplasm by 14-3-3-Mediated Translocation. J. Virol. 2005, 79, 11476–11486. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State-Chain | GDT-TS (AS2S) | TM Score/L (US-Align Server) | Superimposed RMSD < 5 Å (AS2S) |
|---|---|---|---|
| NTD-A (7CDZ) | 91.99 × (128/131) = 89.88 | *L1: 0.93398, L2: 0.91375 | 1.293/128 |
| NTD-B (7CDZ) | 90.35 × (127/131) = 87.59 | 0.92756, 0.90091 | 1.366/127 |
| NTD-C (7CDZ) | 95.24 × (126/131) = 91.60 | 0.95983, 0.92447 | 0.941/126 |
| NTD-D (7CDZ) | 94.26 × (122/131) = 87.78 | 0.93300, 0.90592 | 1.1108/122 |
| CTD-A (7CE0) | 69.73 × (109/110) = 69.10 | 0.74156 | 2.508/109 |
| CTD-B (7CE0) | 69.50 × (109/110) = 68.87 | 0.73968 | 2.519/109 |
| CTD-C (7CE0) | 69.39 × (107/110) = 67.50 | 0.73391 | 2.476/107 |
| CTD-D (7CE0) | 68.93 × (107/110) = 67.05 | 0.73302 | 2.490/107 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.A.; Caetano-Anollés, G. AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Nucleocapsid Protein Variants. Viruses 2024, 16, 1358. https://doi.org/10.3390/v16091358
Ali MA, Caetano-Anollés G. AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Nucleocapsid Protein Variants. Viruses. 2024; 16(9):1358. https://doi.org/10.3390/v16091358
Chicago/Turabian StyleAli, Muhammad Asif, and Gustavo Caetano-Anollés. 2024. "AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Nucleocapsid Protein Variants" Viruses 16, no. 9: 1358. https://doi.org/10.3390/v16091358
APA StyleAli, M. A., & Caetano-Anollés, G. (2024). AlphaFold2 Reveals Structural Patterns of Seasonal Haplotype Diversification in SARS-CoV-2 Nucleocapsid Protein Variants. Viruses, 16(9), 1358. https://doi.org/10.3390/v16091358