
Advances in Antiviral Delivery Systems and Chitosan-Based Polymeric and Nanoparticulate Antivirals and Antiviral Carriers



Abstract
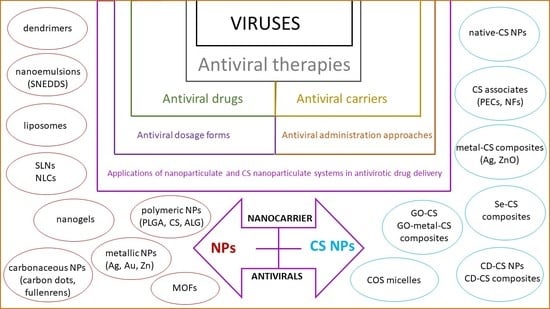
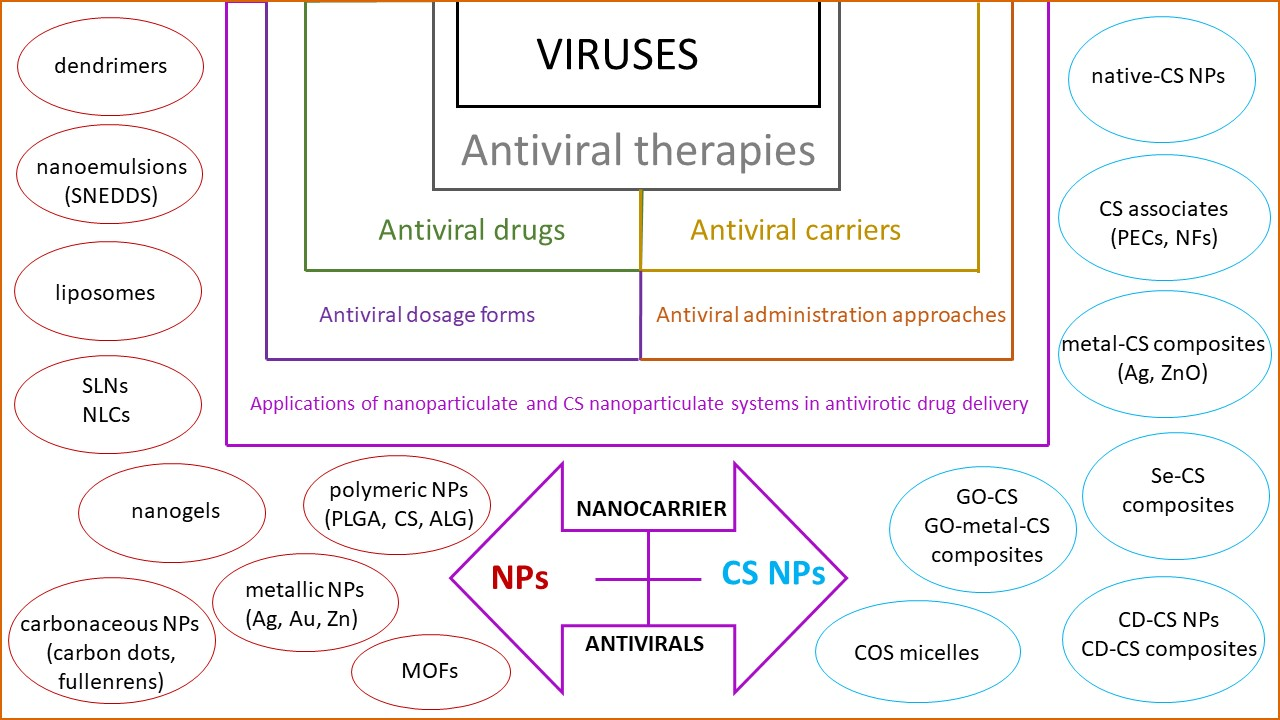
1. Introduction
2. Approaches in Current Antiviral Therapy
2.1. Antivirals’ Classification According to the Basis of Their Target
2.2. Limitations on the Use of Antivirals as a Task for Further Development
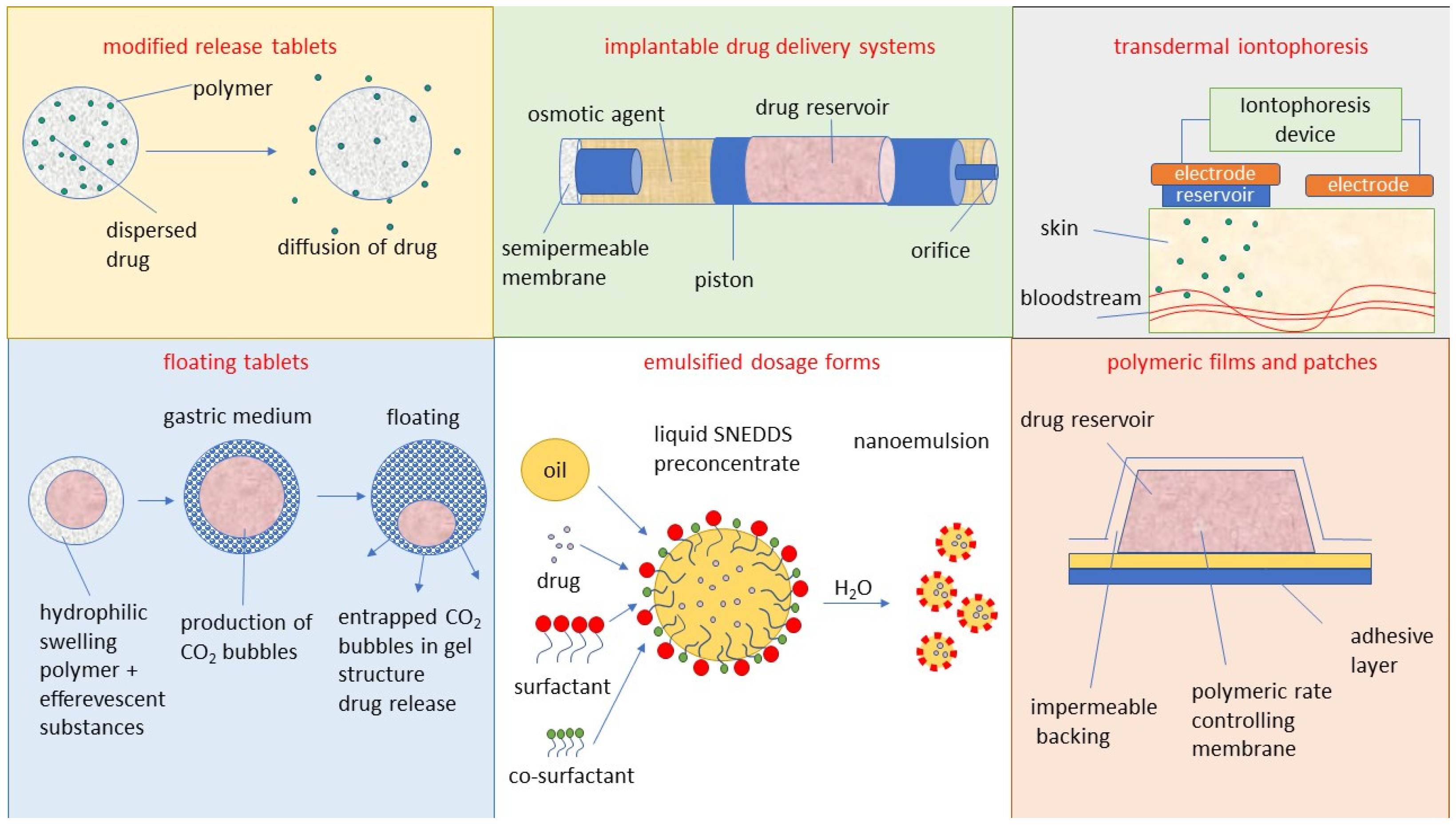
3. Dosage Forms and Drug Delivery Systems for Antivirals
3.1. Modified-Release Tablets
3.2. Floating Delivery Systems
3.3. Implantable Delivery Systems
3.4. Transdermal Iontophoresis Delivery Systems
3.5. Polymeric Films and Patches
3.6. Emulsified Dosage Forms
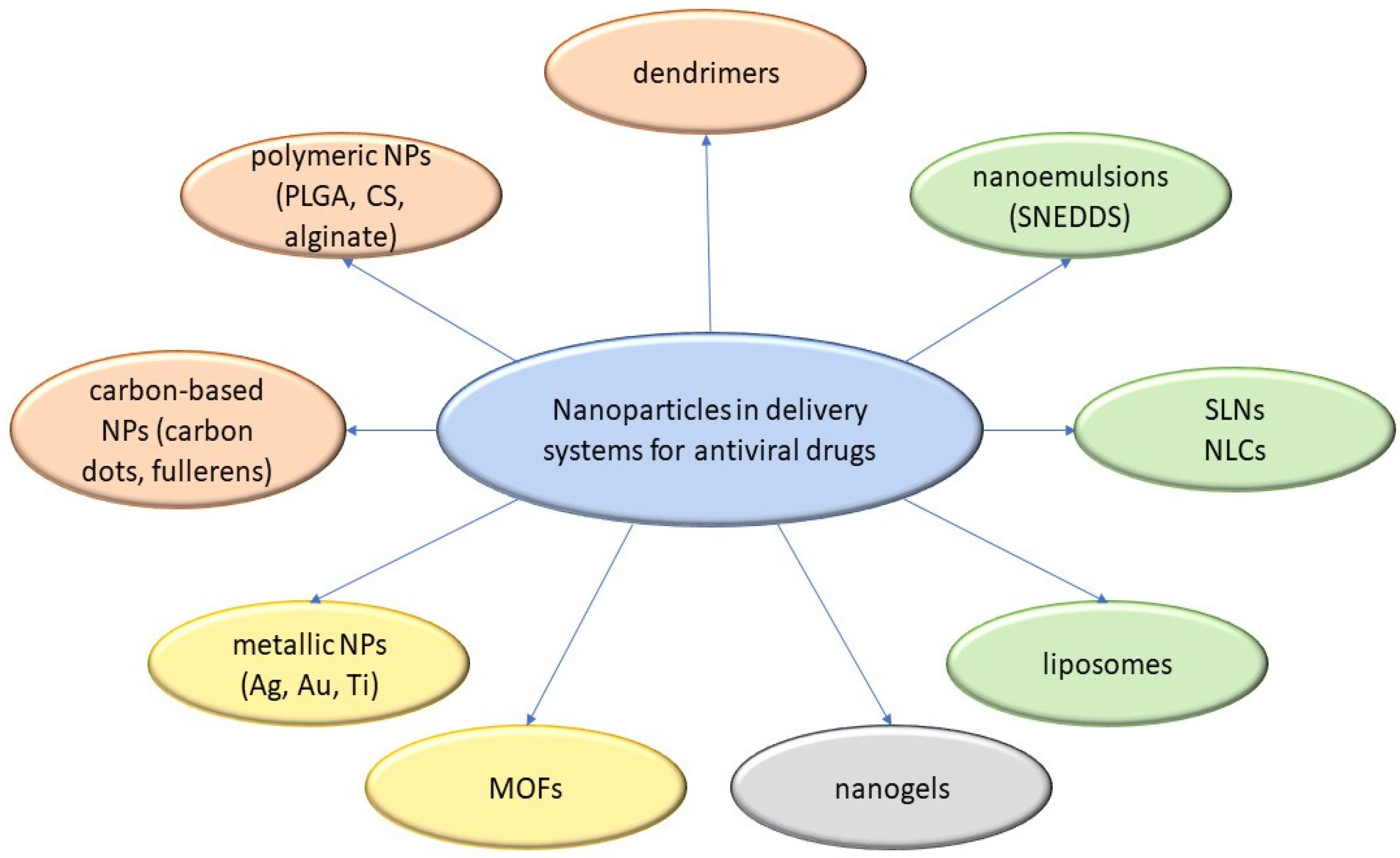
3.7. Micro- and Nanoparticulate Delivery Systems
4. CS-Based Dosage Forms and Drug Delivery Systems for Antivirals
4.1. CS and Its Derivatives as Polymeric Systems in Antiviral Use
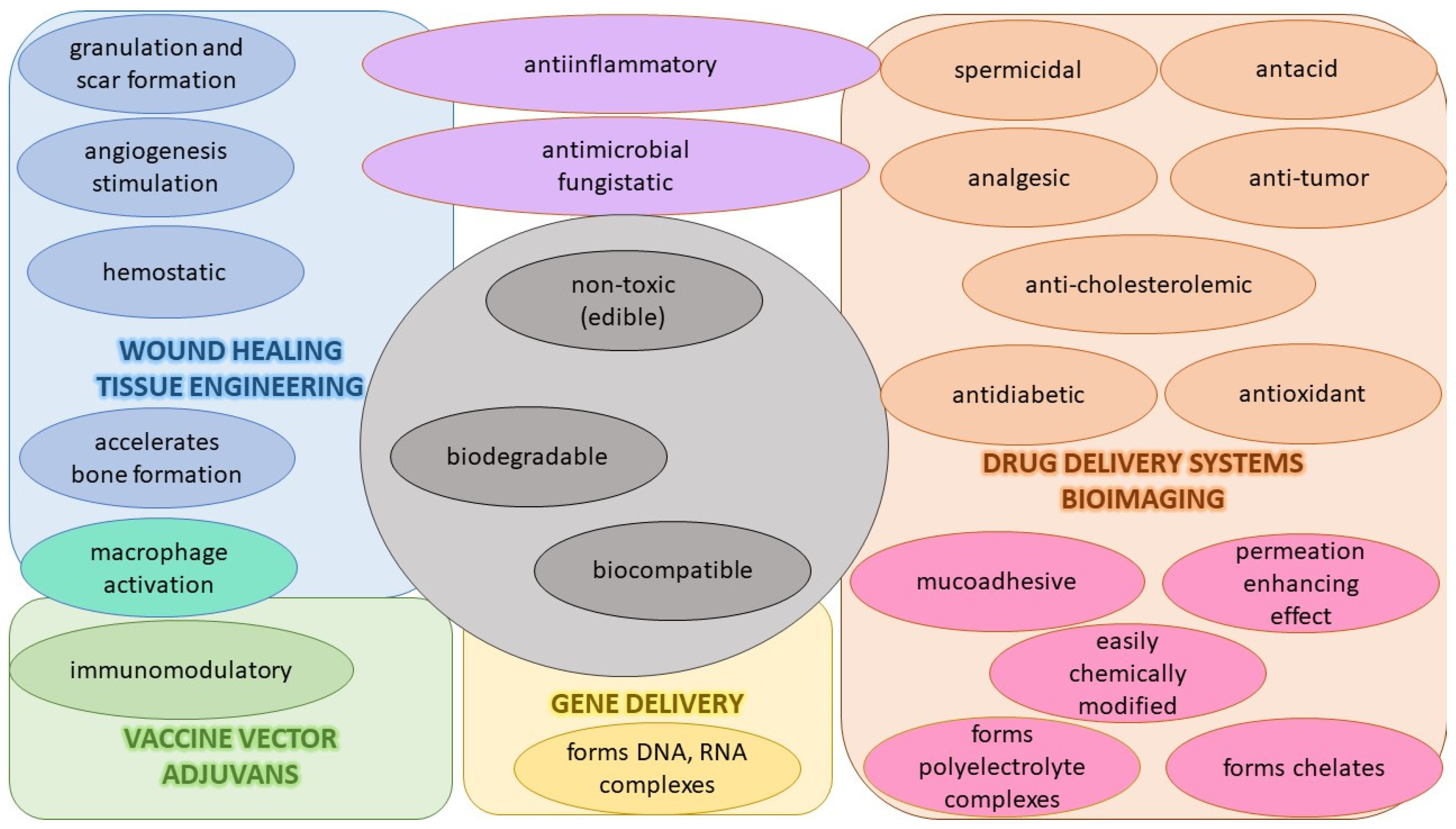
4.1.1. Properties, Activities, and Interactions of CS
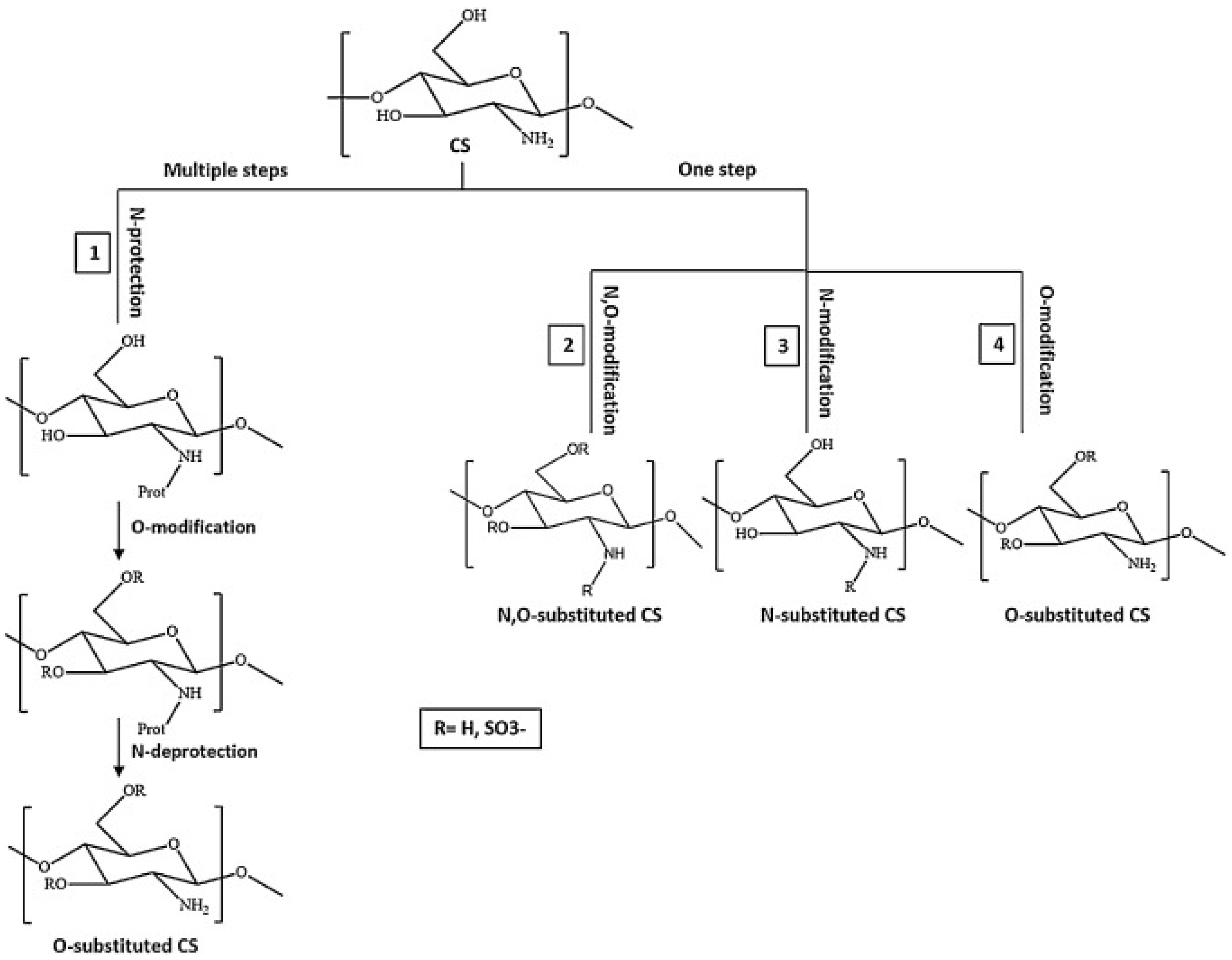
4.1.2. Preparations, Modified Properties, and Use of CS Derivatives
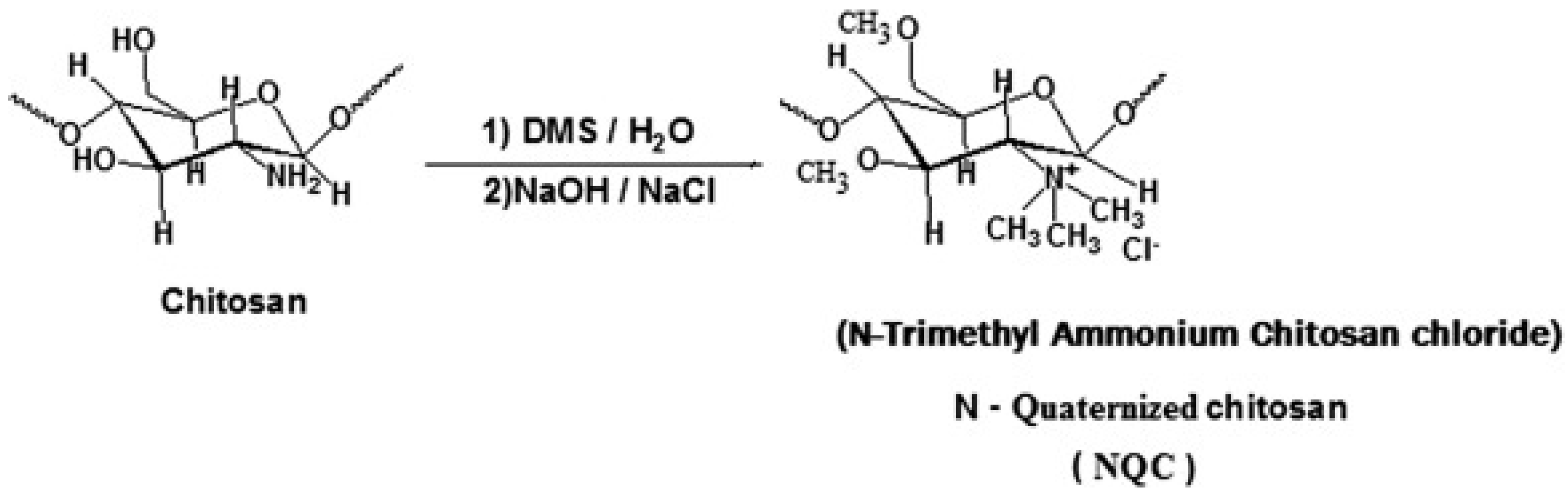
Quaternized CS
Sulfated CS

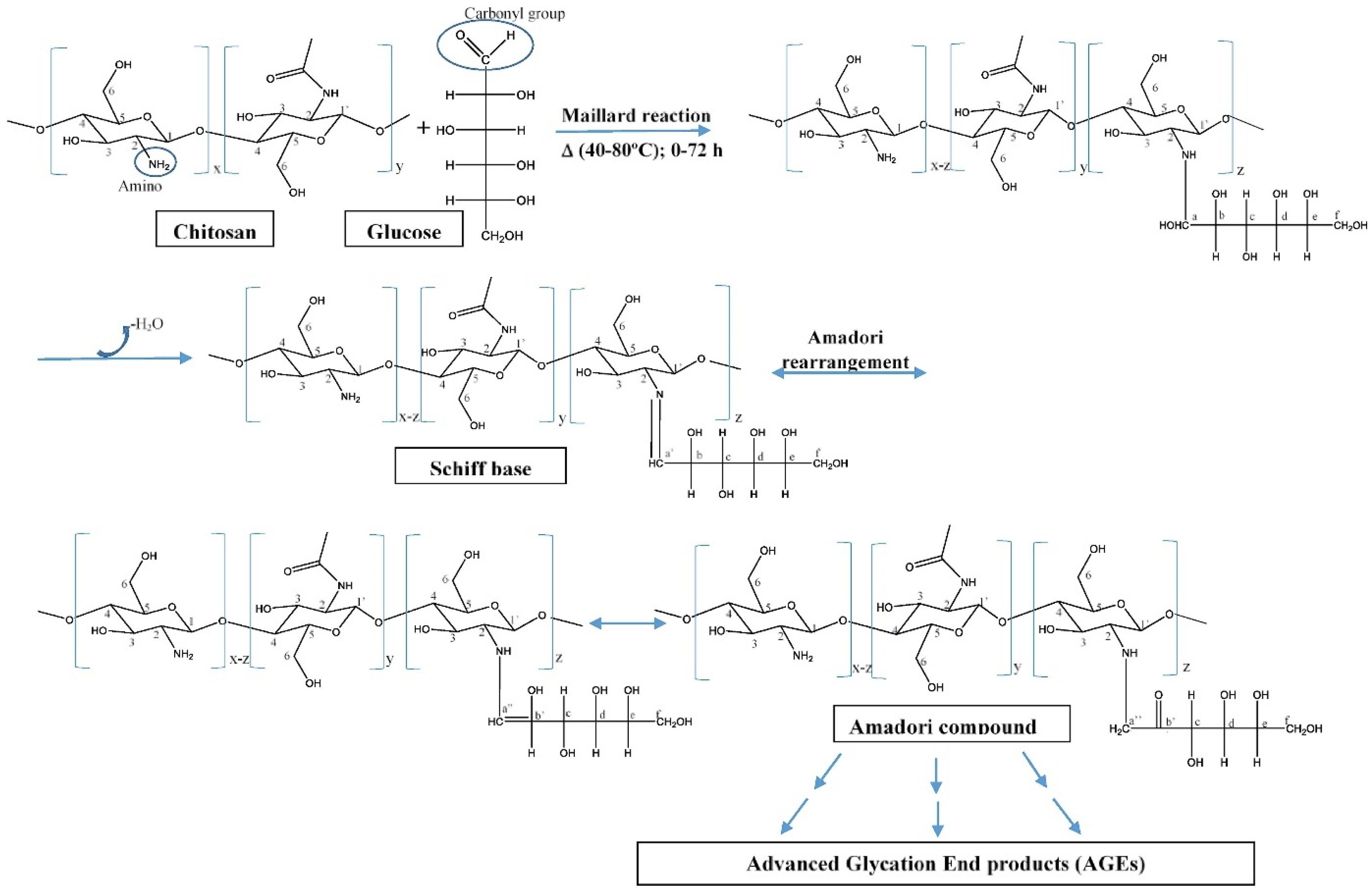
CS Derivatives with Sugar Part
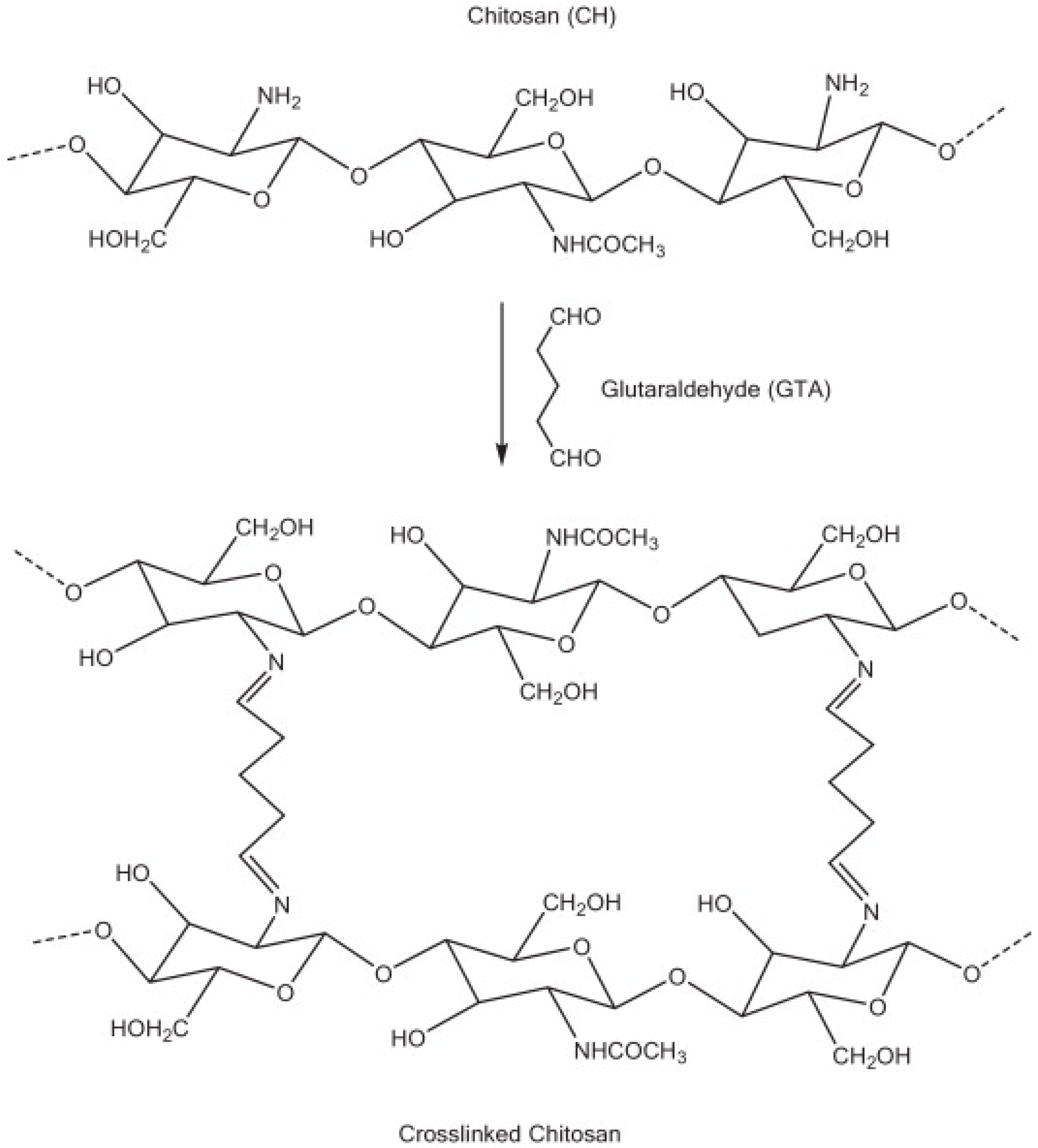
CS Glutaraldehyde Crosslinked Polymer
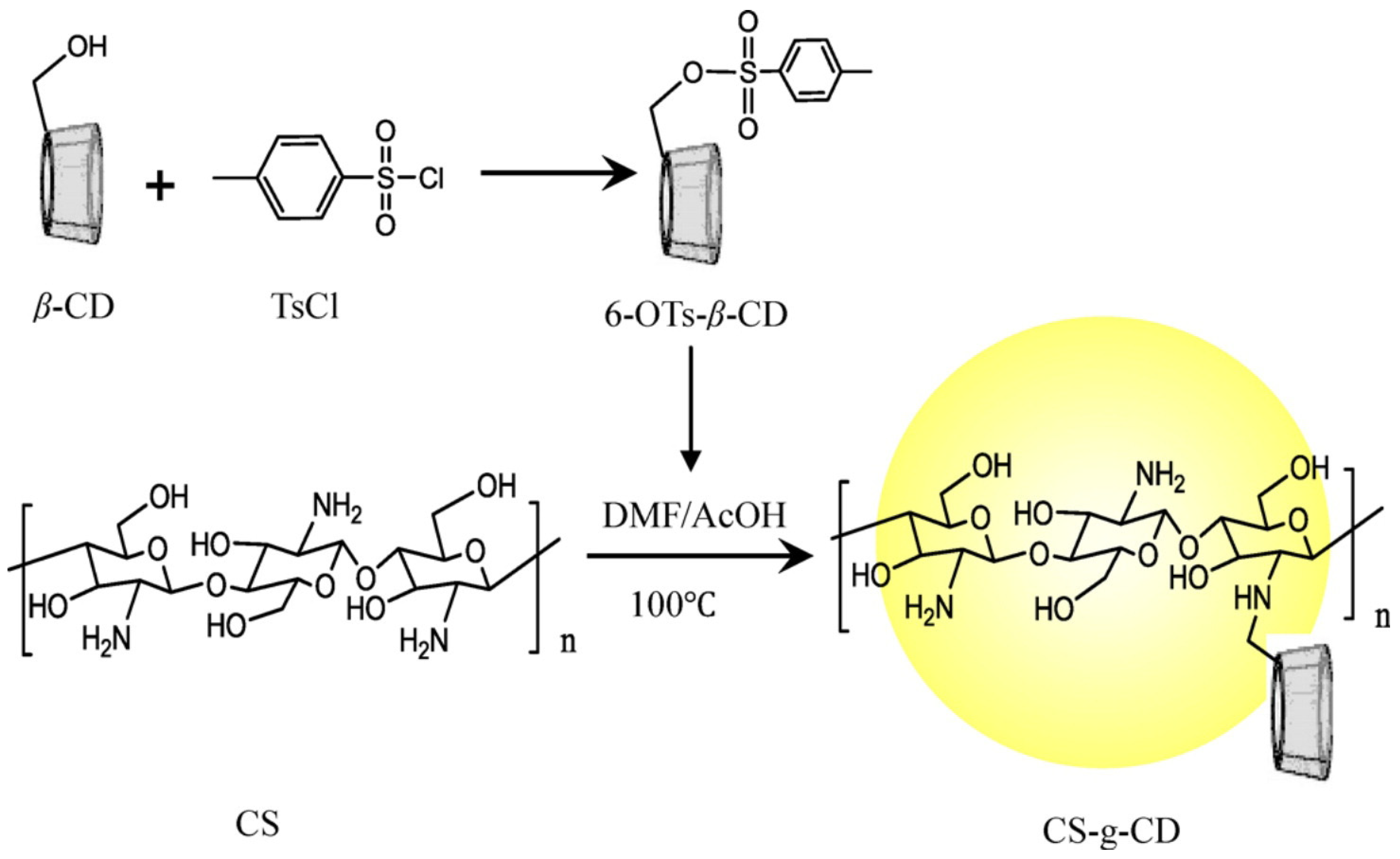
CS Cyclodextrin
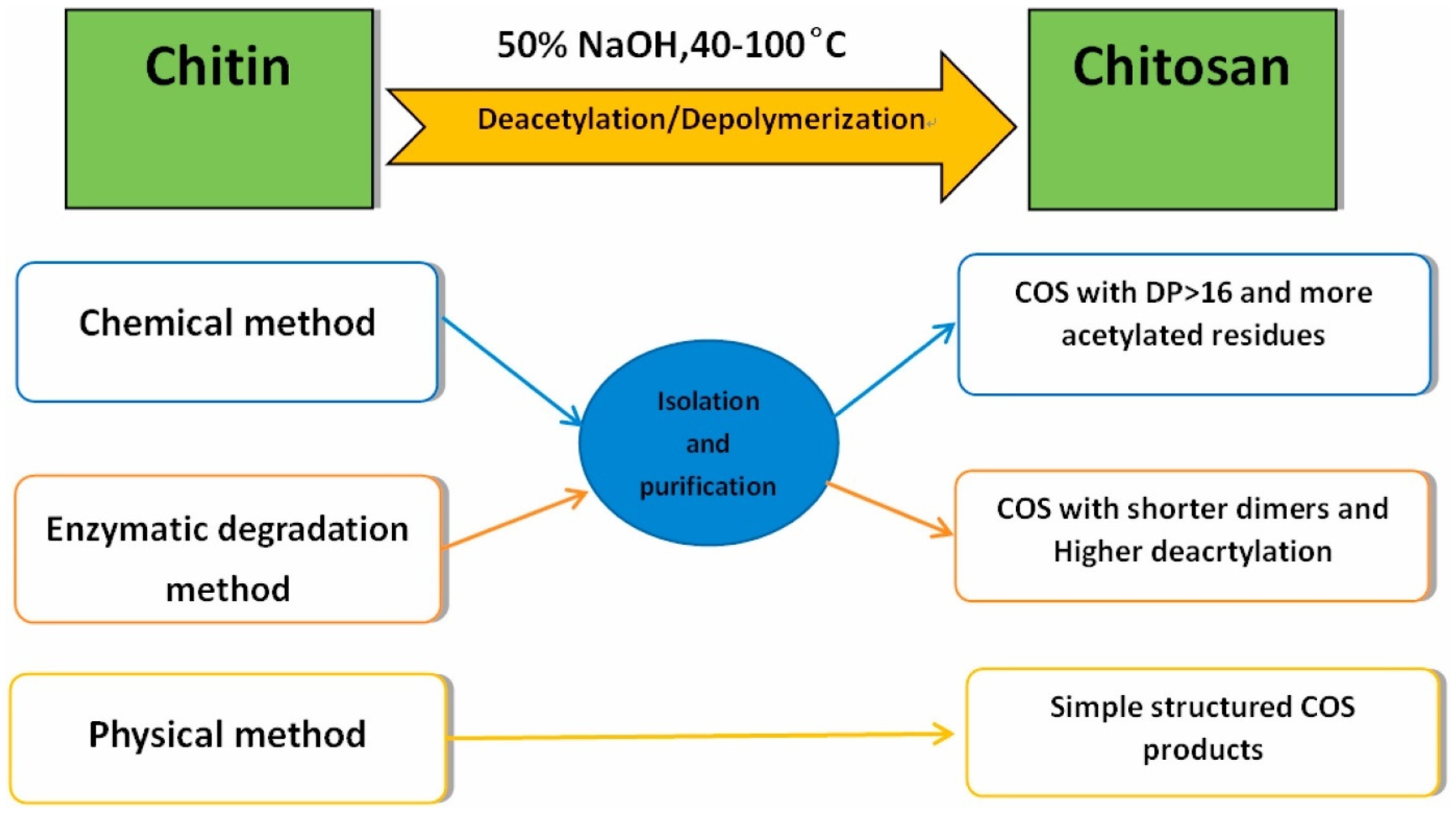
CS Oligomers
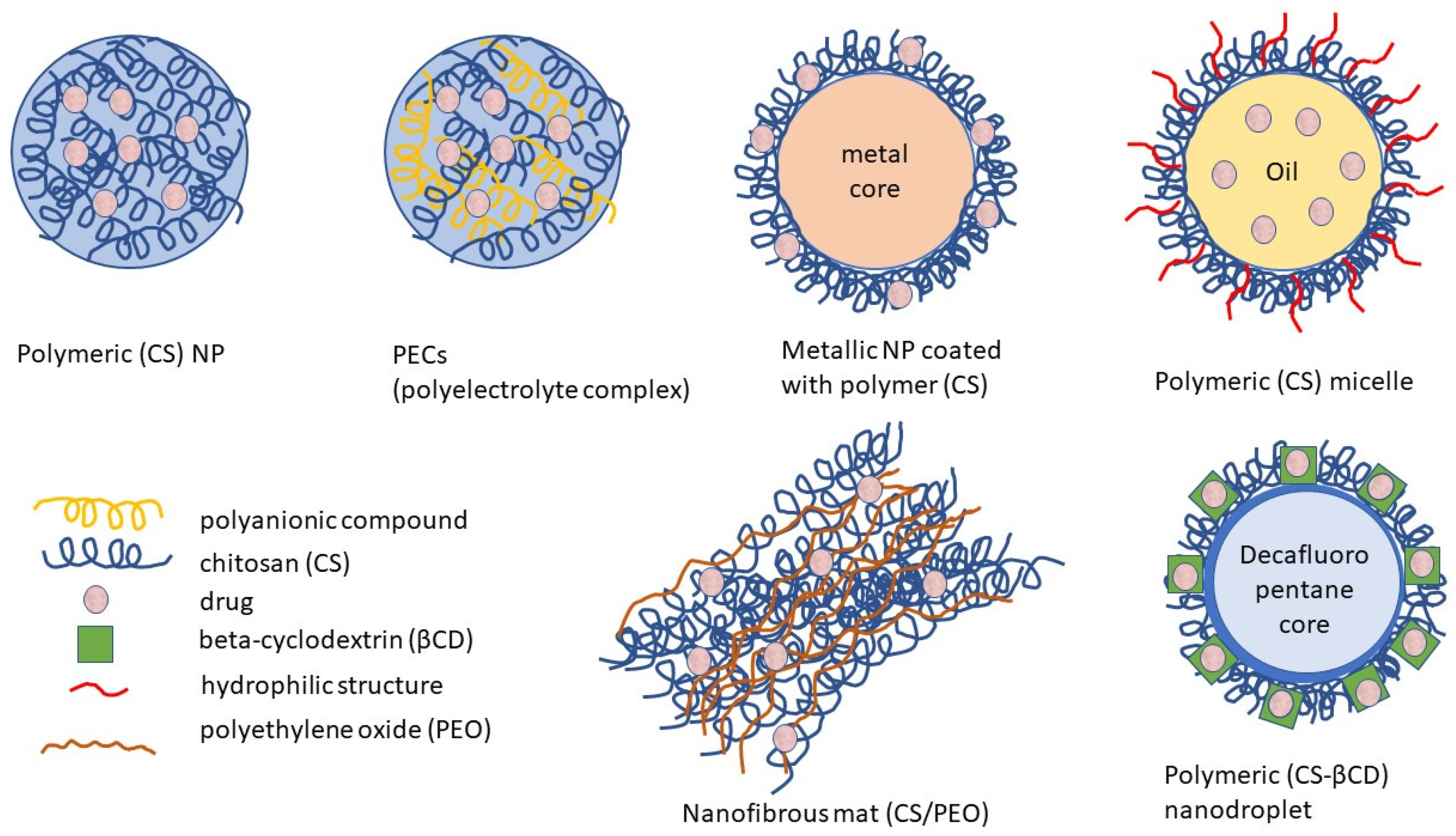
4.2. CS and Its Derivatives in Nanoparticulate Antivirotic Drug Delivery Systems
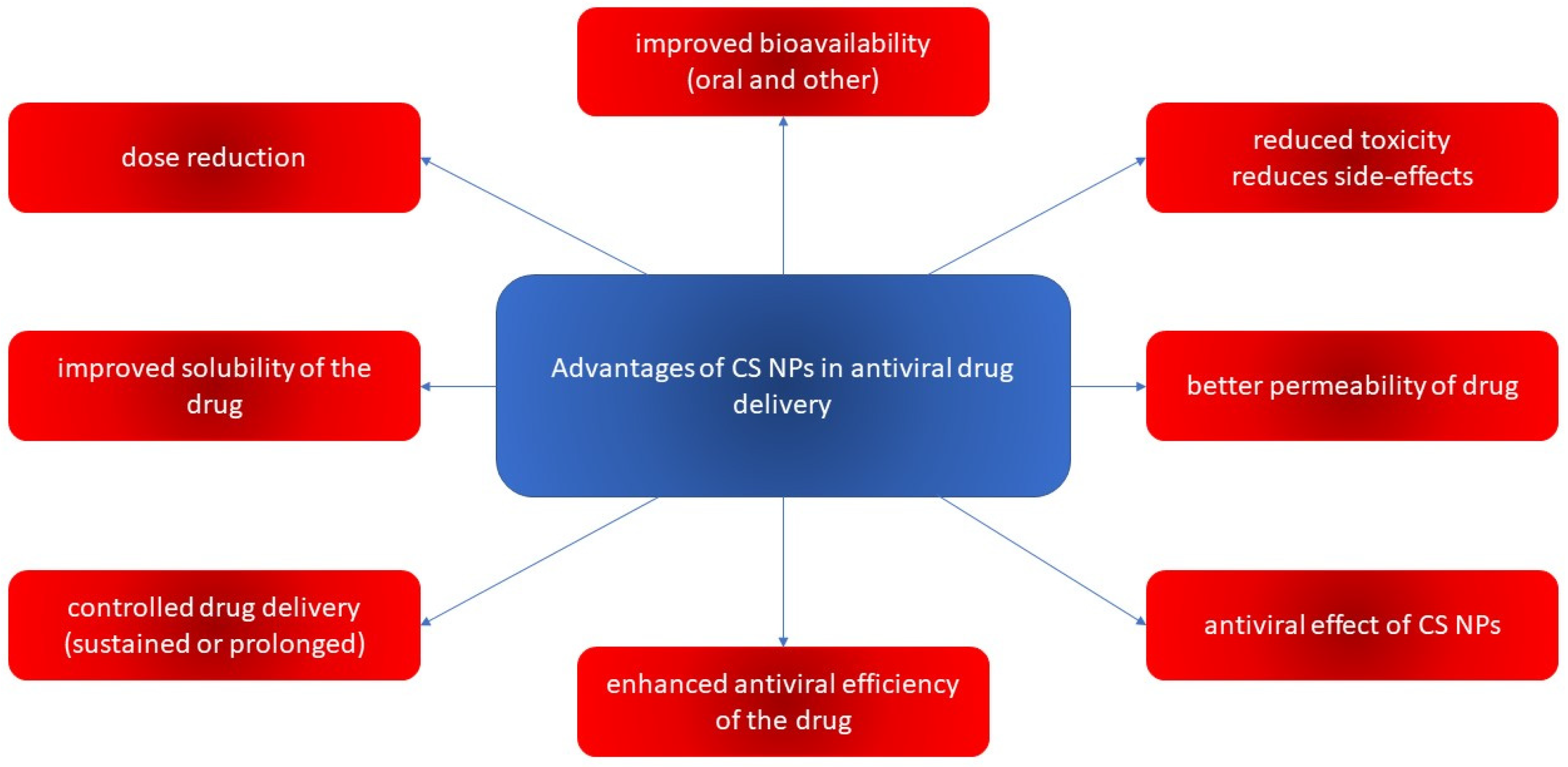
4.2.1. Properties and Activities of CS-Based Nanoparticulate Systems
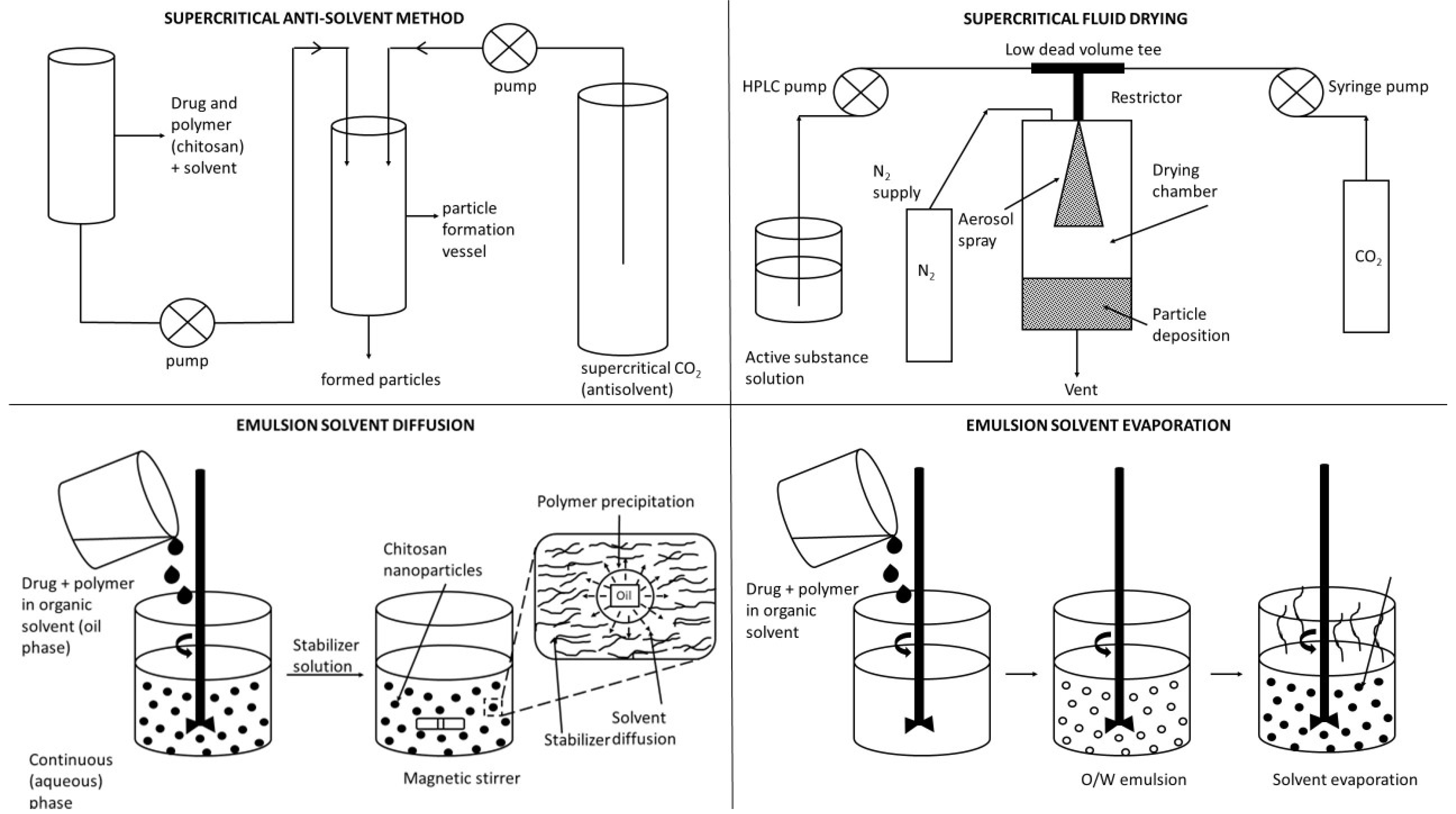
4.2.2. Preparations of CS-Based Nanoparticulate Systems
- (i)
- Solvent evaporation method: In this method, the polymer is dissolved in an organic solvent such as dichloromethane, chloroform, or ethyl acetate. The drug is dissolved or dispersed into the preformed polymer solution, and this mixture is then emulsified into an aqueous solution to make an O/W emulsion by using a surfactant/emulsifying agent such as gelatin, poly(vinyl alcohol), polysorbate-80, poloxamer-188, etc. After the formation of a stable emulsion, the organic solvent is evaporated by increasing the temperature, raising the pressure, or continuously stirring. The W/O/W method has also been used to prepare the water-soluble drug-loaded NPs. Both the above methods use high-speed homogenization or sonication. However, these procedures are good for a laboratory scale only [263].
- (ii)
- Spontaneous emulsification/solvent diffusion method: The water-soluble solvent such as acetone or methanol along with the water-insoluble organic solvent such as dichloromethane or chloroform are used in an oil phase. Due to the spontaneous diffusion of water-soluble solvent (acetone or methanol), interfacial turbulence is created between two phases, leading to the formation of smaller particles. As the concentration of water-soluble solvent (acetone) increases, a considerable decrease in particle size can be achieved [9,264].
- (iii)
- Production of NPs using supercritical fluid: Supercritical fluids have now become attractive alternatives because these are environment-friendly solvents and the method can be profitably used to process particles at a high purity and without any trace amount of the organic solvent. Rapid expansion of supercritical solution (RESS) method is a method where the solute of interest is solubilized in a supercritical fluid and the solution is expanded through a nozzle. Thus, the solvent power of supercritical fluid dramatically decreases and the solute eventually precipitates. This technique is clean because the precipitated solute is completely solvent-free. Unfortunately, most polymers exhibit little or no solubility in supercritical fluids, thus leaving the technique of little practical interest [16].
- (iv)
- Supercritical anti-solvent (SAS) method: The solution is charged with the supercritical fluid in the precipitation vessel containing a solute of interest in an organic solvent. At high pressures, enough anti-solvent enters the liquid phase that the solvent power is lowered and the solute precipitates. After precipitation, when the final operating pressure is reached, the anti-solvent flows through the vessel to strip the residual solvent. When the solvent content has been reduced to the desired level, the vessel is depressurized and the solid product is collected. In a modified version of the SAS technique, the solid of interest is first dissolved in a suitable solvent and then this solution is rapidly introduced into the supercritical fluid through a narrow nozzle. The supercritical fluid completely extracts the solvent, causing the supercritical fluid insoluble solid to precipitate as fine particles. This method, also called the gas anti-solvent (GAS) technique, has been successfully used to produce micro-particles as well as NPs [248].
4.2.3. Applications of CS-Based Nanoparticulate Systems in Antiviral Drug Delivery
Nanoparticles with Native CS
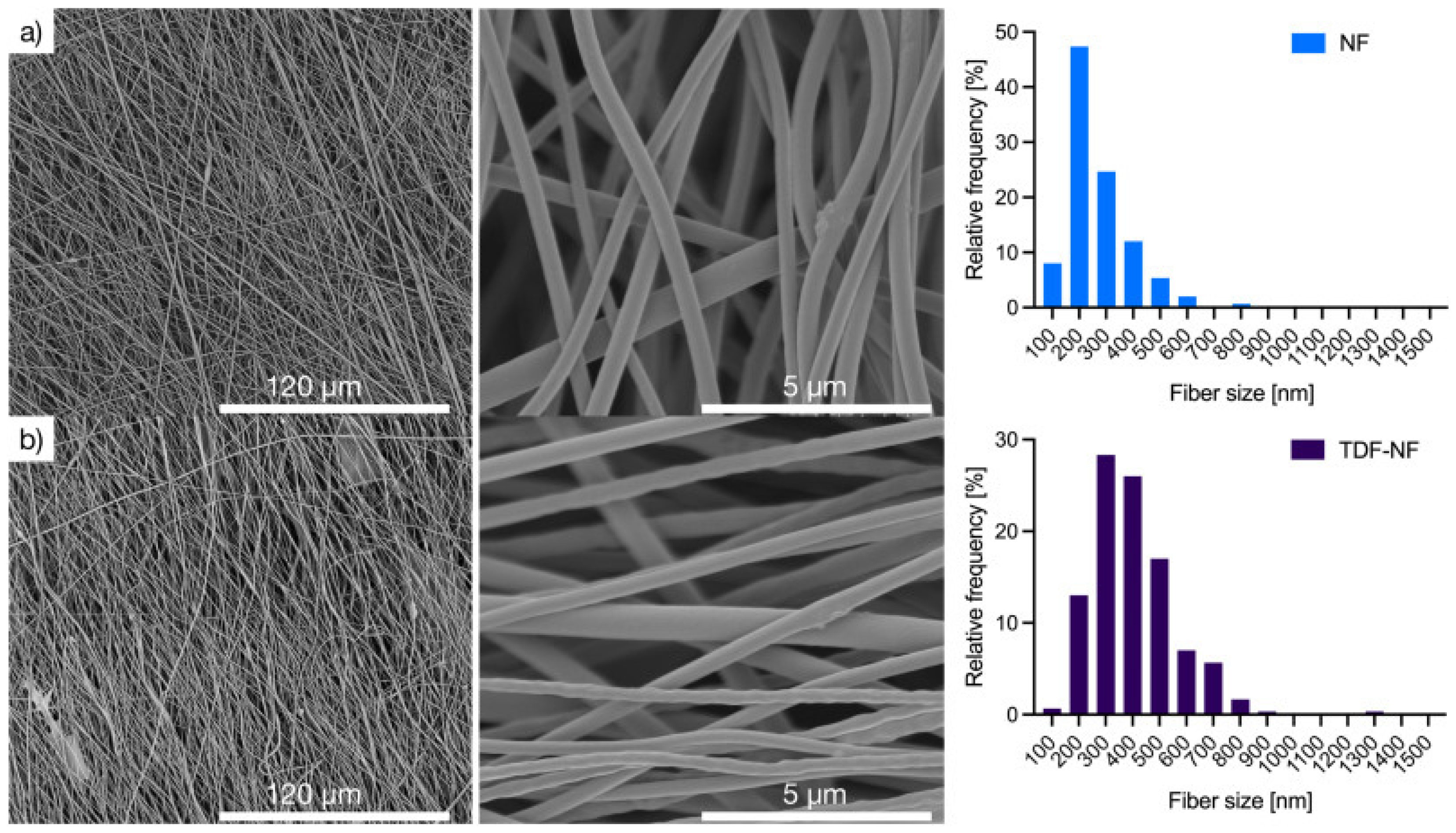
Nanosystems Based on Polymeric Associates with Native CS (PECs, NFs)
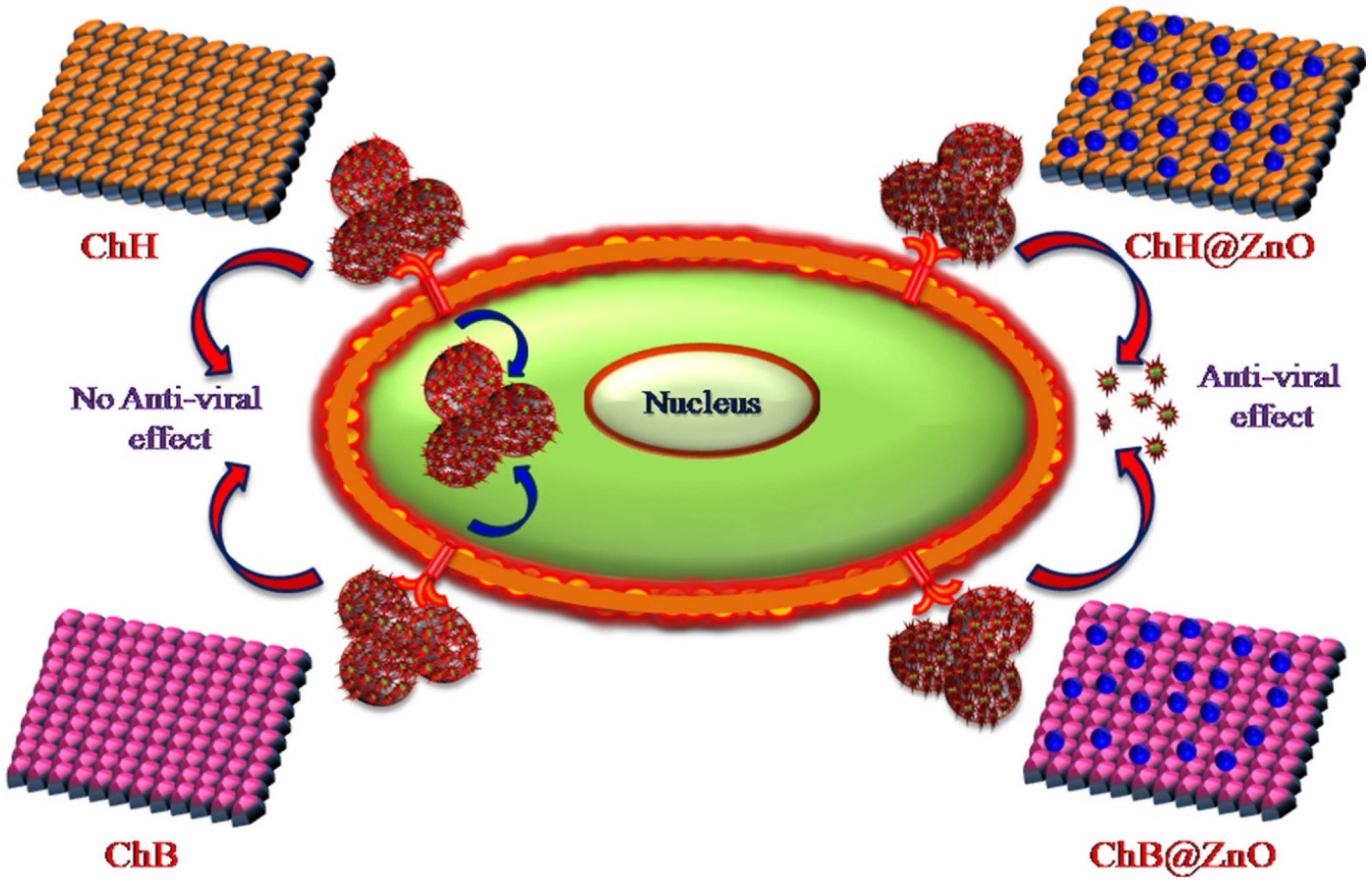
CS–Metal Nanocomposites
CS–Selenium Nanocomposites
CS–Graphene Oxide Nanocomposites
Nanoparticles Based on Cyclodextrin Derivatives and Composites with Native CS
COS Nanosystems
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saxena, S.K.; Mishra, N.; Saxena, R. Advances in antiviral drug discovery and development: Part I: Advancements in antiviral drug discovery. Future Virol. 2009, 4, 101–107. [Google Scholar] [CrossRef]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef]
- Menéndez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol. Sci. 2022, 43, 6–29. [Google Scholar] [CrossRef]
- De Clercq, E. Chemotherapy of Viral Infections. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Maus, A.; Strait, L.; Zhu, D. Nanoparticles as delivery vehicles for antiviral therapeutic drugs. J. Eng. Regen. Med. 2021, 2, 31–46. [Google Scholar] [CrossRef]
- Sharma, P.; Chawla, A.; Arora, S.; Pawar, P. Novel drug delivery approaches on antiviral and antiretroviral agents. J. Adv. Pharm. Tech. Res. 2012, 3, 147–159. [Google Scholar] [CrossRef]
- Durai, R. Drug delivery approaches of an antiviral drug: A comprehensive review. Asian J. Pharm. 2015, 9, 1–12. [Google Scholar] [CrossRef]
- Čierna, M.; Mučaji, P.; Špaglová, M.; Čuchorová, M.; Macho, O. Chitosan and Sodium Alginate Implementation as Pharmaceutical Excipients in Multiple-Unit Particulate Systems. Polymers 2022, 14, 2822. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Ramvignesh, T.R.; Sundaramoorthy, K.; Ayyappan, T.; Vetrichelvan, T. Research Design and evaluation of novel ophthalmic delivery system of acyclovir for herpes simplex infection. J. Pharm. Biol. Chem. Scien. 2011, 2, 803–814. [Google Scholar]
- Sanna, V.; Satta, S.; Hsiai, T.; Sechi, M. Development of targeted nanoparticles loaded with antiviral drugs for SARS-CoV-2 inhibition. Eur. J. Med. Chem. 2022, 231, 114121. [Google Scholar] [CrossRef]
- Jain, S.; Tiwary, A.K.; Sapra, B.; Jain, N.K. Formulation and evaluation of ethosomes for transdermal delivery of lamivudine. AAPS PharmSciTech 2007, 8, E111. [Google Scholar] [CrossRef]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharmacol. 2015, 495, 439–446. [Google Scholar] [CrossRef]
- Dostalova, S.; Moulick, A.; Milosavljevic, V.; Guran, R.; Kominkova, M.; Cihalova, K.; Heger, Z.; Blazkova, L.; Kopel, P.; Hynek, D.; et al. Antiviral activity of fullerene C60 nanocrystals modified with derivatives of anionic antimicrobial peptide maximin H5. Chem. Mon. 2016, 147, 905–918. [Google Scholar] [CrossRef]
- Sur, S.; Rathore, A.; Dave, V.; Reddy, K.R.; Chouhan, R.S.; Sadhu, V. Recent developments in functionalized polymer nanoparticles for efficient drug delivery system. Nano-Struct. Nano-Objects 2019, 20, 100397. [Google Scholar] [CrossRef]
- Garg, U.; Chauhan, S.; Nagaich, U.; Jain, N. Current Advances in Chitosan Nanoparticles Based Drug Delivery and Targeting. Adv. Pharm. Bull. 2019, 9, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Mikušová, V.; Mikuš, P. Advances in Chitosan-Based Nanoparticles for Drug Delivery. Int. J. Mol. Sci. 2021, 22, 9652. [Google Scholar] [CrossRef] [PubMed]
- FDA/CDER. Guidance for Industry, Drug Products, Including Biological Products, That Contain Nanomaterials; Center for Drug Evaluation and Research: Maryland, MD, USA, 2022. [Google Scholar]
- Marques, C.; Maurizi, L.; Borchard, G.; Jordan, O. Characterization Challenges of Self-Assembled Polymer-SPIONs Nanoparticles: Benefits of Orthogonal Methods. Int. J. Mol. Sci. 2022, 23, 16124. [Google Scholar] [CrossRef] [PubMed]
- Delshadi, R.; Bahrami, A.; McClements, D.J.; Moore, M.D.; Williams, L. Development of nanoparticle-delivery systems for antiviral agents: A review. J. Control. Release Off J. Control. Release Soc. 2021, 331, 30–44. [Google Scholar] [CrossRef]
- Lembo, D.; Donalisio, M.; Civra, A.; Argenziano, M.; Cavalli, R. Nanomedicine formulations for the delivery of antiviral drugs: A promising solution for the treatment of viral infections. Expert Opin. Drug Deliv. 2018, 15, 93–114. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Z.; Cao, L.; Ge, K. Constructive strategies for drug delivery systems in antivirus disease therapy by safety materials. Biosaf. Health 2022, 4, 61–170. [Google Scholar] [CrossRef]
- Yang, D. Application of Nanotechnology in the COVID-19 Pandemic. Int. J. Nanomed. 2021, 16, 623–649. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, J.; Raichura, Z.; Khan, T.; Momin, M.; Omri, A. Chitosan Nanoparticles-Insight into Properties, Functionalization and Applications in Drug Delivery and Theranostics. Molecules 2021, 26, 272. [Google Scholar] [CrossRef] [PubMed]
- Bowman, K.; Leong, K.W. Chitosan nanoparticles for oral drug and gene delivery. Int. J. Nanomed. 2006, 1, 117–128. [Google Scholar] [CrossRef]
- Negm, N.A.; Hefni, H.H.H.; Abd-Elaal, A.A.A.; Badr, E.A.; Abou Kana, M.T.H. Advancement on modification of chitosan biopolymer and its potential applications. Int. J. Biol. Macromol. 2020, 152, 681–702. [Google Scholar] [CrossRef]
- Wang, W.; Meng, Q.; Li, Q.; Liu, J.; Zhou, M.; Jin, Z.; Zhao, K. Chitosan Derivatives and Their Application in Biomedicine. Int. J. Mol. Sci. 2020, 21, 487. [Google Scholar] [CrossRef]
- Patrulea, V.; Ostafe, V.; Borchard, G.; Jordan, O. Chitosan as a starting material for wound healing applications. Eur. J. Pharm. Biopharm. Part B 2015, 97, 417–426. [Google Scholar] [CrossRef]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Khan, A.H.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002620. [Google Scholar] [CrossRef]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef] [PubMed]
- LiverTox. Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012. [Google Scholar]
- Kaushik, I.; Ramachandran, S.; Prasad, S.; Srivastava, S.K. Drug rechanneling: A novel paradigm for cancer treatment. Semin. Cancer Biol. 2021, 68, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Scholar, E. Idoxuridine. In XPharm Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Suita, K.; Ohnuki, Y.; Mototani, Y.; Ishikawa, M.; Ito, A.; Nariyama, M.; Morii, A.; Kiyomoto, K.; Tsunoda, M.; et al. Vidarabine, an anti-herpes agent, prevents occlusal-disharmony-induced cardiac dysfunction in mice. J. Physiol. Sci. 2022, 72, 2. [Google Scholar] [CrossRef]
- Taylor, M.; Gerriets, V. Acyclovir. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Shehata, H. Drugs and Drug Therapy. In Basic Science in Obstetrics and Gynaecology, 4th ed.; Bennett, P., Williamson, C., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2010; pp. 259–277. [Google Scholar] [CrossRef]
- Hoth, A.B.; Rawls, N.; Shorr, R.I. (Eds.) Drugs for the Geriatric Patient; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–115. [Google Scholar] [CrossRef]
- Mondal, D. Cidofovir. In XPharm Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Foscarnet. Meyler’s Side Effects of Drugs, 16th ed.; Aronson, J.K., Ed.; Elsevier: Oxford, UK, 2016; pp. 450–451. [Google Scholar] [CrossRef]
- Villemin, D.; Moreau, B.; Bar, N. MCR under Microwave Irradiation: Synthesis in Water of New 2-Amino-bis(2-phosphonoacetic) Acids. Organics 2021, 2, 98–106. [Google Scholar] [CrossRef]
- Bule, M.; Khan, F.; Niaz, K. Antivirals: Past, Present and Future. In Recent Advances in Animal Virology; Malik, Y.S., Singh, R.K., Yadav, M.P., Eds.; Springer: Singapore, 2019; pp. 425–446. [Google Scholar] [CrossRef]
- Waller, D.G.; Sampson, A.P. (Eds.) Chemotherapy of Infections. In Medical Pharmacology and Therapeutics, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 581–629. [Google Scholar] [CrossRef]
- Aliyu, S. Viral, Fungal, Protozoal and Helminthic Infections. In Clinical Pharmacology, 12th ed.; Bennett, P.N., Brown, M.J., Sharma, P., Eds.; Churchill Livingstone: Philadelphia, PA, USA, 2012; pp. 221–247. [Google Scholar] [CrossRef]
- Yee, J.; Preuss, C.V. Efavirenz. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Paintsil, E.; Cheng, Y.C. Antiviral Agents. In Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Academic Press: Cambridge, MA, USA, 2009; pp. 223–257. [Google Scholar] [CrossRef]
- Eckhardt, B.J.; Gulick, R.M. Drugs for HIV Infection. In Infectious Diseases, 4th ed.; Cohen, J., Powderly, W.G., Opal, S.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1293–1308. [Google Scholar] [CrossRef]
- Taylor, K.; Fritz, K.; Parmar, M. Lamivudine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Anderson, P.L.; Kakuda, T.N.; Lichtenstein, K.A. The cellular pharmacology of nucleoside- and nucleotide-analogue reverse-transcriptase inhibitors and its relationship to clinical toxicities. Clin. Infect. Dis. 2004, 38, 743–753. [Google Scholar] [CrossRef]
- de Vries-Sluijs, T.E.M.S.; Reijnders, J.G.P.; Hansen, B.E.; Zaaijer, H.L.; Prins, J.M.; Pas, S.D.; Schutten, M.; Hoepelman, A.I.M.; Richter, C.; Mulder, J.W.; et al. Long-term therapy with tenofovir is effective for patients co-infected with human immunodeficiency virus and hepatitis B virus. Gastroenterology 2010, 139, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K. (Ed.) Adefovir. In Meylers Side Effects of Drugs, 6th ed.; Elsevier: Oxford, UK, 2016; p. 72. [Google Scholar] [CrossRef]
- Rehman, N.; Nguyen, H. Nevirapine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Scott, L.J.; Perry, C.M. Delavirdine: A review of its use in HIV infection. Drugs 2000, 60, 1411–1444. [Google Scholar] [CrossRef] [PubMed]
- Mallayasamy, S.; Penzak, S.R. Pharmacogenomic Considerations in the Treatment of HIV Infection. In Pharmacogenomics, 2nd ed.; Lam, Y.W.F., Scott, S.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 227–245. [Google Scholar] [CrossRef]
- Bodiwala, H.S.; Sabde, S.; Gupta, P.; Mukherjee, R.; Kumar, R.; Garg, P.; Bhutani, K.K.; Mitra, D.; Singh, I.P. Design and synthesis of caffeoyl-anilides as portmanteau inhibitors of HIV-1 integrase and CCR5. Bioorg. Med. Chem. Lett. 2011, 19, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS—Res. Palliat. Care 2015, 7, 95–104. [Google Scholar] [CrossRef]
- Pires, D.; Valente, S.; Calado, M.; Mandal, M.; Azevedo-Pereira, M.; Anes, E. Repurposing Saquinavir for Host-Directed Therapy to Control Mycobacterium Tuberculosis Infection. Front. Immunol. 2021, 12, 647728. [Google Scholar] [CrossRef]
- Pollak, E.B.; Parmar, M. Indinavir. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Markland, W.; Rao, B.G.; Parsons, J.D.; Black, J.; Zuchowski, L.; Tisdale, M.; Tung, R. Structural and Kinetic Analyses of the Protease from an Amprenavir-Resistant Human Immunodeficiency Virus Type 1 Mutant Rendered Resistant to Saquinavir and Resensitized to Amprenavir. J. Virol. 2000, 74, 7636–7641. [Google Scholar] [CrossRef]
- Chapman, T.M.; Plosker, G.L.; Perry, C.M. Fosamprenavir: A review of its use in the management of antiretroviral therapy-naive patients with HIV infection. Drugs 2004, 64, 2101–2124. [Google Scholar] [CrossRef] [PubMed]
- Subeha, M.R.; Telleria, C.M. The Anti-Cancer Properties of the HIV Protease Inhibitor Nelfinavir. Cancers 2020, 12, 3437. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Innao, V.; Allegra, A.G.; Pulvirenti, N.; Pugliese, M.; Musolino, C. Antitumorigenic action of nelfinavir: Effects on multiple myeloma and hematologic malignancies (Review). Oncol. Rep. 2020, 43, 1729–1736. [Google Scholar] [CrossRef]
- Information on COVID-19 Treatment, Prevention and Research. COVID-19 Treat Guidel n.d. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 29 January 2023).
- Hung, I.F.-N.; Lung, K.-C.; Tso, E.Y.-K.; Liu, R.; Chung, T.W.-H.; Chu, M.-Y.; Ng, Y.-Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiong, N.; Li, C.; Gong, Y.; Liu, L.; Yang, H.; Tan, X.; Jiang, N.; Zong, Q.; Wang, J.; et al. Efficacy of ribavirin and interferon-α therapy for hospitalized patients with COVID-19: A multicenter, retrospective cohort study. Int. J. Infect. Dis. 2021, 104, 641–648. [Google Scholar] [CrossRef] [PubMed]
- McHutchison, J.G.; Dev, A.T. Future trends in managing hepatitis C. Gastroenterol. Clin. N. Am. 2004, 33, 51–61. [Google Scholar] [CrossRef]
- Lin, C.; Yeh, L.-T.; Vitarella, D.; Hong, Z. Viramidine, a prodrug of ribavirin, shows better liver-targeting properties and safety profiles than ribavirin in animals. Antivir. Chem. Chemother. 2003, 14, 145–152. [Google Scholar] [CrossRef]
- Zarrouk, K.; Piret, J.; Boivin, G. Herpesvirus DNA polymerases: Structures, functions and inhibitors. Virus Res. 2017, 234, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Yan, H.; Wang, J.; Liu, K.; Liu, B.; Shi, Y. Current scenario on non-nucleoside reverse transcriptase inhibitors (2018-present). Arab. J. Chem. 2022, 15, 104378. [Google Scholar] [CrossRef]
- Gill, M.S.A.; Hassan, S.S.; Ahemad, N. Evolution of HIV-1 reverse transcriptase and integrase dual inhibitors: Recent advances and developments. Eur. J. Med. Chem. 2019, 179, 423–448. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, R.; Hruby, V. Antiviral Drugs. In Synthesis of Best-Seller Drugs; Vardanyan, R., Hruby, V., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 687–736. [Google Scholar] [CrossRef]
- Kim, J.; De Jesus, O. Medication Routes of Administration. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Poole, C.L.; James, S.H. Antiviral Therapies for Herpesviruses: Current Agents and New Directions. Clin. Ther. 2018, 40, 1282–1298. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, T.; Song, J.; Pu, D.; He, D.; Li, J.; Yang, J.; Li, K.; Zhong, C.; Zhang, J. Antiviral Drug Delivery System for Enhanced Bioactivity, Better Metabolism and Pharmacokinetic Characteristics. Int. J. Nanomed. 2021, 16, 4959–4984. [Google Scholar] [CrossRef] [PubMed]
- Richelsen, R.K.B.; Jensen, S.B.; Nielsen, H. Incidence and predictors of intravenous acyclovir-induced nephrotoxicity. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1965–1971. [Google Scholar] [CrossRef]
- Gonda, I. Systemic delivery of drugs to humans via inhalation. J. Int. Soc. Aerosols Med. 2006, 19, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Quiñones-Mateu, M.E.; Das, S.C. Inhaled therapy for COVID-19: Considerations of drugs, formulations and devices. Int. J. Pharm. 2022, 624, 122042. [Google Scholar] [CrossRef]
- Razonable, R.R. Antiviral Drugs for Viruses Other Than Human Immunodeficiency Virus. Mayo Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-Y.; Chen, Y.; Huang, Y.-Y.; Cheng, C.-M. Transdermal drug delivery systems for fighting common viral infectious diseases. Drug Deliv. Transl. Res. 2021, 11, 1498–1508. [Google Scholar] [CrossRef]
- Hmingthansanga, V.; Singh, N.; Banerjee, S.; Manickam, S.; Velayutham, R.; Natesan, S. Improved Topical Drug Delivery: Role of Permeation Enhancers and Advanced Approaches. Pharmaceutics 2022, 14, 2818. [Google Scholar] [CrossRef]
- Kim, J.; Yeom, M.; Lee, T.; Kim, H.-O.; Na, W.; Kang, A.; Lim, J.-W.; Park, G.; Park, C.; Song, D.; et al. Porous gold nanoparticles for attenuating infectivity of influenza A virus. J. Nanobiotechnol. 2020, 18, 54. [Google Scholar] [CrossRef]
- Muñoz, A.; Sigwalt, D.; Illescas, B.M.; Luczkowiak, J.; Rodríguez-Pérez, L.; Nierengarten, I.; Holler, M.; Remy, J.-S.; Buffet, K.; Vincent, S.P.; et al. Synthesis of giant globular multivalent glycofullerenes as potent inhibitors in a model of Ebola virus infection. Nat. Chem. 2016, 8, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Lembo, D.; Swaminathan, S.; Donalisio, M.; Civra, A.; Pastero, L.; Aquilano, D.; Vavia, P.; Trotta, F.; Cavalli, R. Encapsulation of Acyclovir in new carboxylated cyclodextrin-based nanosponges improves the agent’s antiviral efficacy. Int. J. Pharm. 2013, 443, 262–272. [Google Scholar] [CrossRef]
- Cautela, M.P.; Moshe, H.; Sosnik, A.; Sarmento, B.; das Neves, J. Composite films for vaginal delivery of tenofovir disoproxil fumarate and emtricitabine. Eur. J. Pharm. Biopharm. 2019, 138, 3–10. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Taher, A.; Park, B.K.; Kwon, H.-J.; Al-Nazawi, M. A pilot study of the antiviral activity of anionic and cationic polyamidoamine dendrimers against the Middle East respiratory syndrome coronavirus. J. Med. Virol. 2020, 92, 1665–1670. [Google Scholar] [CrossRef]
- Horcajada, P.; Chalati, T.; Serre, C.; Gillet, B.; Sebrie, C.; Baati, T.; Eubank, J.F.; Heurtaux, D.; Clayette, P.; Kreuz, C.; et al. Porous metal–organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat. Mater. 2009, 9, 172–178. [Google Scholar] [CrossRef]
- Reolon, J.B.; Brustolin, M.; Accarini, T.; Viçozzi, G.P.; Sari, M.H.M.; Bender, E.A.; Haas, S.E.; Brum, M.C.S.; Gündel, A.; Colomé, L.M. Co-encapsulation of acyclovir and curcumin into microparticles improves the physicochemical characteristics and potentiates in vitro antiviral action: Influence of the polymeric composition. Eur. J. Pharm. Sci. 2019, 131, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Manyarara, T.E.; Khoza, S.; Dube, A.; Maponga, C.C. Formulation and characterization of a paediatric nanoemulsion dosage form with modified oral drug delivery system for improved dissolution rate of nevirapine. MRS Adv. 2018, 3, 2203. [Google Scholar] [CrossRef]
- Hosny, K.M.; Sindi, A.M.; Alkhalidi, H.M.; Kurakula, M.; Alruwaili, N.K.; Alhakamy, N.A.; Abualsunun, W.A.; Bakhaidar, R.B.; Bahmdan, R.H.; Rizg, W.Y.; et al. Oral gel loaded with penciclovir-lavender oil nanoemulsion to enhance bioavailability and alleviate pain associated with herpes labialis. Drug Deliv. 2021, 28, 1043–1054. [Google Scholar] [CrossRef]
- Jha, M. Modified release formulations to achieve the quality target product profile (QTPP). Int. J. Pharm. Sci. Res. 2012, 3, 2376. [Google Scholar]
- Karpe, M.; Mali, N.; Kadam, V. Formulation Development and Evaluation of Acyclovir Orally Disintegrating Tablets. J. Appl. Pharm. Sci. 2012, 2, 101–105. [Google Scholar]
- Gröning, R.; Berntgen, M.; Georgarakis, M. Acyclovir serum concentrations following peroral administration of magnetic depot tablets and the influence of extracorporal magnets to control gastrointestinal transit. Eur. J. Pharm. Biopharm. 1998, 46, 285–291. [Google Scholar] [CrossRef]
- Zielenkiewicz, W.; Koźbiał, M.; Golankiewicz, B.; Poznanski, J. Enhancement of aqueous solubility of tricyclic acyclovir derivatives by their complexation with hydroxypropyl-β-cyclodextrin. J. Therm. Anal. Calorim. 2010, 101, 555–560. [Google Scholar] [CrossRef]
- Bencini, M.; Ranucci, E.; Ferruti, P.; Trotta, F.; Donalisio, M.; Cornaglia, M.; Lembo, D.; Cavalli, R. Preparation and in vitro evaluation of the antiviral activity of the Acyclovir complex of a beta-cyclodextrin/poly(amidoamine) copolymer. J. Control. Release 2008, 126, 17–25. [Google Scholar] [CrossRef]
- Chaudhari, K.D.; Nimbalwar, M.G.; Singhal, N.S.; Panchale, W.A.; Manwar, J.V.; Bakal, R.L. Comprehensive review on characterizations and application of gastro-retentive floating drug delivery system. GSC Adv. Res. Rev. 2021, 7, 35–44. [Google Scholar] [CrossRef]
- Ahmed, F.J.; Drabu, S.; Khatri, S.; Babu, S. Formulation and evaluation of acyclovir capsules. Int. J. Drug Dev. Res. 2011, 3, 162–167. [Google Scholar]
- Ahmed, F.; Drabu, S.; Khatri, S.; Babu, S. Development and evaluation of floating matrix tablets of acyclovir. Res. J. Pharm. Biol. Chem. Sci. 2011, 2, 547–553. [Google Scholar]
- Vinodbhai, P.K.; Gohel, D.M.C.; Parikh, D.R.K.; Bariya, D.S.; Suthar, R.N. Sustained release floating microspheres of acyclovir: Formulation, optimization, characterization and in vitro evaluation. Int. J. Drug Dev. Res. 2011, 3, 242–251. [Google Scholar]
- Singhal, P.; Kumar, K.; Pandey, M.; Saraf, S.A. Evaluation of acyclovir loaded oil entrapped calcium alginate beads prepared by ionotropic gelation method. Int. J. Chem. Technol. Res. 2010, 2, 2076–2085. [Google Scholar]
- Kleiner, L.W.; Wright, J.C.; Wang, Y. Evolution of implantable and insertable drug delivery systems. J. Control. Release 2014, 181, 1–10. [Google Scholar] [CrossRef]
- Kumar, A.; Pillai, J. Nanostructures for the Engineering of Cells, Tissues and Organs. In Implantable Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2018; pp. 473–511. [Google Scholar]
- Jacob, J.; Haponiuk, J.T.; Thomas, S.; Gopi, S. Biopolymer based nanomaterials in drug delivery systems: A review. Mater. Today Chem. 2018, 9, 43–55. [Google Scholar] [CrossRef]
- Stewart, S.A.; Domínguez-Robles, J.; Donnelly, R.F.; Larrañeta, E. Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications. Polymers 2018, 12, 1379. [Google Scholar] [CrossRef]
- Dash, A.K.; Cudworth, G.C. Therapeutic applications of implantable drug delivery systems. J. Pharmacol. Toxicol. Methods 1998, 40, 1–12. [Google Scholar] [CrossRef]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef]
- Alam, A.; Gamal, S.; Naggar, V. Formulation and evaluation of acyclovir ophthalmic inserts. Asian J. Pharm. Sci. 2008, 3, 58–67. [Google Scholar]
- Kerur, S.; Dangari, P.; Deshpande, P. Controlled release polymeric ocular inserts for delivery of acyclovir. Turk. J. Pharm. Sci. 2010, 7, 75–90. [Google Scholar]
- Khan, S.; Ali, A.; Singhavi, D.; Yeole, P. Controlled ocular delivery of acyclovir through rate controlling ocular insert of Eudragit: A technical note. AAPS PharmSciTech 2008, 9, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.B.; Dandagi, P.; Udupa, N.; Gopal, S.V.; Jain, S.S.; Vasanth, S.G. Controlled release polymeric ocular delivery of acyclovir. Pharm. Dev. Technol. 2010, 15, 369–378. [Google Scholar] [CrossRef]
- Han, T.; Das, D.B. Potential of combined ultrasound and microneedles for enhanced transdermal drug permeation: A review. Eur. J. Pharm. Biopharm. 2015, 89, 312–328. [Google Scholar] [CrossRef]
- Schoellhammer, C.M.; Blankschtein, D.; Langer, R. Skin permeabilization for transdermal drug delivery: Recent advances and future prospects. Expert Opin. Drug Deliv. 2014, 11, 393–407. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Singh, T.R.R.; Morrow, D.I.J.; Woolfson, A.D. Transdermal Delivery Applications. In Microneedle-Mediated Transdermal and Intradermal Drug Delivery; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 1–19. [Google Scholar] [CrossRef]
- Kretsos, K.; Kasting, G.B. A Geometrical Model of Dermal Capillary Clearance. Math. Biosci. 2007, 208, 430–453. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Prausnitz, M.R.; Mitragotri, S. Micro-Scale Devices for Transdermal Drug Delivery. Int. J. Pharm. 2008, 364, 227–236. [Google Scholar] [CrossRef]
- Tuan-Mahmood, T.; McCrudden, M.T.; Torrisi, B.M.; McAlister, E.; Garland, M.J.; Singh, T.R.R.; Donnelly, R.F. Microneedles for Intradermal and Transdermal Drug Delivery. Eur. J. Pharm. Sci. 2013, 50, 623–637. [Google Scholar] [CrossRef]
- Prausnitz, M.R.; Langer, R. Transdermal Drug Delivery. Nat. Biotechnol. 2008, 26, 1261–1268. [Google Scholar] [CrossRef]
- Ita, K. Transdermal Drug Delivery: Progress and Challenges. J. Drug Deliv. Sci. Technol. 2014, 24, 245–250. [Google Scholar] [CrossRef]
- Lashmar, U.T.; Manger, J. Investigation into the potential for iontophoresis facilitated transdermal delivery of acyclovir. Int. J. Pharm. 1994, 111, 73–82. [Google Scholar] [CrossRef]
- Gonsho, A.; Imanidis, G.; Vogt, P.; Kern, E.R.; Tsuge, H.; Su, M.-H.; Choi, S.-H.; Higuchi, W.I. Controlled (trans) dermal delivery of an antiviral agent (acyclovir). I: An in vivo animal model for efficacy evaluation in cutaneous HSV-1 infections. Int. J. Pharm. 1990, 65, 183–194. [Google Scholar] [CrossRef]
- Saxena, A.; Tewari, G.; Saraf, S.A. Formulation and evaluation of mucoadhesive buccal patch of acyclovir utilizing inclusion phenomenon. Braz. J. Pharm. Sci. 2011, 47, 887–897. [Google Scholar] [CrossRef]
- Tallury, P.; Alimohammadi, N.; Kalachandra, S. Poly(ethylene-co-vinyl acetate) copolymer matrix for delivery of chlorhexidine and acyclovir drugs for use in the oral environment: Effect of drug combination, copolymer composition and coating on the drug release rate. Dent. Mater. 2007, 23, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.K.; Majithiya, R.J.; Umrethia, M.L.; Murthy, R.S.R. Design and development of microemulsion drug delivery system of acyclovir for improvement of oral bioavailability. AAPS PharmSciTech 2006, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Shishu; Rajan, S.; Kamalpreet. Development of novel microemulsion-based topical formulations of acyclovir for the treatment of cutaneous herpetic infections. AAPS PharmSciTech 2009, 10, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Tamura, M.; Tahara, Y.; Kamiya, N.; Goto, M. Ionic liquid-in-oil microemulsion as a potential carrier of sparingly soluble drug: Characterization and cytotoxicity evaluation. Int. J. Pharm. 2010, 400, 243–250. [Google Scholar] [CrossRef]
- Jain, S.; Jain, V.; Mahajan, S.C. Lipid Based Vesicular Drug Delivery Systems. Adv. Pharm. 2014, 2014, 574673. [Google Scholar] [CrossRef]
- Chen, L.; Liang, J. An overview of functional nanoparticles as novel emerging antiviral therapeutic agents. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 112, 110924. [Google Scholar] [CrossRef]
- Attia, I.A.; El-Gizawy, S.A.; Fouda, M.A.; Donia, A.M. Influence of a niosomal formulation on the oral bioavailability of acyclovir in rabbits. AAPS PharmSciTech 2007, 8, E106. [Google Scholar] [CrossRef]
- Alsarra, I.A.; Hamed, A.Y.; Alanazi, F.K. Acyclovir liposomes for intranasal systemic delivery: Development and pharmacokinetics evaluation. Drug Deliv. 2008, 15, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Chetoni, P.; Rossi, S.; Burgalassi, S.; Monti, D.; Mariotti, S.; Saettone, M.F. Comparison of Liposome-Encapsulated Acyclovir with Acyclovir Ointment: Ocular Pharmacokinetics in Rabbits. J. Ocul. Pharmacol. Ther. 2004, 20, 169–177. [Google Scholar] [CrossRef]
- Garg, M.; Jain, N.K. Reduced hematopoietic toxicity, enhanced cellular uptake and altered pharmacokinetics of azidothymidine loaded galactosylated liposomes. J. Drug Target. 2006, 14, 1–11. [Google Scholar] [CrossRef]
- Javan, F.; Vatanara, A.; Azadmanesh, K.; Nabi-Meibodi, M.; Shakouri, M. Encapsulation of ritonavir in solid lipid nanoparticles: In-vitro anti-HIV-1 activity using lentiviral particles. J. Pharm. Pharmacol. 2017, 69, 1002–1009. [Google Scholar] [CrossRef]
- Seyfoddin, A.; Al-Kassas, R. Development of solid lipid nanoparticles and nanostructured lipid carriers for improving ocular delivery of acyclovir. Drug Dev. Ind. Pharm. 2013, 39, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Mura, P. Advantages of the combined use of cyclodextrins and nanocarriers in drug delivery: A review. Int. J. Pharm. 2020, 579, 119181. [Google Scholar] [CrossRef]
- Perret, F.; Duffour, M.; Chevalier, Y.; Parrot-Lopez, H. Design, synthesis, and in vitro evaluation of new amphiphilic cyclodextrin-based nanoparticles for the incorporation and controlled release of acyclovir. Eur. J. Pharm. Biopharm. 2013, 83, 25–32. [Google Scholar] [CrossRef]
- Nanjwade, B.K.; Bechra, H.M.; Derkar, G.K.; Manvi, F.V.; Nanjwade, V.K. Dendrimers: Emerging polymers for drug-delivery systems. Eur. J. Pharm. Sci. 2009, 38, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Bono, N.; Pennetta, C.; Bellucci, M.C.; Sganappa, A.; Malloggi, C.; Tedeschi, G.; Candiani, G.; Volonterio, A. Role of Generation on Successful DNA Delivery of PAMAM–(Guanidino)Neomycin Conjugates. ACS Omega 2019, 4, 6796–6807. [Google Scholar] [CrossRef]
- Kawano, Y.; Jordan, O.; Hanawa, T.; Borchard, G.; Patrulea, V. Are Antimicrobial Peptide Dendrimers an Escape from ESKAPE? Adv. Wound Care 2020, 9, 378–395. [Google Scholar] [CrossRef]
- Heitz, M.; Kwok, A.; Eggimann, G.A.; Hollfelder, F.; Darbre, T.; Reymond, J.L. Peptide Dendrimer–Lipid Conjugates as DNA and siRNA Transfection Reagents: Role of Charge Distribution Across Generations. Chimia 2017, 71, 220–225. [Google Scholar] [CrossRef]
- Esfand, R.; Tomalia, D.A. Poly(amidoamine) (PAMAM) dendrimers: From biomimicry to drug delivery and biomedical applications. Drug Discov. Today 2001, 6, 427–436. [Google Scholar] [CrossRef]
- Bourne, N.; Stanberry, L.R.; Kern, E.R.; Holan, G.; Matthews, B.; Bernstein, D.I. Dendrimers, a new class of candidate topical microbicides with activity against herpes simplex virus infection. Antimicrob. Agents Chemother. 2000, 44, 2471–2474. [Google Scholar] [CrossRef]
- Oka, H.; Onaga, T.; Koyama, T.; Guo, C.-T.; Suzuki, Y.; Esumi, Y.; Hatano, K.; Terunuma, D.; Matsuoka, K. Sialyl alpha(2-->3) lactose clusters using carbosilane dendrimer core scaffolds as influenza hemagglutinin blockers. Bioorg. Med. Chem. Lett. 2008, 18, 4405–4408. [Google Scholar] [CrossRef] [PubMed]
- Witvrouw, M.; Fikkert, V.; Pluymers, W.; Matthews, B.; Mardel, K.; Schols, D.; Raff, J.; Debyser, Z.; De Clercq, E.; Holan, G.; et al. Polyanionic (i.e., polysulfonate) dendrimers can inhibit the replication of human immunodeficiency virus by interfering with both virus adsorption and later steps (reverse transcriptase/integrase) in the virus replicative cycle. Mol. Pharmacol. 2000, 58, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.L.; Sidwell, R.W.; Gage, T.L.; Okleberry, K.M.; Matthews, B.; Holan, G. Anti-Respiratory Syncytial Virus Activity of Dendrimer Polyanions. Antivir. Res. 1997, 2, A88. [Google Scholar] [CrossRef]
- Rodríguez-Izquierdo, I.; Natalia, C.; García, F.; Muñoz-Fernandez, M.D.L. G2-S16 sulfonate dendrimer as new therapy for treatment failure in HIV-1 entry inhibitors. Nanomedicine 2019, 14, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yoshida, D.; Kanamoto, T.; Nakashima, H.; Uryu, T.; Yoshida, T. Sulfated oligosaccharide cluster with polylysine core scaffold as a new anti-HIV dendrimer. Carbohydr. Polym. 2010, 80, 1111–1115. [Google Scholar] [CrossRef]
- Borges, A.R.; Wieczorek, L.; Johnson, B.; Benesi, A.J.; Brown, B.K.; Kensinger, R.D.; Krebs, F.C.; Wigdahl, B.; Blumenthal, R.; Puri, A.; et al. Multivalent dendrimeric compounds containing carbohydrates expressed on immune cells inhibit infection by primary isolates of HIV-1. Virology 2010, 408, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Doménech, R.; Abian, O.; Bocanegra, R.; Correa, J.; Sousa-Herves, A.; Riguera, R.; Mateu, M.G.; Fernandez-Megia, E.; Velázquez-Campoy, A.; Neira, J.L. Dendrimers as Potential Inhibitors of the Dimerization of the Capsid Protein of HIV-1. Biomacromolecules 2010, 11, 2069–2078. [Google Scholar] [CrossRef]
- Stella, B.; Arpicco, S.; Rocco, F.; Burgalassi, S.; Nicosia, N.; Tampucci, S.; Chetoni, P.; Cattel, L. Nonpolymeric nanoassemblies for ocular administration of acyclovir: Pharmacokinetic evaluation in rabbits. Eur. J. Pharm. Biopharm. 2012, 80, 39–45. [Google Scholar] [CrossRef]
- Lin, Z.; Li, Y.; Guo, M.; Xu, T.; Wang, C.; Zhao, M.; Wang, H.; Chen, T.; Zhu, B. The inhibition of H1N1 influenza virus-induced apoptosis by silver nanoparticles functionalized with zanamivir. RSC Adv. 2017, 7, 742–750. [Google Scholar] [CrossRef]
- Ferdous, Z.; Nemmar, A. Health Impact of Silver Nanoparticles: A Review of the Biodistribution and Toxicity Following Various Routes of Exposure. Int. J. Mol. Sci. 2020, 21, 2375. [Google Scholar] [CrossRef]
- Ramanathan, R.; Jiang, Y.; Read, B.; Golan-Paz, S.; Woodrow, K. Biophysical characterization of small molecule antiviral-loaded nanolipogels for HIV-1 chemoprophylaxis and topical mucosal application. Acta Biomater. 2016, 36, 122–131. [Google Scholar] [CrossRef]
- Jansen, C.; Tran-Cong, N.M.; Schlüsener, C.; Schmitz, A.; Proksch, P.; Janiak, C. MOF@chitosan Composites with Potential Antifouling Properties for Open-Environment Applications of Metal-Organic Frameworks. Solids 2022, 3, 35–54. [Google Scholar] [CrossRef]
- Batten, S.R.; Champness, N.R.; Chen, X.-M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Paik Suh, M.; Reedijk, J. Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef]
- Zhou, H.-C.J.; Kitagawa, S. Metal-organic frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. [Google Scholar] [CrossRef] [PubMed]
- Farha, O.K.; Hupp, J.T. Rational design, synthesis, purification, and activation of metal-organic framework materials. Acc. Chem. Res. 2010, 43, 1166–1175. [Google Scholar] [CrossRef]
- Taddei, M. When defects turn into virtues: The curious case of zirconium-based metal-organic frameworks. Coord. Chem. Rev. 2017, 343, 1–24. [Google Scholar] [CrossRef]
- Keskin, S.; Kızılel, S. Biomedical Applications of Metal Organic Frameworks. Ind. Eng. Chem. Res. 2011, 50, 1799–1812. [Google Scholar] [CrossRef]
- Agostoni, V.; Chalati, T.; Horcajada, P.; Willaime, H.; Anand, R.; Semiramoth, N.; Baati, T.; Hall, S.; Maurin, G.; Chacun, H.; et al. Towards an improved anti-HIV activity of NRTI via metal-organic frameworks nanoparticles. Adv. Health Mater. 2013, 2, 1630–1637. [Google Scholar] [CrossRef]
- Akbari, M.; Ghasemzadeh, M.A.; Fadaeian, M. Synthesis and Application of ZIF-8 MOF Incorporated in a TiO2@Chitosan Nanocomposite as a Strong Nanocarrier for the Drug Delivery of Acyclovir. Chem. Sel. 2020, 5, 14564–14571. [Google Scholar] [CrossRef]
- Franklyne, J.S.; Gopinath, P.M.; Mukherjee, A.; Chandrasekaran, N. Nanoemulsions: The rising star of antiviral therapeutics and nanodelivery system-current status and prospects. Curr. Opin. Colloid. Interface Sci. 2021, 54, 101458. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Elahimehr, Z.; Mahboobian, M.M. Acyclovir-Loaded Nanoemulsions: Preparation, Characterization and Irritancy Studies for Ophthalmic Delivery. Curr. Eye Res. 2021, 46, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Nemade, S.M.; Kakad, S.P.; Kshirsagar, S.J.; Padole, T.R. Development of nanoemulsion of antiviral drug for brain targeting in the treatment of neuro-AIDS. Beni-Suef Univ. J. Basic Appl. Sci. 2022, 11, 1–10. [Google Scholar] [CrossRef]
- Cherniakov, I.; Domb, A.J.; Hoffman, A. Self-nano-emulsifying drug delivery systems: An update of the biopharmaceutical aspects. Expert Opin. Drug Deliv. 2015, 12, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Charman, W.N.; Rogge, M.C.; Boddy, A.W.; Berger, B.M. Effect of food and a monoglyceride emulsion formulation on danazol bioavailability. J. Clin. Pharmacol. 1993, 33, 381–386. [Google Scholar] [CrossRef]
- Shahba, A.A.-W.; Mohsin, K.; Alanazi, F.K. Novel self-nanoemulsifying drug delivery systems (SNEDDS) for oral delivery of cinnarizine: Design, optimization, and in-vitro assessment. AAPS PharmSciTech 2012, 13, 967–977. [Google Scholar] [CrossRef]
- Khan, A.A.; Akhtar, S.; Yadav, Y.; Akhtar, A.; Alelwani, W.; Bannunah, A.M.; Mahmood, S. Lopinavir-Loaded Self-Nanoemulsifying Drug Delivery System for Enhanced Solubility: Development, Characterisation and Caco-2 Cell Uptake. Curr. Drug Deliv. 2022. [Google Scholar] [CrossRef]
- Aranaz, I.; Alcántara, A.R.; Civera, M.C.; Arias, C.; Elorza, B.; Caballero, A.H.; Acosta, N. Chitosan: An Overview of Its Properties and Applications. Polymers 2021, 13, 3256. [Google Scholar] [CrossRef]
- Parhi, R. Drug delivery applications of chitin and chitosan: A review. Environ. Chem. Lett. 2020, 18, 577–594. [Google Scholar] [CrossRef]
- Mahmoud, M.G.; Em El Kady, E.; Asker, M.S. Chitin, Chitosan and Glucan, Properties and Applications. World J. Agri. Soil Sci. 2019, 3, 1–19. [Google Scholar] [CrossRef]
- Shariatinia, Z. Pharmaceutical applications of chitosan. Adv. Colloid. Interface Sci. 2019, 263, 131–194. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Gómez, C.P.; Cecilia, J.A. Chitosan: A Natural Biopolymer with a Wide and Varied Range of Applications. Molecules 2020, 25, 3981. [Google Scholar] [CrossRef]
- Karava, A.; Lazaridou, M.; Nanaki, S.; Michailidou, G.; Christodoulou, E.; Kostoglou, M.; Iatrou, H.; Bikiaris, D.N. Chitosan Derivatives with Mucoadhesive and Antimicrobial Properties for Simultaneous Nanoencapsulation and Extended Ocular Release Formulations of Dexamethasone and Chloramphenicol Drugs. Pharmaceutics 2020, 12, 594. [Google Scholar] [CrossRef]
- Yan, D.; Li, Y.; Liu, Y.; Li, N.; Zhang, X.; Yan, C. Antimicrobial Properties of Chitosan and Chitosan Derivatives in the Treatment of Enteric Infections. Molecules 2021, 26, 7136. [Google Scholar] [CrossRef] [PubMed]
- Perinelli, D.R.; Fagioli, L.; Campana, R.; Lam, J.K.W.; Baffone, W.; Palmieri, G.F.; Casettari, L.; Bonacucina, G. Chitosan-based nanosystems and their exploited antimicrobial activity. Eur. J. Pharm. Sci. 2018, 117, 8–20. [Google Scholar] [CrossRef]
- Alqahtani, F.; Aleanizy, F.; El Tahir, E.; Alhabib, H.; Alsaif, R.; Shazly, G.; AlQahtani, H.; Alsarra, I.; Mahdavi, J. Antibacterial Activity of Chitosan Nanoparticles Against Pathogenic N. gonorrhoea. Int. J. Nanomed. 2020, 15, 7877–7887. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, S.; Khan, J.A.; Rabbani, F.; Latif, U.; Arfan, M.; Yameen, M.A. Biocompatible biodegradable polymeric antibacterial nanoparticles for enhancing the effects of a third-generation cephalosporin against resistant bacteria. J. Med. Microbiol. 2017, 66, 318–327. [Google Scholar] [CrossRef]
- Safdar, R.; Omar, A.A.; Arunagiri, A.; Regupathi, I.; Thanabalan, M. Potential of Chitosan and its derivatives for controlled drug release applications—A review. J. Drug Deliv. Sci. Technol. 2019, 49, 642–659. [Google Scholar] [CrossRef]
- Hamed, I.; Özogul, F.; Regenstein, J.M. Industrial applications of crustacean by-products (chitin, chitosan, and chitooligosaccharides): A review. Trends Food Sci. Technol. 2016, 48, 40–50. [Google Scholar] [CrossRef]
- Peniche, C.; Argüelles-Monal, W.; Goycoolea, F.M. Chitin and Chitosan: Major Sources, Properties and Applications. In Monomers, Polymers and Composites from Renewable Resources; Belgacem, M.N., Gandini, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 517–542. [Google Scholar] [CrossRef]
- Nwe, N.; Furuike, T.; Tamura, H. Isolation and characterization of chitin and chitosan from marine origin. Adv. Food Nutr. Res. 2014, 72, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jaber, N.; Al-Remawi, M.; Al-Akayleh, F.; Al-Muhtaseb, N.; Al-Adham, I.S.I.; Collier, P.J. A review of the antiviral activity of Chitosan, including patented applications and its potential use against COVID-19. J. Appl. Microbiol. 2022, 132, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Morin-Crini, N.; Lichtfouse, E.; Torri, G.; Crini, G. Fundamentals and Applications of Chitosan. In Sustainable Agriculture Reviews; Crini, G., Lichtfouse, E., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; Volume 35, pp. 49–123. [Google Scholar] [CrossRef]
- Sashiwa, H.; Aiba, S. Chemically modified chitin and chitosan as biomaterials. Progr. Polym. Sci. 2004, 29, 887–908. [Google Scholar] [CrossRef]
- Mourya, V.K.; Inamdar, N.N. Trimethyl chitosan and its applications in drug delivery. J. Mater. Sci. Mater. Med. 2009, 20, 1057–1079. [Google Scholar] [CrossRef]
- Tan, R.S.L.; Hassandarvish, P.; Chee, C.F.; Chan, L.W.; Wong, T.W. Chitosan and its derivatives as polymeric anti-viral therapeutics and potential anti-SARS-CoV-2 nanomedicine. Carbohydr. Polym. 2022, 290, 119500. [Google Scholar] [CrossRef]
- Pathak, K.; Misra, S.; Sehgal, A.; Singh, S.; Bungau, S.; Najda, A.; Gruszecki, R.; Behl, T. Biomedical Applications of Quaternized Chitosan. Polymers 2021, 13, 2514. [Google Scholar] [CrossRef]
- Chung, Y.-C.; Yeh, J.-Y.; Tsai, C.-F. Antibacterial Characteristics and Activity of Water-Soluble Chitosan Derivatives Prepared by the Maillard Reaction. Molecules 2011, 16, 8504–8514. [Google Scholar] [CrossRef]
- Park, I.K.; Yang, J.; Jeong, H.J.; Bom, H.S.; Harada, I.; Akaike, T.; Kim, S.; Cho, C. Galactosylated chitosan as a synthetic extracellular matrix for hepatocytes attachment. Biomaterials 2003, 24, 2331–2337. [Google Scholar] [CrossRef]
- Monteiro, O.A.; Airoldi, C. Some studies of crosslinking chitosan-glutaraldehyde interaction in a homogeneous system. Int. J. Biol. Macromol. 1999, 26, 119–128. [Google Scholar] [CrossRef]
- Ding, W.-Y.; Zheng, S.-D.; Qin, Y.; Yu, F.; Bai, J.-W.; Cui, W.-Q.; Yu, T.; Chen, X.-R.; Bello-Onaghise, G.; Li, Y.-H. Chitosan Grafted With β-Cyclodextrin: Synthesis, Characterization, Antimicrobial Activity, and Role as Absorbefacient and Solubilizer. Front. Chem. 2019, 6, 657. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Biswas, S.; Ghosh, T. Preparation and Evaluation of Silymarin β-cyclodextrin Molecular Inclusion Complexes. J. Young Pharm. 2011, 3, 205–210. [Google Scholar] [CrossRef]
- El-Tahlawy, K.; Gaffar, M.A.; El-Rafie, S. Novel method for preparation of β-cyclodextrin/grafted chitosan and it’s application. Carbohydr. Polym. 2006, 3, 385–392. [Google Scholar] [CrossRef]
- Sun, T.; Zhou, D.; Xie, J.; Mao, F. Preparation of chitosan oligomers and their antioxidant activity. Eur. Food Res. Technol. 2007, 225, 451–456. [Google Scholar] [CrossRef]
- Hastarini, E.; Ayudiarti, D.L. Addition of Chitosan Oligomers to Improve Bread Texture. IOP Conf. Ser. Earth Environ. Sci. 2019, 278, 012034. [Google Scholar] [CrossRef]
- Anraku, M.; Gebicki, J.M.; Iohara, D.; Tomida, H.; Uekama, K.; Maruyama, T.; Hirayama, F.; Otagiri, M. Antioxidant activities of chitosans and its derivatives in in vitro and in vivo studies. Carbohydr. Polym. 2018, 199, 141–149. [Google Scholar] [CrossRef]
- Poecheim, J.; Patrulea, V.; Reichert, C.; Borchard, G. Characterization of pDNA-TMC Nanoparticle Interaction and Stability. Curr. Drug Deliv. 2016, 13, 301–308. [Google Scholar] [CrossRef]
- Muzzarelli, R.; Jeuniaux, C.; Gooday, G.W. (Eds.) International Conference on Chitin and Chitosan; Plenum Press: Senigallia, Italy, 1986; pp. 303–306. [Google Scholar] [CrossRef]
- Xu, T.; Xin, M.; Li, M.; Huang, H.; Zhou, S.; Liu, J. Synthesis, characterization, and antibacterial activity of N,O-quaternary ammonium chitosan. Carbohydr. Res. 2011, 346, 2445–2450. [Google Scholar] [CrossRef]
- Seong, H.S.; Whang, H.S.; Ko, S.W. Synthesis of a Quaternary Ammonium Derivative of Chito-oligosaccharide as Antimicrobial Agent for Cellulosic Fibers. J. Appl. Polym. Sci. 2000, 76, 2009–2015. [Google Scholar] [CrossRef]
- Mohamed, R.R.; Abu Elella, M.H.; Sabaa, M.W. Synthesis, characterization and applications of N-quaternized chitosan/poly(vinyl alcohol) hydrogels. Int. J. Biol. Macromol. 2015, 80, 149–161. [Google Scholar] [CrossRef]
- Doncel-Pérez, E.; Aranaz, I.; Bastida, A.; Revuelta, J.; Camacho, C.; Acosta, N.; Garrido, L.; Civera, C.; García-Junceda, E.; Heras, A.; et al. Synthesis, physicochemical characterization and biological evaluation of chitosan sulfate as heparan sulfate mimics. Carbohydr. Polym. 2018, 191, 225–233. [Google Scholar] [CrossRef]
- Revuelta, J.; Fraile, I.; Monterrey, D.T.; Peña, N.; Benito-Arenas, R.; Bastida, A.; Fernández-Mayoralas, A.; García-Junceda, E. Heparanized chitosans: Towards the third generation of chitinous biomaterials. Mater. Horiz. 2021, 8, 2596–2614. [Google Scholar] [CrossRef]
- Sila, A.; Bougatef, H.; Capitani, F.; Krichen, F.; Mantovani, V.; Amor, I.B.; Galeotti, F.; Maccari, F.; Nedjar, N.; Volpi, N.; et al. Studies on European eel skin sulfated glycosaminoglycans: Recovery, structural characterization and anticoagulant activity. Int. J. Biol. Macromol. 2018, 115, 891–899. [Google Scholar] [CrossRef]
- Valcarcel, J.; Novoa-Carballal, R.; Pérez-Martín, R.I.; Reis, R.L.; Vázquez, J.A. Glycosaminoglycans from marine sources as therapeutic agents. Biotechnol. Adv. 2017, 35, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Krichen, F.; Ghlissi, Z.; Amor, I.B.; Sayari, N.; Kallel, R.; Gargouri, J.; Sahnoun, Z.; Boudawara, T.; Bougatef, A. In vitro and in vivo anti-coagulant activity and toxicological studies of marine sulfated glycosaminoglycans. Exp. Toxicol. Pathol. 2017, 69, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Campelo, C.S.; Chevallier, P.; Vaz, J.M.; Vieira, R.S.; Mantovani, D. Sulfonated chitosan and dopamine based coatings for metallic implants in contact with blood. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 72, 682–691. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, J.; Chen, R.; Cao, L.; Tang, W.; Lin, D.; Wang, J.; Liu, C. Enhancement of VEGF-Mediated Angiogenesis by 2-N,6-O-Sulfated Chitosan-Coated Hierarchical PLGA Scaffolds. ACS Appl. Mater. Interfaces 2015, 7, 9982–9990. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Wang, Y.; Wang, H.; Yuan, L.; Tan, M.; Shi, X.; Lyu, Z.; Liu, Y.; Chen, H. 6-O-Sulfated Chitosan Promoting the Neural Differentiation of Mouse Embryonic Stem Cells. ACS Appl. Mater. Interfaces 2014, 6, 20043–20050. [Google Scholar] [CrossRef]
- Nishimura, S.-I.; Kai, H.; Shinada, K.; Yoshida, T.; Tokura, S.; Kurita, K.; Nakashima, H.; Yamamoto, N.; Uryu, T. Regioselective syntheses of sulfated polysaccharides: Specific anti-HIV-1 activity of novel chitin sulfates. Carbohydr. Res. 1998, 306, 427–433. [Google Scholar] [CrossRef]
- Vikhoreva, G.; Bannikova, G.; Stolbushkina, P.; Panov, A.; Drozd, N.; Makarov, V.; Varlamov, V.; Gal’Braikh, L. Preparation and anticoagulant activity of a low-molecular-weight sulfated chitosan. Carbohydr. Polym. 2005, 62, 327–332. [Google Scholar] [CrossRef]
- Huang, R.; Du, Y.; Zheng, L.; Liu, H.; Fan, L. A new approach to chemically modified chitosan sulfates and study of their influences on the inhibition of Escherichia coli and Staphylococcus aureus growth. React. Funct. Polym. 2004, 59, 41–51. [Google Scholar] [CrossRef]
- Zhang, K.; Helm, J.; Peschel, D.; Groth, T.; Fischer, S. NMR and FT Raman characterisation of regioselectively sulfated chitosan regarding the distribution of sulfate groups and the degree of substitution. Polymer 2010, 51, 4698–4705. [Google Scholar] [CrossRef]
- Zhou, H.; Qian, J.; Wang, J.; Yao, W.; Liu, C.; Chen, J.; Cao, X. Enhanced bioactivity of bone morphogenetic protein-2 with low dose of 2-N, 6-O-sulfated chitosan in vitro and in vivo. Biomaterials 2009, 30, 1715–1724. [Google Scholar] [CrossRef]
- Wang, R.; Huang, J.; Wei, M.; Zeng, X. The synergy of 6-O-sulfation and N- or 3-O-sulfation of chitosan is required for efficient inhibition of P-selectin-mediated human melanoma A375 cell adhesion. Biosci. Biotechnol. Biochem. 2010, 74, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Karadeniz, F.; Karagozlu, M.Z.; Pyun, S.-Y.; Kim, S.-K. Sulfation of chitosan oligomers enhances their anti-adipogenic effect in 3T3-L1 adipocytes. Carbohydr. Polym. 2011, 86, 666–671. [Google Scholar] [CrossRef]
- Bianculli, R.H.; Mase, J.D.; Schulz, M.D. Antiviral polymers: Past approaches and future possibilities. Macromolecules 2020, 53, 9158–9186. [Google Scholar] [CrossRef]
- Witvrouw, M.; De Clercq, E. Sulfated polysaccharides extracted from sea algae as potential antiviral drugs. Gen. Pharmacol. 1997, 29, 497–511. [Google Scholar] [CrossRef]
- Ito, M.; Baba, M.; Sato, A.; Pauwels, R.; De Clercq, E.; Shigeta, S. Inhibitory effect of dextran sulfate and heparin on the replication of human immunodeficiency virus (HIV) in vitro. Antivir. Res. 1987, 7, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Dimassi, S.; Tabary, N.; Chai, F.; Blanchemain, N.; Martel, B. Sulfonated and sulfated chitosan derivatives for biomedical applications: A review. Carbohydr. Polym. 2018, 202, 382–396. [Google Scholar] [CrossRef]
- Sacco, P.; Cok, M.; Scognamiglio, F.; Pizzolitto, C.; Vecchies, F.; Marfoglia, A.; Marsich, E.; Donati, I. Glycosylated-Chitosan Derivatives: A Systematic Review. Molecules 2020, 25, 1534. [Google Scholar] [CrossRef]
- Yalpani, M.; Hall, L.D. Some chemical and analytical aspects of polysaccharide modifications. III. Formation of branched-chain, soluble chitosan derivatives. Macromolecules 1984, 17, 272–281. [Google Scholar] [CrossRef]
- Donati, I.; Stredanska, S.; Silvestrini, G.; Vetere, A.; Marcon, P.; Marsich, E.; Mozetic, P.; Gamini, A.; Paoletti, S.; Vittur, F. The aggregation of pig articular chondrocyte and synthesis of extracellular matrix by a lactose-modified chitosan. Biomaterials 2005, 26, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Ilina, A.V.; Varlamov, V.P. Galactosylated derivatives of low-molecular-weight chitosan: Obtaining and properties. Appl. Biochem. Microbiol. 2007, 43, 73–77. [Google Scholar] [CrossRef]
- Chung, T.W.; Yang, J.; Akaike, T.; Cho, K.Y.; Nah, J.W.; Kim, S.I.; Cho, C.S. Preparation of alginate/galactosylated chitosan scaffold for hepatocyte attachment. Biomaterials 2002, 23, 2827–2834. [Google Scholar] [CrossRef]
- Ruiz Matute, A.I.; Cardelle-Cobas, A.; García-Bermejo, A.B.; Montilla, A.; Olano, A.; Corzo, N. Synthesis, characterization and functional properties of galactosylated derivatives of chitosan through amide formation. Food Hydrocoll. 2013, 33, 245–255. [Google Scholar] [CrossRef]
- Hodge, J.E. Dehydrated Foods, Chemistry of Browning Reactions in Model Systems. J. Agric. Food Chem. 1953, 1, 928–943. [Google Scholar] [CrossRef]
- Gullón, B.; Montenegro, M.I.; Ruiz-Matute, A.I.; Cardelle-Cobas, A.; Corzo, N.; Pintado, M.E. Synthesis, optimization and structural characterization of a chitosan–glucose derivative obtained by the Maillard reaction. Carbohydr. Polym. 2016, 137, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, P.; Gao, G.F.; Cheng, S. Carbohydrate-functionalized chitosan fiber for influenza virus capture. Biomacromolecules 2011, 12, 3962–3969. [Google Scholar] [CrossRef] [PubMed]
- Grenha, A. Chitosan nanoparticles: A survey of preparation methods. J. Drug Target. 2012, 20, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Baldino, L.; Concilio, S.; Cardea, S.; De Marco, I.; Reverchon, E. Complete glutaraldehyde elimination during chitosan hydrogel drying by SC-CO2 processing. J. Supercrit. Fluids 2015, 103, 70–76. [Google Scholar] [CrossRef]
- Berger, J.; Reist, M.; Mayer, J.M.; Felt, O.; Peppas, N.A.; Gurny, R. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur. J. Pharm. Biopharm. 2004, 57, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gao, X.; Wang, Y.; Zhang, X.; Tong, Z. Comparison of chitosan/starch composite film properties before and after cross-linking. Int. J. Biol. Macromol. 2013, 52, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. New way to crosslink chitosan in aqueous solution. Eur. Polym. J. 2010, 46, 1537–1544. [Google Scholar] [CrossRef]
- Jain, H.; Jain, V.; Jain, S.K.; Khangar, P.K. Development and evaluation of acyclovir loaded chitosan microspheres and cross linked with glutaraldehyde. J. Drug Deliv. Ther. 2021, 11, 110–114. [Google Scholar] [CrossRef]
- Machín, R.; Isasi, J.R.; Vélaz, I. β-Cyclodextrin hydrogels as potential drug delivery systems. Carbohydr. Polym. 2012, 87, 2024–2030. [Google Scholar] [CrossRef]
- Jiang, X.; Qi, Y.; Wang, S.; Tian, X. New amphoteric flocculant containing beta-cyclodextrin, synthesis, characterization and decolorization properties. J. Hazard. Mater. 2010, 173, 298–304. [Google Scholar] [CrossRef]
- Pinho, E.; Grootveld, M.; Soares, G.; Henriques, M. Cyclodextrin-based hydrogels toward improved wound dressings. Crit. Rev. Biotechnol. 2014, 34, 328–337. [Google Scholar] [CrossRef]
- Yuan, Z.; Ye, Y.; Gao, F.; Yuan, H.; Lan, M.; Lou, K.; Wang, W. Chitosan-graft-β-cyclodextrin nanoparticles as a carrier for controlled drug release. Int. J. Pharm. 2013, 446, 191–198. [Google Scholar] [CrossRef]
- Malik, N.S.; Ahmad, M.; Alqahtani, M.S.; Mahmood, A.; Barkat, K.; Khan, M.T.; Tulain, U.R.; Rashid, A. β-cyclodextrin chitosan-based hydrogels with tunable pH-responsive properties for controlled release of acyclovir: Design, characterization, safety, and pharmacokinetic evaluation. Drug Deliv. 2021, 28, 1093–1108. [Google Scholar] [CrossRef]
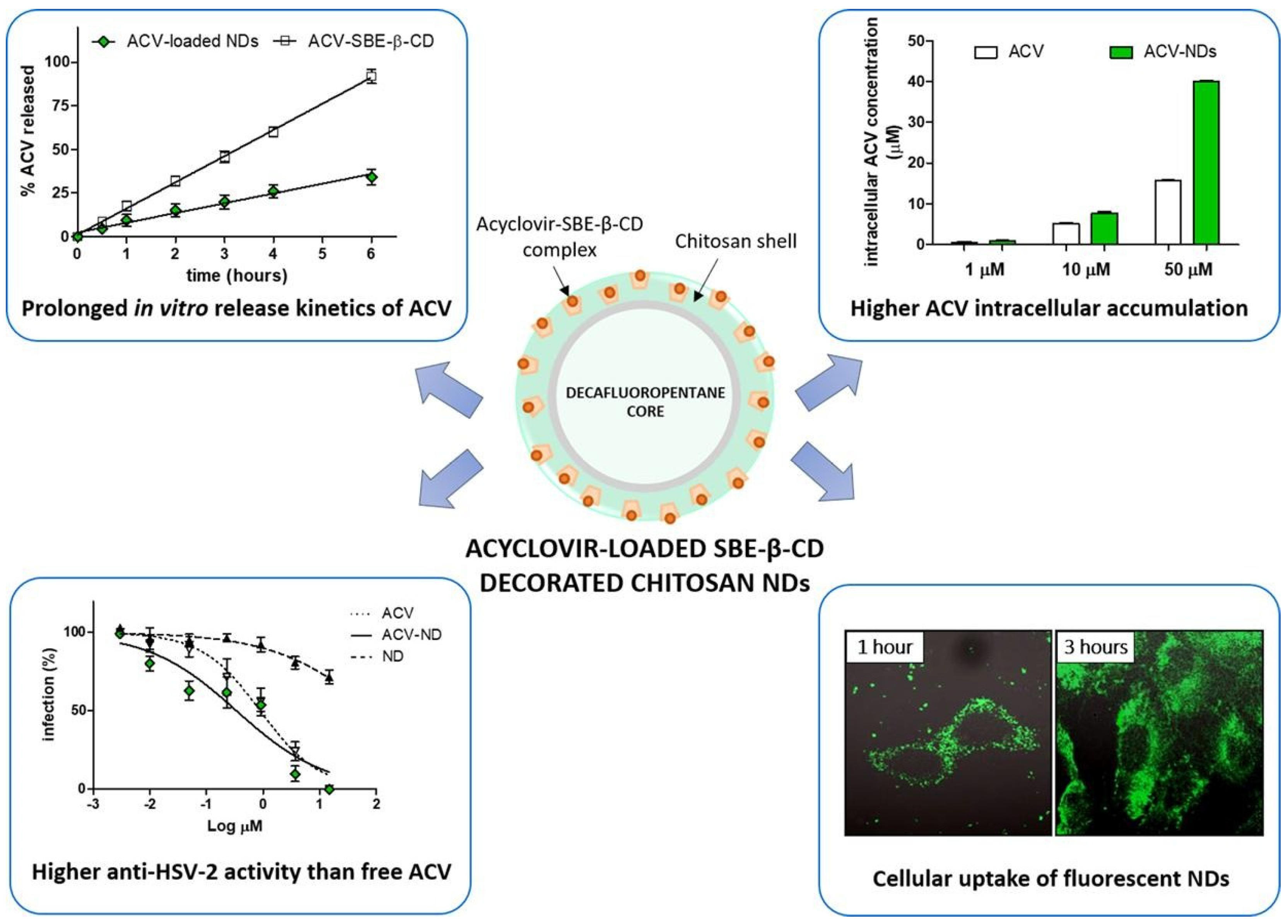
- Donalisio, M.; Argenziano, M.; Rittà, M.; Bastiancich, C.; Civra, A.; Lembo, D.; Cavalli, R. Acyclovir-loaded sulfobutyl ether-β-cyclodextrin decorated chitosan nanodroplets for the local treatment of HSV-2 infections. Int. J. Pharm. 2020, 587, 119676. [Google Scholar] [CrossRef]
- Muanprasat, C.; Chatsudthipong, V. Chitosan oligosaccharide: Biological activities and potential therapeutic applications. Pharmacol. Ther. Oxf. 2017, 170, 80–97. [Google Scholar] [CrossRef]
- Bonin, M.; Sreekumar, S.; Cord-Landwehr, S.; Moerschbacher, B.M. Preparation of Defined Chitosan Oligosaccharides Using Chitin Deacetylases. Int. J. Mol. Sci. 2020, 21, 7835. [Google Scholar] [CrossRef]
- Benchamas, G.; Huang, G.; Huang, S.; Huang, H. Preparation and biological activities of chitosan oligosaccharides. Trends Food Sci. Technol. 2021, 107, 38–44. [Google Scholar] [CrossRef]
- Liang, Z.; Gong, T.; Sun, X.; Tang, J.Z.; Zhang, Z. Chitosan oligomers as drug carriers for renal delivery of zidovudine. Carbohydr. Polym. 2012, 87, 2284–2290. [Google Scholar] [CrossRef]
- Klausner, E.A.; Zhang, Z.; Chapman, R.L.; Multack, R.F.; Volin, M.V. Ultrapure chitosan oligomers as carriers for corneal gene transfer. Biomaterials 2010, 31, 1814–1820. [Google Scholar] [CrossRef]
- Charelli, L.E.; de Mattos, G.C.; de Jesus Sousa-Batista, A.; Pinto, J.C.; Balbino, T.A. Polymeric nanoparticles as therapeutic agents against coronavirus disease. J. Nanoparticle Res. Interdiscip Forum Nanoscale Sci. Technol. 2022, 24, 12. [Google Scholar] [CrossRef] [PubMed]
- Jana, B.; Chatterjee, A.; Roy, D.; Ghorai, S.; Pan, D.; Pramanik, S.K.; Chakraborty, N.; Ganguly, J. Chitosan/benzyloxy-benzaldehyde modified ZnO nano template having optimized and distinct antiviral potency to human cytomegalovirus. Carbohydr. Polym. 2022, 278, 118965. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ensinas, A.; Verrier, B.; Primard, C.; Cuvillier, A.; Champier, G.; Paul, S.; Delair, T. Zinc-Stabilized Chitosan-Chondroitin Sulfate Nanocomplexes for HIV-1 Infection Inhibition Application. Mol. Pharm. 2016, 13, 3279–3291. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Ono, T.; Miyahira, Y.; Nguyen, V.Q.; Matsui, T.; Ishihara, M. Antiviral activity of silver nanoparticle/chitosan composites against H1N1 influenza A virus. Nanoscale Res. Lett. 2013, 8, 1–6. [Google Scholar] [CrossRef]
- Sofy, A.R.; Hmed, A.A.; Abd El Haliem, N.F.; Zein, M.A.-E.; Elshaarawy, R.F.M. Polyphosphonium-oligochitosans decorated with nanosilver as new prospective inhibitors for common human enteric viruses. Carbohydr. Polym. 2019, 226, 115261. [Google Scholar] [CrossRef]
- Milewska, A.; Chi, Y.; Szczepanski, A.; Barreto-Duran, E.; Dabrowska, A.; Botwina, P.; Obloza, M.; Liu, K.; Liu, D.; Guo, X.; et al. HTCC as a Polymeric Inhibitor of SARS-CoV-2 and MERS-CoV. J. Virol. 2021, 95, e01622-20. [Google Scholar] [CrossRef]
- Pyrc, K.; Milewska, A.; Duran, E.B.; Botwina, P.; Lopes, R.; Arenas-Pinto, A.; Badr, M.; Mellor, R.; Kalber, T.; Fernandez-Reyes, D.; et al. SARS-CoV-2 inhibition in human airway epithelial cells using a mucoadhesive, amphiphilic chitosan that may serve as an anti-viral nasal spray. BioRxiv 2020. [Google Scholar] [CrossRef]
- Iacob, A.-T.; Drăgan, M.; Ghețu, N.; Pieptu, D.; Vasile, C.; Buron, F.; Routier, S.; Giusca, S.E.; Caruntu, I.-D.; Profire, L. Preparation, Characterization and Wound Healing Effects of New Membranes Based on Chitosan, Hyaluronic Acid and Arginine Derivatives. Polymers 2018, 10, 607. [Google Scholar] [CrossRef]
- Cánepa, C.; Imperiale, J.C.; Berini, C.A.; Lewicki, M.; Sosnik, A.; Biglione, M.M. Development of a Drug Delivery System Based on Chitosan Nanoparticles for Oral Administration of Interferon-α. Biomacromolecules 2017, 18, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Du, Y.-Z.; Yuan, H.; Zhang, X.-G.; Miao, J.; Cui, F.-D.; Hu, F.-Q. Synthesis of lamivudine stearate and antiviral activity of stearic acid-g-chitosan oligosaccharide polymeric micelles delivery system. Eur. J. Pharm. Sci. 2010, 41, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-K.; Wang, Y.-Y.; Qiu, W.-Y.; Wu, J.-Y. Construction and characterization of nanosized curdlan sulfate/chitosan polyelectrolyte complex toward drug release of zidovudine. Carbohydr. Polym. 2017, 174, 209–216. [Google Scholar] [CrossRef] [PubMed]
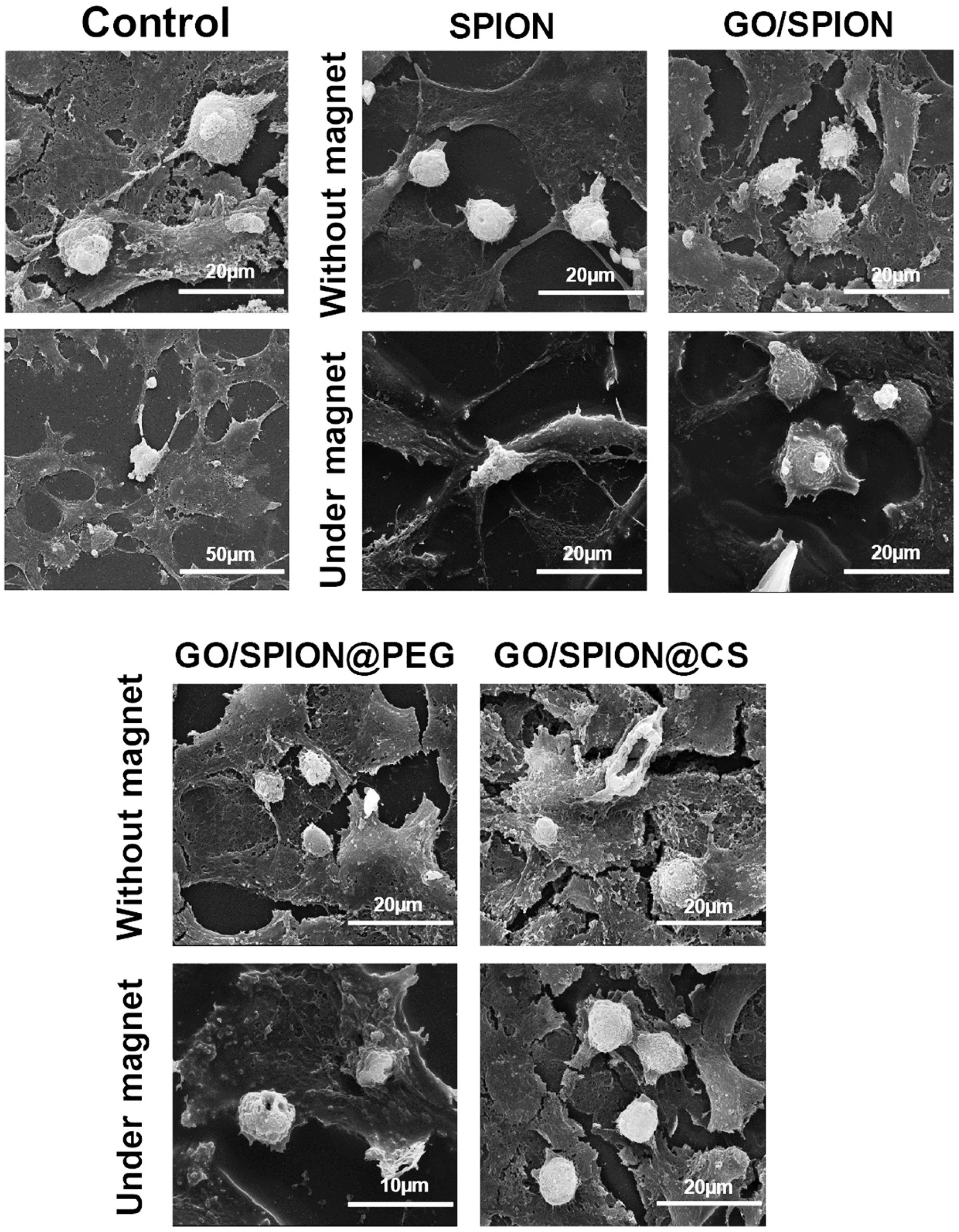
- Kohzadi, S.; Najmoddin, N.; Baharifar, H.; Shabani, M. Functionalized SPION immobilized on graphene-oxide: Anticancer and antiviral study. Diam. Relat. Mater. 2022, 127, 109149. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Gaglianone, N.; Baldassari, S.; Parodi, B.; Cafaggi, S.; Zibana, C.; Donalisio, M.; Cagno, V.; Lembo, D.; Caviglioli, G. Preparation, characterization and in vitro antiviral activity evaluation of foscarnet-chitosan nanoparticles. Colloids Surf. B Biointerfaces 2014, 118, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Loutfy, S.A.; Abdel-SAlam, A.I.; Moatasim, Y.; Gomaa, M.R.; Gomaa, M.R.; Abdel Fattah, N.F.; Emam, M.H.; Ali, F.; ElShehaby, H.A.; Ragab, E.A.; et al. Antiviral activity of chitosan nanoparticles encapsulating silymarin (Sil–CNPs) against SARS-CoV-2 (in silico and in vitro study). RSC Adv. 2022, 12, 15775–15786. [Google Scholar] [CrossRef]
- Szymańska, E.; Wojasiński, M.; Dąbrowska, J.; Krzyżowska, M.; Nowicka, M.; Ciach, T.; Winnicka, K. Chitosan-poly(ethylene oxide) nanofibrous mat as a vaginal platform for tenofovir disoproxyl fumarate—The effect of vaginal pH on drug carrier performance. Int. J. Biol. Macromol. Part A 2022, 222, 856–867. [Google Scholar] [CrossRef]
- Shao, C.; Yu, Z.; Luo, T.; Zhou, B.; Song, Q.; Li, Z.; Yu, X.; Jiang, S.; Zhou, Y.; Dong, W.; et al. Chitosan-Coated Selenium Nanoparticles Attenuate PRRSV Replication and ROS/JNK-Mediated Apoptosis in vitro. Int. J. Nanomed. 2022, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Rehman, N.; Park, T.J.; Basit, M.A. Antiviral role of nanomaterials: A material scientist’s perspective. RSC Adv. 2023, 13, 47–79. [Google Scholar] [CrossRef]
- Rahman, S.S.; Ahmed, A.B. Preparation, characterization and statistical optimization of Nevirapine loaded chitosan nanoparticle for vaginal delivery. J. Pharm. Sci. 2019, 11, 348–358. [Google Scholar]
- Shoueir, K.R.; El-Desouky, N.; Rashad, M.M.; Ahmed, M.K.; Janowska, I.; El-Kemary, M. Chitosan based-nanoparticles and nanocapsules: Overview, physicochemical features, applications of a nanofibrous scaffold, and bioprinting. Int. J. Biol. Macromol. 2021, 167, 1176–1197. [Google Scholar] [CrossRef] [PubMed]
- Essa, D.; Choonara, Y.E.; Kondiah, P.P.D.; Pillay, V. Comparative Nanofabrication of PLGA-Chitosan-PEG Systems Employing Microfluidics and Emulsification Solvent Evaporation Techniques. Polymers 2020, 12, 1882. [Google Scholar] [CrossRef] [PubMed]
- Krishna Sailaja, A.; Amareshwar, P. Preparation of bovine serum albumin loaded chitosan nanoparticles using reverse micelle method. Res. J. Pharm. Biol. Chem. Sci. 2011, 2, 837–846. [Google Scholar]
- Kumari, G.D.; Raksha, G.; Deepak, K.; Anjana, G.; Mary, C.S. A Review on Chitosan Nanoparticle as a Drug delivery system. Asian J. Pharm. Res. 2020, 10, 299–306. [Google Scholar] [CrossRef]
- Gérard, L.; Salmon-Céron, D. Pharmacology and clinical use of foscarnet. Int. J. Antimicrob. Agents 1995, 5, 209–217. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Quispe, C.; Butnariu, M.; Rotariu, L.S.; Sytar, O.; Sestito, S.; Rapposelli, S.; Akram, M.; Iqbal, M.; Krishna, A.; et al. Chitosan nanoparticles as a promising tool in nanomedicine with particular emphasis on oncological treatment. Cancer Cell Int. 2021, 21, 318. [Google Scholar] [CrossRef]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef]
- Li, X.; Chan, W.K. Transport, metabolism and elimination mechanisms of anti-HIV agents. Adv. Drug Deliv. Rev. 1999, 39, 81–103. [Google Scholar] [CrossRef]
- Kim, A.E.; Dintaman, J.M.; Waddell, D.S.; Silverman, J.A. Saquinavir, an HIV protease inhibitor, is transported by P-glycoprotein. J. Pharmacol. Exp. Ther. 1998, 286, 1439–1445. [Google Scholar] [PubMed]
- Kim, R.B.; Fromm, M.F.; Wandel, C.; Leake, B.; Wood, A.J.; Roden, D.M.; Wilkinson, G.R. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J. Clin. Investig. 1998, 101, 289–294. [Google Scholar] [CrossRef]
- Ramana, L.N.; Sharma, S.; Sethuraman, S.; Ranga, U.; Krishnan, U.M. Evaluation of chitosan nanoformulations as potent anti-HIV therapeutic systems. Biochim. Biophys. Acta 2014, 1840, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, V.H.B. In-vivo pharmacokinetic studies of Dolutegravir loaded spray dried Chitosan nanoparticles as milk admixture for paediatrics infected with HIV. Sci. Rep. 2022, 12, 13907. [Google Scholar] [CrossRef]
- Global Health Sector Strategy on Sexually Transmitted Infections. Available online: https://www.who.int/publications-detail-redirect/WHO-RHR-16.09 (accessed on 29 January 2023).
- Pandey, M.; Choudhury, H.; Abdul-Aziz, A.; Bhattamisra, S.K.; Gorain, B.; Carine, T.; Wee Toong, T.; Yi, N.J.; Win Yi, L. Promising Drug Delivery Approaches to Treat Microbial Infections in the Vagina: A Recent Update. Polymers 2020, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Jeevanandam, J.; Krishnan, S.; Hii, Y.S.; Pan, S.; Chan, Y.S.; Acquah, C.; Danquah, M.K.; Rodrigues, J. Synthesis approach-dependent antiviral properties of silver nanoparticles and nanocomposites. J. Nanostruct. Chem. 2022, 12, 809–831. [Google Scholar] [CrossRef] [PubMed]
- Travan, A.; Marsich, E.; Donati, I.; Paoletti, S. Silver Nanocomposites and Their Biomedical Applications. In Nanotechnologies for the Life Sciences; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar] [CrossRef]
- Aliyev, E.; Filiz, V.; Khan, M.M.; Lee, Y.J.; Abetz, C.; Abetz, V. Structural Characterization of Graphene Oxide: Surface Functional Groups and Fractionated Oxidative Debris. Nanomaterials 2019, 9, 1180. [Google Scholar] [CrossRef]
- Ege, D.; Kamali, A.R.; Boccaccini, A.R. Graphene Oxide/Polymer-Based Biomaterials. Adv. Eng. Mater. 2017, 19, 1700627. [Google Scholar] [CrossRef]
- Pichon, C.; Billiet, L.; Midoux, P. Chemical vectors for gene delivery: Uptake and intracellular trafficking. Curr. Opin. Biotechnol. 2010, 21, 640–645. [Google Scholar] [CrossRef]
- Donalisio, M.; Leone, F.; Civra, A.; Spagnolo, R.; Ozer, O.; Lembo, D.; Cavalli, R. Acyclovir-Loaded Chitosan Nanospheres from Nano-Emulsion Templating for the Topical Treatment of Herpesviruses Infections. Pharmaceutics 2018, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Sajomsang, W.; Nuchuchua, O.; Saesoo, S.; Gonil, P.; Chaleawlert-Umpon, S.; Pimpha, N.; Sramala, I.; Soottitantawat, A.; Puttipipatkhachorn, S.; Ruktanonchai, U.R. A comparison of spacer on water-soluble cyclodextrin grafted chitosan inclusion complex as carrier of eugenol to mucosae. Carbohydr. Polym. 2013, 92, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Belgamwar, A.; Khan, S.; Yeole, P. Intranasal chitosan-g-HPβCD nanoparticles of efavirenz for the CNS targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Čalija, B.; Milić, J.; Cekić, N.; Krajišnik, D.; Daniels, R.; Savić, S. Chitosan oligosaccharide as prospective cross-linking agent for naproxen-loaded Ca-alginate microparticles with improved pH sensitivity. Drug Dev. Ind. Pharm. 2013, 39, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xu, Y.; Liang, W.; Leung, G.P.H.; Cheung, K.-H.; Zheng, C.; Chen, F.; Lam, J. DNA-loaded chitosan oligosaccharide nanoparticles with enhanced permeability across Calu-3 cells. J. Drug Target. 2013, 21, 474–486. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fusion (Attachment) Inhibitors | ||||||
|---|---|---|---|---|---|---|
| Antiviral Drug | Mechanism of Action | Administration/Dosage Form | Diseases | Advantages | Limitations | References |
Enfuvirtide | -interferes with the entry of HIV-1 into cells by inhibiting the fusion of viral and cellular membranes -binds to the first heptad-repeat (HR1) in the gp41 subunit of the viral envelope glycoprotein and prevents the conformational changes required for the fusion of viral and cellular membranes | Subcutaneous route/injection | AIDS | -increases the number of CD4 cells -reducing the amount of HIV in the blood reduces the risk of death or infections due to low immunity | -adverse effects: depression, nervousness, tiredness, muscle pain, nausea, loss of appetite, weight loss, diarrhea, constipation, flu-like symptoms, swollen glands, or painful, red, or teary eyes | [29,30] |
| DNA Polymerase Inhibitors (DPIs) | ||||||
Idoxuridine | Nucleoside inhibitor -inhibits viral replication by substituting itself for thymidine in viral DNA. This, in turn, inhibits thymidylate phosphorylase and viral DNA polymerases from properly functioning. The effect of idoxuridine results in the inability of the virus to reproduce or infect/destroy tissue | Ocular route/ointment, solution | Feline herpetic keratitis and conjunctivitis | -potential anti-cancer effects thanks to its cytotoxicity | -cardiotoxicity, just for local use -burning, stinging, pain, irritation, itching, redness, blurred vision, eyelid itching, eyelid swelling, or sensitivity to light | [31,32] |
Vidarabine | -stops replication of herpes viral DNA in two ways: (1) competitive inhibition of viral DNA polymerase, and consequently, (2) incorporation into and termination of the growing viral DNA chain | Ocular route/ointment | Herpes and acyclovir-resistant viruses Varicella zoster | -reduces lesion formation and the duration of viral shedding -less susceptible to the development of drug-resistant strains than other antivirals | -burning, stinging, pain, irritation, itching, redness, swelling, blurred vision, tearing, feeling like something is in the eye, or sensitivity to light | [33] |
Acyclovir | -acyclovir triphosphate competitively inhibits viral DNA polymerase by acting as an analog to deoxyguanosine triphosphate (dGTP) -incorporation of acyclovir triphosphate into DNA results in chain termination since the absence of a 3′ hydroxyl group prevents the attachment of additional nucleosides | Oral route/tablet Intravenous route/injection Transdermal route/ointment, cream | Herpes simplex virus infections Herpes zoster infection Varicella zoster virus infection Cytomegalovirus infection | -prevention of recurrent genital herpes infections -helps relieve the herpes pain and discomfort and helps the sores heal faster | -nausea, vomiting, burning, stinging, pruritus, rash, urticaria, headache, diarrhea, occasionally renal insufficiency and neurotoxicity -absorbed drug reaches the breast milk, placenta, and amniotic fluid | [34,35,36] |
Cidofovir | Nucleotide inhibitor -acts as a competitive inhibitor and an alternate substrate for cytomegalovirus (CMV) DNA polymerase | Intravenous route/infusion | Cytomegaloviral retinitis in people with AIDS | -used with probenecid to treat a certain viral eye infection -lowers the risk of blindness and other vision problems | -nausea, vomiting, diarrhea, loss of appetite, white patches or sores inside mouth or on lips, headache, skin rash, hair loss, or cough | [37] |
Foscarnet | Pyrophosphate analog -interacts with the enzymatic action of polymerases and inhibits the cleavage of pyrophosphate from the nucleoside triphosphate -a non-competitive inhibitor of herpesvirus DNA polymerase, hepatitis B virus DNA polymerase, and reverse transcriptases | Intravenous route/infusion | Cytomegalovirus infection | -successful in the treatment of limited numbers of immunocompromised patients with CMV-associated gastrointestinal (improvement in over 67% of patients) and other infections | -mineral and electrolyte imbalances, neurotoxicity, nausea, vomiting, anemia, bone marrow suppression, decreased creatinine clearance, or conjunctivitis | [30,38] |
Phosphonoacetic acid | Intravenous route/infusion | Herpes simplex 1 infection Herpes simplex 2 infections Epstein–Barr virus infection Cytomegalovirus infection | -lack of toxicity toward many animal cells | -nausea, vomiting, anemia, bone marrow suppression, decreased creatinine clearance | [39,40] | |
| Reverse Transcriptase Inhibitors | ||||||
Zidovudine | Nucleoside analog -active against HIV, a retrovirus. The drugs inhibit RNA virus replication by reversible inhibition of viral HIV reverse transcriptase, which reverse transcribes viral RNA into DNA for insertion into the host DNA sequence | Oral route/tablet Intravenous route/injection | AIDS | -will not cure or prevent HIV infection or AIDS; however, it helps keep HIV from reproducing and appears to slow down the destruction of the immune system | -monotherapy is recommended only in the initial management of HIV-1-infected patients -bone marrow suppression -combination therapy in advanced disease (zidovudine in combination with lamuvidine as combivir and with lamuvidine and abacavir as trizvir | [41] |
Didanosine | Oral route/capsule | AIDS | -helps to decrease the amount of HIV in the body so the immune system can work better. -this lowers the chance of getting HIV complications | -peripheral neuropathy -pancreatitis, lactic acidosis, hepatomegaly, hyperuricaemia -similar activity to zidovudine | [30,42,43] | |
Zalcitabine | Oral route/tablet | AIDS | -blocking the growth of HIV -used in combination with other medicines | -peripheral neuropathy, nausea, vomiting, headache, hepatotoxicity, or cardiomyopathy | [44] | |
Stavudine | Oral route/capsule, solution | AIDS | -helps to decrease the amount of HIV in the body so your immune system can work better | -numbness, tingling, pain in hands or feet, weakness, liver problems, stomach pain, loss of appetite, dark urine, clay-colored stools, jaundice (yellowing of the skin or eyes), pancreatitis, fever, nausea, or vomiting | [45] | |
Lamivudine | Oral route/tablet | AIDS Chronic hepatitis B | -lamivudine therapy is associated with a significant improvement in hepatic histology, normalization of hepatic enzymes, and suppression of plasma HBV DNA | -headache, nausea, fatigue, dizziness, neutropenia, or skin rash | [46,47] | |
Tenofovir | Nucleotide analog -inhibits HIV-1 reverse transcriptase and the hepatitis B polymerase through direct binding competition with the natural deoxyribonucleotide substrate (deoxyadenosine 5′-triphosphate) and, after integration into DNA, causes viral DNA chain termination | Oral route/tablet | AIDS Chronic hepatitis B infection | -in HBeAg-negative patients, tenofovir was the most effective in inducing undetectable levels of HBV DNA (94%) and improving liver histology (65%); it ranked second for normalization of ALT levels (73%) | -side effects: diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash | [46,48] |
Adefovir | Oral route/tablet | Chronic hepatitis B infection | -provides sustained suppression of the virus and improvement in liver disease | -weakness, headache, fever, increased cough, nausea, vomiting, diarrhea -risk of lactic acidosis and hepatomegaly with steatosis -patients with renal dysfunction since chronic administration may result in nephrotoxicity | [49] | |
Efavirenz | Nonnucleoside analog -directly inhibits the HIV-1 reverse transcriptase by binding in a reversible and non-competitive manner to the enzyme | Oral route/capsule, tablet | AIDS | -improves the function of the immune system by decreasing the amount of HIV in the body | -neuropsychiatric side effects: dizziness, headache, insomnia, impaired concentration, or abnormal dreams | [43,49] |
Nevirapine | Oral route/tablet | AIDS | -prevents mother-to-child transmission of HIV-1 in pregnant women | -induces the metabolism of warfarin -side effects: nausea, headache, or rash | [45,50] | |
Delavirdine | Oral route/tablet | AIDS | -is generally well tolerated -patients achieve marked improvements in virological and immunological markers -favorably increases plasma concentrations of several protease inhibitors, and the drug may also be beneficial as a component of salvage therapy in combination with protease inhibitors | -associated with a low rate of transient serum aminotransferase elevations during therapy and is a rare cause of clinically apparent acute liver injury -maculopapular rash | [30,51] | |
Etravirine | Oral route/tablet | AIDS | -does not need metabolic activation -decreases the amount of HIV in the blood | -side effects: rash, headache, nausea, diarrhea, fatigue, hypertension, abdominal pain, or peripheral neuropathy | [30] | |
| Integrase Inhibitors | ||||||
Raltegravir | -stops integrase from working, which stops HIV from entering CD4 cells -the medication does not cure HIV, but it keeps the virus from multiplying -as part of an antiretroviral treatment plan, it helps reduce the amount of HIV in the body to undetectable levels | Oral route/tablet, chewable tablet, granules for oral suspension | AIDS | -lowers the chance of getting HIV complications -supports the immune system by decreasing the amount of HIV in the blood so it can work better | -metabolized by glucuronidation -adverse effects: nausea, diarrhea, or headache -myopathy and rhabdomyolysis are connected with the drug associated with muscle toxicity | [30,52] |
| Portmanteau Inhibitors | ||||||
Caffeoyl-anilide compounds | -function as a reverse transcriptase inhibitor as well as an integrase inhibitor | Intravenous route/injection | AIDS | -strategy to reduce the pill burden -dual action in inhibiting HIV integrase and blocking the CCR5 receptor-mediated entry | -price -high in toxicity because many drugs are taken at one time | [53] |
| Protease Inhibitors | ||||||
Saquinavir | -inhibits the cleavage of the polyprotein into functional proteins -protease is a protein-based enzyme that normally breaks the polyprotein into functional proteins, so blocking or inhibiting protease prevents this essential step of viral reproduction | Oral route/capsule, tablet | AIDS | -in combination with ritonavir and other HIV medications, is used to help control HIV infection -it helps to decrease the amount of HIV in the body so the immune system can work better -only four percent of patients receiving saquinavir had side effects | -potential side effects: insulin resistance, nausea, diarrhea, development of gallstones or kidney stones, changes in how things taste, insomnia, elevated numbers in liver function tests, rash or dry skin, or elevated cholesterol | [54,55] |
Indinavir | Oral route/capsule | AIDS | -recent studies have analyzed the effects of indinavir as an anti-cancer agent (human papillomavirus is induced by increased expression of eukaryotic translation initiation factor 4E (eIF4E), potentially leading to cervical cancer) | -side effects: nausea, vomiting, fatigue, diarrhea, kidney stones, nephrolithiasis (for solubility only in acidic conditions), or kidney inflammation | [56] | |
Amprenavir | Oral route/capsule | AIDS | -greatly increased water solubility and improved oral bioavailability -this allows a reduction in the daily dose | -general adverse effects: nausea, vomiting, diarrhea, epigastric pain, flatulence, paresthesia, headache, rash, or fatigue | [57,58] | |
Nelfinavir | Oral route/tablet, powder | AIDS | -has also shown anti-cancer effects in in vitro and in vivo studies -supports the immune system with a lower amount of HIV in the blood | -low white blood cell counts, nausea, diarrhea, gas, stomach pain, loss of appetite, rash, or changes in the shape or location of body fat | [59,60] | |
Ritonavir | Oral route/tablet, soft gel capsule, oral suspension | AIDS SARS-CoV-2 infection | -The COVID-19 Treatment Guidelines Panel recommends against the use of lopinavir/ritonavir and other HIV protease inhibitors for the treatment of COVID-19 in hospitalized and non-hospitalized patients | -general side effects: drowsiness, diarrhea, gas, heartburn, change in the ability to taste food, headache, numbness, burning, tingling of the hands, feet, or area around the mouth, muscle or joint pain, stomach pain, nausea, or vomiting | [61] | |
| Signaling Inhibitors | ||||||
Ribavirin  | -directly inhibits viral mRNA polymerase by binding to the nucleotide-binding site of the enzyme -prevents the binding of the correct nucleotides, leading to a reduction in viral replication or the production of defective virions -administered as a combination therapy -inhibits inosine monophosphate dehydrogenase | Oral route/tablet Intravenous route/injection Inhalation route/aerosol | SARS-CoV-2 infection Hepatitis C Severe respiratory syncytial virus infection | -improves the signs and symptoms of viral bronchiolitis in infancy | -not effective when used alone; the potential is tripled when in combination with lopinavir/ritonavir and interferon-beta-1b, which alleviated symptoms of SARS-CoV-2 completely within four days -common side effects: cough, upset stomach, vomiting, diarrhea, constipation, heartburn, loss of appetite, or weight loss | [62,63] |
Viramidine | Oral route/tablet | Chronic hepatitis C | -viramidine is taken up into hepatocytes by a mechanism distinct from that of ribavirin, with greater affinity for the liver | -side effects: hemolytic anemia, vomiting, diarrhea, or loss of appetite | [64,65] | |
| CS Derivatives | Formula | Physical Properties | Biological Properties | Preparation Method | Advantages/Limitations/Potential Uses | References |
|---|---|---|---|---|---|---|
| Ionic Derivatives | ||||||
| Quarternized derivatives |  | -cationic derivatives -water-soluble at neutral pH -N,N, N-trimethyl CS chloride (TMC) ↑ aqueous solubility than CS | -biocompatibility -biodegradability -mucoadhesion | -direct quaternary ammonium substitution -epoxy derivative open loop -N-alkylation | -antifungal, antibacterial, antituberculosis -enzyme inhibition -permeation enhancers -gene transfection and delivery -good moisture retention and absorption -mucoadhesivity ↓ with ↑ degree of quaternization -↑ degree of quaternization ↓ intrinsic viscosity -pH 7.4 CS and salts failed to increase the permeability -absorption enhancer for intestinal lumen with pH close to its pKa -TMC collects and delivers more negatively charged DNA/genes than plain CS -quaternized CS ↑ hydroxyl radical scavenging activity in comparison to other CS -pH-sensitive targeting | [184] |
| Sulfated derivatives |  | -water-soluble sulfoethylated CS with improved swelling property -during sulfation, some amino groups of CS are converted to anionic centers, which leads to better polyelectrolyte properties -N-alkyl-O-sulfated CS has amphiphilicity since it carries long-chain alkyl groups with a hydrophobic nature and sulfated groups with a hydrophilic nature | -biocompatibility -biodegradability | -sulfation of 2-chloroethane sulfonic acid sodium salt in alkaline media -sulfur-containing derivatives were obtained by reacting CS with CS2, formaldehyde, and primary amine | -antisclerotic, antioxidant, antibacterial, anti-HIV, antiviral, and enzyme inhibition -blood anticoagulant -hemagglutination inhibition activity -amphiphilic polymer enables the formation of micelles with physical entrapment of water-insoluble drugs such as taxol -structural similarity of CS salt with heparin -high sorption capacities -a great advantage for metal ion recovery | [25] |
| CS derivatives with sugar part |  | -water-soluble -investigated mainly for rheological studies | -biodegradability -biocompatibility -nontoxic | -reductive N-alkylation (using NaCNBH3 and unmodified sugar, a sugar-aldehyde derivative, or N-alkylation of CS performed in aqueous methanol with various aldehydes, monosaccharides, and disaccharides) | -antibacterial and antimicrobial activity -this type of modification has usually been used to introduce cell-specific sugars into CS -synthesis of sugar-bound CS, such as those with D- and L-fucose, and their specific interactions with lectin and cells -lactose-modified CS for a potential application in the repair of the articular cartilage -galactosylated CS prepared from lactobionic acid and CS with 1-ethyl-3-(3-dimethyl aminopropyl)- carbodiimide (EDC) and N-hydroxysuccinimide (NHS) showed promise as a synthetic extracellular matrix for hepatocyte attachment -graft copolymers of galactosylated CS with poly(ethylene glycol) or poly(vinyl pyrrolidone) were useful as hepatocyte-targeting DNA carriers | [181,185,186] |
| CS glutaraldehyde crosslinked polymer |  | -improved permeability, mechanical properties, wetting, and chemical resistance -gelation temperature: 32–33 °C -viscosity of the hydrogel increased quickly after gelation | -not cytotoxic to human corneal epithelial cells at a low concentration -biocompatibility -biodegradability | -CS dissolved in acetic acid with glutaraldehyde performed in a short time | -N-trimethylated CS crosslinked with glutaraldehyde has been used to fabricate hollow microspheres for drug loading | [187] |
| CS cyclodextrin derivatives |  | -can enhance the solubility of sulfadiazine, sulfamonomethoxine, and sulfamethoxazole | -antimicrobial activity -antifungal activity -bioavailability -nontoxic -biodegradability | -physical mixing, kneading, co-precipitation, and solvent evaporation | -increases dissolution -improves stability | [188,189,190] |
| CS oligomers |  | -soluble over a wide pH range, from acidic to basic -water-soluble | -antioxidant activity -anti-inflammatory activity -antiviral activity -nontoxic -antifungal activity -biodegradability -biocompatibility -immunological and antibacterial activities | -prepared from the degradation of CS -oxidative degradation method involving hydrogen peroxide (H2O2) and combined degradation using hydrogen peroxide and microwave radiation | -inhibit the expression of TNF-α, IL-6, and iNOS, which are associated with an inflammatory response -depress oxidative stress -decrease induced cell apoptosis -alleviate or delay Alzheimer´s disease process | [191,192] |
| Drug Delivery System | Virus | Mechanism of Action | Limitations and Advantages | State of Implementation In Practice | References |
|---|---|---|---|---|---|
| CS NPs (zinc) | HIV-1 | -decrease the viral load -mostly interfere with the active replication of the virus, thereby decreasing its copy number | -greater effectiveness on resistant strains of microbial pathogens -less toxicity and heat resistance -biocompatibility and biocidal effect | -research study | [245,246] |
| CS NPs (silver) | H1N1 influenza A | -binding to the negatively charged protein capsid with subsequent destabilization of the envelope and altered permeability -inhibit the viral contact with host cells and interaction of silver NPs with viral glycoproteins | -no adverse effect of L-B-L coating on the physical properties of the fabric -sustained release of Ag+ from CS-Ag-NP-coated fabric -antibacterial, antifungal, anti-inflammatory, antiviral, anti-angiogenesis, and antiplatelet activities -exhibit a higher rate of cell destruction than common agents used as reference -good biocompatibility and dose-dependant toxicity | -research study | [247] |
| Polyquarternary phosphonium oligo CS NPs (nanosilver) | Hepatitis A | -induce ribonuclease catalyzed by CS to degrade the viral RNA and consequently prevent its transcription and translation | -NBCs are more virucidally active than PQPOCs against the target virus -the antiviral activity of NBCs is concentration- and pH-dependent | -research study | [248] |
| HTTC (N-(2-hydroxypropyl)-3-trimethylammonium CS) NPs | SARS-CoV-2 | -exhibit effective inhibition of SARS-CoV-2 -complex formation between HTCC polymer and S-protein of the virus, which blocks viral entry into the host cell | -efficiently hamper infection of all low-pathogenicity human coronaviruses in vitro and ex vivo -replication of both viruses (SARS-CoV-2 and MERS-CoV) is efficiently hampered | -research study | [249] |
| GCPQ (N-palmitoyl-N-monomethyl-N,N-dimethyl-N,N,N-trimethyl-6-O-glycolCS) | SARS-CoV-2 | -bind electrostatically to coronavirus S proteins to block not only entry to host cells but also viral replication | -mucoadhesive and chemically stable -limit viral cell entry in the nasal cavity, which could have a profound impact on the course and severity of the disease | -research study | [250] |
| CS-arginine derivative NPs | H1N1 influenza A, herpes simplex-1, encephalomyocarditis virus | -increase permeability of virus protein capsid | -good swelling degree, improved hydrophilicity, and biocompatibility in terms of surface-free energy components, which supports their application for tissue regeneration | -research study | [28,251] |
| CS NPs (IFNα-2b) | Human lymphotropic-T virus type 1 Vesicular stomatitis virus | -regulate the expression of many genes involved in the suppression of cell proliferation via Janus kinase/signal transducer and activator of transcription signaling pathway and the inhibition of viral replication | -ionotropic gelation method for the production of the nanodrug delivery system -water-based, simple, reproducible, cost-viable, and eventually scalable in an industrial setting -enhance oral absorption | -preliminary pharmacokinetic study | [252] |
| CS NPs (acyclovir) | HSV-1, HSV-2, CMV | -competitively inhibit viral DNA polymerase by acting as an analog to deoxyguanosine triphosphate (dGTP) | -use crosslinked CS with tripolyphosphate (TPP) | -research study | [19] |
| CS NPs (saquinavir) | HIV | -control viral proliferation as measured using two viral strains, NL4-3 and Indie-C1, and two target T-cells, Jurkat and CEM-CCR5 -bind to the active site of the viral protease and prevent cleavage of viral polyproteins, thus preventing maturation of the virus | -biopolymer-based NPs -ionic gelation technique -superior drug-loading potential with greater cell-targeting efficiency, leading to efficient control of the viral proliferation in target T-cells | -research study | [19] |
| CS-g-HPβCD NPs (efavirenz) | HIV | -non-nucleoside reverse transcriptase (RT) inhibitor of human immunodeficiency virus type 1 (HIV-1) -non-competitive inhibition of HIV-1 RT | -NPs showed sustained efavirenz release (99.03 ± 0.30% in 8 h) and followed the Fickian diffusion mechanism -NPs showed 4.76 times greater permeability than un-encapsulated efavirenz solution through the porcine nasal mucosa -development of intranasal mucoadhesive EFV-NPs for CNS targeting | -research study | [19] |
| Lamivudinestearate (LAS) loaded to stearic acid-g-CSoligosaccharide (CSO-SA) micelles (CSO-SA/LAS) | HBV | -must be converted intracellularly to its triphosphate form, which then competes with cytosine triphosphate for incorporation into the developing viral DNA strand | -faster release behavior of LA from CSO-SA micelles at low pH presents the potential for efficient antiviral agent delivery -exhibit relatively low cytotoxicity, high uptake, and conspicuous in vitro anti-HBV activities, while the blank CSO-SA micelles conduct some antiviral activities, as well -promising carriers for effective therapy of anti-HBV drugs | -research study | [253] |
| CS nanodroplets (sulfobutyl ether-β-cyclo-dextrin inclused with acyclovir) | HSV-2 | -competitively inhibit viral DNA polymerase by acting as an analog to deoxyguanosine triphosphate (dGTP) | -ACV loaded into nanodroplets shows higher antiviral activity against HSV-2 in cell cultures compared to the free drug -higher intracellular concentration of the drug delivered by the nanodroplets -drug was complexed with sulfobutyl ether-β-cyclo-dextrin and then incorporated in the nanodroplet CS shell via electrostatic interaction | -in vitro study | [238] |
| Curdlan sulfate/CS polyelectrolyte complexes (CRDS/CS PECs) NPs (zidovudine) | HIV | -inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis | -fabricated CRDS/CSPECs could be explored as potential nanocarriers for delivering antiviral drugs -cytotoxicity and antiviral activity in vitro of the AZT-loaded PECs are currently being studied | -in vitro study | [254] |
| GO/SPION/CS (graphene oxide/superparamagnetic iron oxide NPs/CS) | SARS-CoV-2 | -GO could directly interact with viruses via electrostatic interactions, hydrogen bonding, and redox reactions -GO can absorb charged lipids and destroy membranes, suggesting the possibility of interaction with enveloped viruses such as the SARS-CoV-2 virus -inactivate viruses by attaching to the tail fiber and dissociating a virus, interfering with the viral replication process, binding to the viral ligands, and preventing their interaction with the host cell receptors | -capable of neutralizing the SARS-CoV-2 virus effectively and could be considered a promising material against COVID-19 -with decreasing sample magnetization, the relative magnetic cytotoxicity percentage shows a downward trend -no sign of antiviral activity is observed for SPION NPs -GO/SPION reveals merely 23% viral inhibition -the highest level of inhibition is for GO/SPION@CS, with more than 86% viral inhibition | -in vitro study | [255] |
| CS NPs (foscarnet) | CMV, HIV | -block the pyrophosphate binding site, preventing cleavage of pyrophosphate from deoxynucleotide triphosphates | -NPs showed no toxicity on non-infected HELF cells -crosslinked NPs showed controlled drug release | -in vitro study | [256] |
| CS NPs (sylimarine) | SARS-CoV-2 | blocki viral host receptor ACE2, thus preventing viral attachment and its entry into cells | -take the form of intranasal drug delivery to inhibit viral entry -mucoadhesive property healing in its application as intranasal vaccine targeting mucosal pathway inducing local humoral and cellular immune responses | -in silico and in vitro study | [257] |
| CS/PEO (polyethylene oxide) nanofibrous mat (tenofovir) | HSV-2 | -reduces TDF uptake and saturation of the enzymes involved in prodrug hydrolysis | -uniform drug distribution -mucoadhesive and swelling activity -better retention time with low leakage from the vaginal cavity -reduced TDF uptake and saturation of the enzymes involved in prodrug hydrolysis -fluctuations in the physiological pH range of 3.8–5.0 substantially affect mucoadhesive behavior and drug dissolution rate from the nanofibrous carrier -more acidic vaginal environment favors nanofibrous mat adherence to the human vaginal tissue and speeds up drug diffusion from the polymer matrix, which, in turn, may increase the drug residence time with mucosal tissue and help to initiate microbicide absorption onset | -in vitro studies | [258] |
| CS-SeNPs | PRRSV (porcine reproductive and respiratory syndrome virus) | -suppress oxidative stress induced by rPRRSV-EGFP infection by increasing GSH-Px activity, promoting GSH production, and inhibiting H2O2 synthesis -enhance the antioxidant capacity and effectively suppress PRRSV-induced apoptosis in Marc-145 cells via the ROS/JNK signaling pathway, thereby inhibiting PRRSV replication | -inhibit ORF5 gene expression, viral titers, and N protein of r-PRRSV-EGFP at 24 and 48 h post-infection (hpi) in Marc-145 cells | -in vitro study | [259] |
| CS-based polymeric NPs (dolutegravir) | HIV | -inhibit HIV integrase by binding to the active site and blocking the strand transfer step of retroviral DNA integration in the host cell | -no toxicity associated with the administration of the NPs through spray-drying technology as a milk admixture for pediatrics -organ biodistribution of the drug was significantly higher when administered as a nanoformulation than the pure drug -coadministration of the drug along with milk does not affect the absorption coefficient in vivo; the time taken to reach the maximum concentration is delayed | -in vivo study using Balb-C mice models | [260] |
| CS NPs (nevirapine) | HIV | -bind directly to reverse transcriptase and blocks the RNA-dependent and DNA-dependent DNA polymerase activities by disrupting the enzyme’s catalytic site | -very good permeation of the drug across the vaginal tissue -increase the bioavailability in the target area -increase the therapeutic effect -reduce the systemic toxicity -pharmacokinetics and pharmacodynamic study and safety and efficacy assessments are not finished | -ex vivo study | [261] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Žigrayová, D.; Mikušová, V.; Mikuš, P. Advances in Antiviral Delivery Systems and Chitosan-Based Polymeric and Nanoparticulate Antivirals and Antiviral Carriers. Viruses 2023, 15, 647. https://doi.org/10.3390/v15030647
Žigrayová D, Mikušová V, Mikuš P. Advances in Antiviral Delivery Systems and Chitosan-Based Polymeric and Nanoparticulate Antivirals and Antiviral Carriers. Viruses. 2023; 15(3):647. https://doi.org/10.3390/v15030647
Chicago/Turabian StyleŽigrayová, Dominika, Veronika Mikušová, and Peter Mikuš. 2023. "Advances in Antiviral Delivery Systems and Chitosan-Based Polymeric and Nanoparticulate Antivirals and Antiviral Carriers" Viruses 15, no. 3: 647. https://doi.org/10.3390/v15030647
APA StyleŽigrayová, D., Mikušová, V., & Mikuš, P. (2023). Advances in Antiviral Delivery Systems and Chitosan-Based Polymeric and Nanoparticulate Antivirals and Antiviral Carriers. Viruses, 15(3), 647. https://doi.org/10.3390/v15030647