
Novel Chaphamaparvovirus in Insectivorous Molossus molossus Bats, from the Brazilian Amazon Region

 , , ,
, , ,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Processing of the Samples
2.3. Bioinformatics Analysis
2.4. Genome Annotation
2.5. Genetic Distance
2.6. Phylogenetic Analysis
2.7. Likelihood Mapping
2.8. Phylogenetic Networks
3. Results
3.1. Characterization of Samples
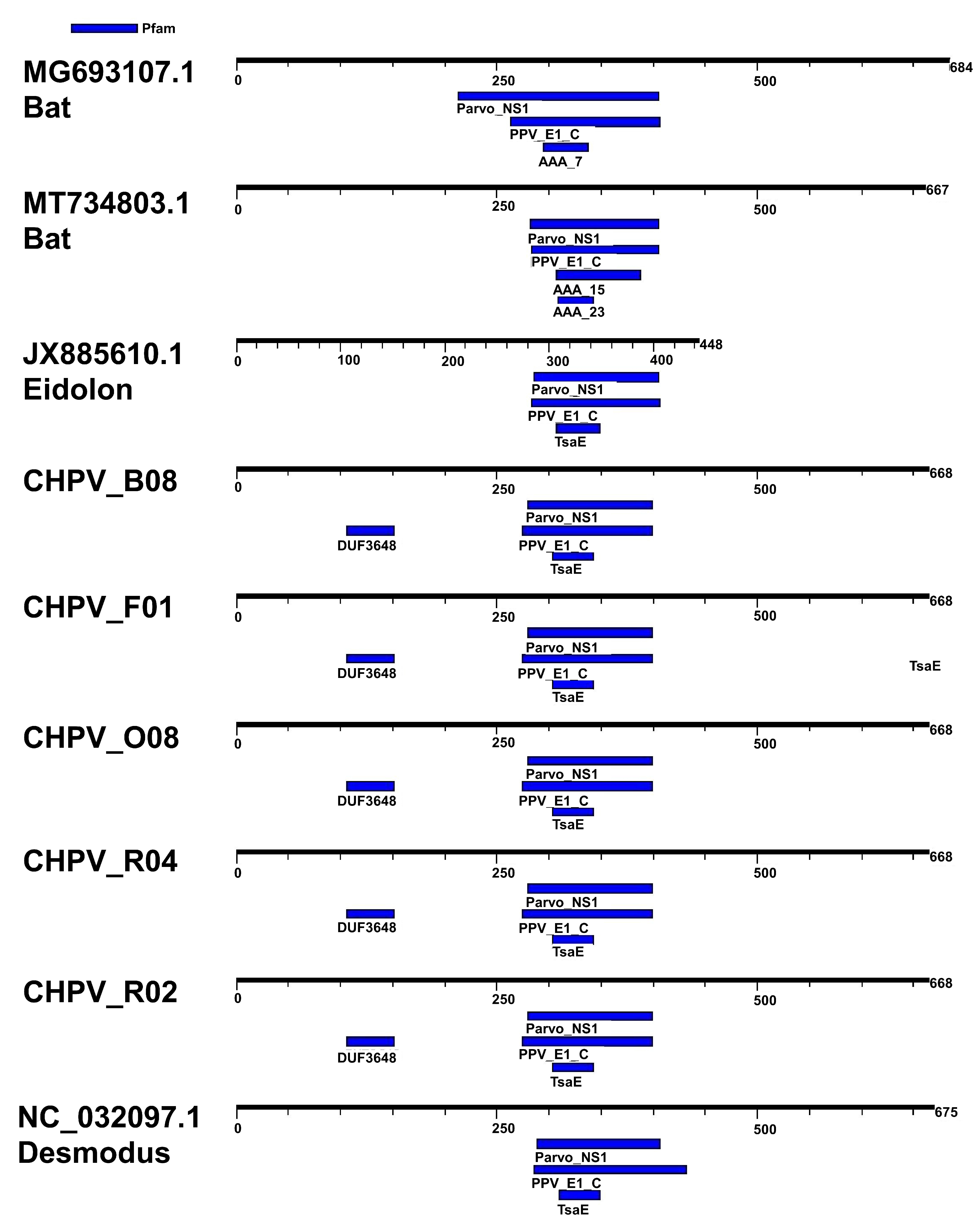
3.2. Comparative Analysis of Genomes
3.3. Phylogenetic Inference
3.4. Reticulated Evolution of CHPV
3.5. Genetic Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Springer, M.S.; Teeling, E.C.; Madsen, O.; Stanhope, M.J.; de Jong, W.W. Integrated Fossil and Molecular Data Reconstruct Bat Echolocation. Proc. Natl. Acad. Sci. USA 2001, 98, 6241–6246. [Google Scholar] [CrossRef]
- Nowak, R.M.; Walker, E.P. Walker’s Bats of the World; Johns Hopkins University Press: Baltimore, MD, USA, 1994; ISBN 9780801849862. [Google Scholar]
- Thomas, S.P.; Suthers, R.A. The Physiology and Energetics of Bat Flight. J. Exp. Biol. 1972, 57, 317–335. [Google Scholar] [CrossRef]
- Zhou, H.; Ji, J.; Chen, X.; Bi, Y.; Li, J.; Wang, Q.; Hu, T.; Song, H.; Zhao, R.; Chen, Y.; et al. Identification of Novel Bat Coronaviruses Sheds Light on the Evolutionary Origins of SARS-CoV-2 and Related Viruses. Cell 2021, 184, 4380–4391.e14. [Google Scholar] [CrossRef]
- Ithete, N.L.; Stoffberg, S.; Corman, V.M.; Cottontail, V.M.; Richards, L.R.; Schoeman, M.C.; Drosten, C.; Drexler, J.F.; Preiser, W. Close Relative of Human Middle East Respiratory Syndrome Coronavirus in Bat, South Africa. Emerg. Infect. Dis. 2013, 19, 1697–1699. [Google Scholar] [CrossRef]
- Šimić, I.; Zorec, T.M.; Lojkić, I.; Krešić, N.; Poljak, M.; Cliquet, F.; Picard-Meyer, E.; Wasniewski, M.; Zrnčić, V.; Ćukušić, A.; et al. Viral Metagenomic Profiling of Croatian Bat Population Reveals Sample and Habitat Dependent Diversity. Viruses 2020, 12, 891. [Google Scholar] [CrossRef]
- Irving, A.T.; Ahn, M.; Goh, G.; Anderson, D.E.; Wang, L.-F. Lessons from the Host Defences of Bats, a Unique Viral Reservoir. Nature 2021, 589, 363–370. [Google Scholar] [CrossRef]
- Smith, I.; Wang, L.-F. Bats and Their Virome: An Important Source of Emerging Viruses Capable of Infecting Humans. Curr. Opin. Virol. 2013, 3, 84–91. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the Bat Virome Catalog to Better Understand the Ecological Diversity of Bat Viruses and the Bat Origin of Emerging Infectious Diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef]
- Nikolay, B.; Salje, H.; Hossain, M.J.; Khan, A.K.M.D.; Sazzad, H.M.S.; Rahman, M.; Daszak, P.; Ströher, U.; Pulliam, J.R.C.; Kilpatrick, A.M.; et al. Transmission of Nipah Virus—14 Years of Investigations in Bangladesh. N. Engl. J. Med. 2019, 380, 1804–1814. [Google Scholar] [CrossRef]
- Olival, K.; Hayman, D. Filoviruses in Bats: Current Knowledge and Future Directions. Viruses 2014, 6, 1759–1788. [Google Scholar] [CrossRef]
- Stoner-Duncan, B.; Streicker, D.G.; Tedeschi, C.M. Vampire Bats and Rabies: Toward an Ecological Solution to a Public Health Problem. PLoS Negl. Trop. Dis. 2014, 8, e2867. [Google Scholar] [CrossRef]
- Pernet, O.; Schneider, B.S.; Beaty, S.M.; LeBreton, M.; Yun, T.E.; Park, A.; Zachariah, T.T.; Bowden, T.A.; Hitchens, P.; Ramirez, C.M.; et al. Evidence for Henipavirus Spillover into Human Populations in Africa. Nat. Commun. 2014, 5, 5342. [Google Scholar] [CrossRef]
- Dovih, P.; Laing, E.D.; Chen, Y.; Low, D.H.W.; Ansil, B.R.; Yang, X.; Shi, Z.; Broder, C.C.; Smith, G.J.D.; Linster, M.; et al. Filovirus-Reactive Antibodies in Humans and Bats in Northeast India Imply Zoonotic Spillover. PLoS Negl. Trop. Dis. 2019, 13, e0007733. [Google Scholar] [CrossRef]
- Selvey, L.A.; Wells, R.M.; McCormack, J.G.; Ansford, A.J.; Murray, K.; Rogers, R.J.; Lavercombe, P.S.; Selleck, P.; Sheridan, J.W. Infection of Humans and Horses by a Newly Described Morbillivirus. Med. J. Aust. 1995, 162, 642–645. [Google Scholar] [CrossRef]
- Chua, K.B.; Chua, B.H.; Wang, C.W. Anthropogenic Deforestation, El Niño and the Emergence of Nipah Virus in Malaysia. Malays. J. Pathol. 2002, 24, 15–21. [Google Scholar]
- Snedden, C.E.; Makanani, S.K.; Schwartz, S.T.; Gamble, A.; Blakey, R.V.; Borremans, B.; Helman, S.K.; Espericueta, L.; Valencia, A.; Endo, A.; et al. SARS-CoV-2: Cross-Scale Insights from Ecology and Evolution. Trends Microbiol. 2021, 29, 593–605. [Google Scholar] [CrossRef]
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic Origins of Human Coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef]
- Fagre, A.C.; Kading, R.C. Can Bats Serve as Reservoirs for Arboviruses? Viruses 2019, 11, 215. [Google Scholar] [CrossRef]
- Vicente-Santos, A.; Moreira-Soto, A.; Soto-Garita, C.; Chaverri, L.G.; Chaves, A.; Drexler, J.F.; Morales, J.A.; AlfarAlarcón, A.; Rodríguez-Herrera, B.; Corrales-Aguilar, E. Neotropical Bats That Co-Habit with Humans Function as Dead-End Hosts for Dengue Virus. PLoS Negl. Trop. Dis. 2017, 11, e0005537. [Google Scholar] [CrossRef]
- de Thoisy, B.; Lacoste, V.; Germain, A.; Muñoz-Jordán, J.; Colón, C.; Mauffrey, J.-F.; Delaval, M.; Catzeflis, F.; Kazanji, M.; Matheus, S.; et al. Dengue Infection in Neotropical Forest Mammals. Vector-Borne Zoonotic Dis. 2009, 9, 157–170. [Google Scholar] [CrossRef]
- Del Ángel, R.M.; Recio-Tótoro, B.; Rodríguez-Moreno, Á.; Lanz, H.; Sánchez-Cordero, V.; Cabrera-Romo, S.; Ludert, J.E.; Alcalá, A.C. Experimental Inoculation of Artibeus Jamaicensis Bats with Dengue Virus Serotypes 1 or 4 Showed No Evidence of Sustained Replication. Am. J. Trop. Med. Hyg. 2014, 91, 1227–1234. [Google Scholar] [CrossRef]
- Perea-Martínez, L.; Moreno-Sandoval, H.N.; Moreno-Altamirano, M.M.; Salas-Rojas, M.; García-Flores, M.M.; Aréchiga-Ceballos, N.; Tordo, N.; Marianneau, P.; Aguilar-Setién, A. Experimental Infection of Artibeus Intermedius Bats with Serotype-2 Dengue Virus. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 193–198. [Google Scholar] [CrossRef]
- Van Brussel, K.; Holmes, E.C. Doença Zoonótica e Diversidade de Virome em Morcegos. Opinião Atual Em Virol. 2022, 52, 192–202. [Google Scholar] [CrossRef]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-Borne Virus Diversity, Spillover and Emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Subudhi, S.; Rapin, N.; Misra, V. Immune System Modulation and Viral Persistence in Bats: Understanding Viral Spillover. Viruses 2019, 11, 192. [Google Scholar] [CrossRef]
- Schountz, T.; Baker, M.L.; Butler, J.; Munster, V. Immunological Control of Viral Infections in Bats and the Emergence of Viruses Highly Pathogenic to Humans. Front. Immunol. 2017, 8, 1098. [Google Scholar] [CrossRef]
- Banerjee, A.; Baker, M.L.; Kulcsar, K.; Misra, V.; Plowright, R.; Mossman, K. Novel Insights Into Immune Systems of Bats. Front. Immunol. 2020, 11, 26. [Google Scholar] [CrossRef]
- Wang, L.-F.; Anderson, D.E. Viruses in Bats and Potential Spillover to Animals and Humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef]
- Han, B.A.; Schmidt, J.P.; Bowden, S.E.; Drake, J.M. Rodent Reservoirs of Future Zoonotic Diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 7039–7044. [Google Scholar] [CrossRef]
- Mollentze, N.; Streicker, D.G. Viral Zoonotic Risk Is Homogenous among Taxonomic Orders of Mammalian and Avian Reservoir Hosts. Proc. Natl. Acad. Sci. USA 2020, 117, 9423–9430. [Google Scholar] [CrossRef]
- Luis, A.D.; Hayman, D.T.S.; O’Shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.C.; Mills, J.N.; Timonin, M.E.; Willis, C.K.R.; Cunningham, A.A.; et al. A Comparison of Bats and Rodents as Reservoirs of Zoonotic Viruses: Are Bats Special? Proc. R. Soc. B. 2013, 280, 20122753. [Google Scholar] [CrossRef]
- Nieto-Rabiela, F.; Wiratsudakul, A.; Suzán, G.; Rico-Chávez, O. Viral Networks and Detection of Potential Zoonotic Viruses in Bats and Rodents: A Worldwide Analysis. Zoonoses Public Health 2019, 66, 655–666. [Google Scholar] [CrossRef]
- Brook, C.E.; Boots, M.; Chandran, K.; Dobson, A.P.; Drosten, C.; Graham, A.L.; Grenfell, B.T.; Müller, M.A.; Ng, M.; Wang, L.-F.; et al. Accelerated Viral Dynamics in Bat Cell Lines, with Implications for Zoonotic Emergence. eLife 2020, 9, e48401. [Google Scholar] [CrossRef]
- Bernstein, A.S.; Ando, A.W.; Loch-Temzelides, T.; Vale, M.M.; Li, B.V.; Li, H.; Busch, J.; Chapman, C.A.; Kinnaird, M.; Nowak, K.; et al. The Costs and Benefits of Primary Prevention of Zoonotic Pandemics. Sci. Adv. 2022, 8, eabl4183. [Google Scholar] [CrossRef]
- Keusch, G.T.; Amuasi, J.H.; Anderson, D.E.; Daszak, P.; Eckerle, I.; Field, H.; Koopmans, M.; Lam, S.K.; Das Neves, C.G.; Peiris, M.; et al. Pandemic Origins and a One Health Approach to Preparedness and Prevention: Solutions Based on SARS-CoV-2 and Other RNA Viruses. Proc. Natl. Acad. Sci. USA 2022, 119, e2202871119. [Google Scholar] [CrossRef]
- Souza, M.B.; de Souza Santos, L.R.; Borges, R.E.; Nunes, H.F.; Vieira, T.B.; Pacheco, S.M.; de Melo e Silva, D. Current Status of Ecotoxicological Studies of Bats in Brazil. Bull. Environ. Contam. Toxicol. 2020, 104, 393–399. [Google Scholar] [CrossRef]
- Nunes, H.; Rocha, F.L.; Cordeiro-Estrela, P. Bats in Urban Areas of Brazil: Roosts, Food Resources and Parasites in Disturbed Environments. Urban Ecosyst. 2017, 20, 953–969. [Google Scholar] [CrossRef]
- Souza, W.; Dennis, T.; Fumagalli, M.; Araujo, J.; Sabino-Santos, G.; Maia, F.; Acrani, G.; Carrasco, A.; Romeiro, M.; Modha, S.; et al. Novel Parvoviruses from Wild and Domestic Animals in Brazil Provide New Insights into Parvovirus Distribution and Diversity. Viruses 2018, 10, 143. [Google Scholar] [CrossRef]
- de Souza, W.M.; Romeiro, M.F.; Fumagalli, M.J.; Modha, S.; de Araujo, J.; Queiroz, L.H.; Durigon, E.L.; Figueiredo, L.T.M.; Murcia, P.R.; Gifford, R.J. Chapparvoviruses Occur in at Least Three Vertebrate Classes and Have a Broad Biogeographic Distribution. J. Gen. Virol. 2017, 98, 225–229. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Abreu, W.U.; Rodrigues, L.R.R. Diversidade Viral Em Morcegos No Brasil: Uma Revisão Sistemática Do Período 2000 a 2020/Viral Diversity in Bats in Brazil: A Systematic Review from 2000 to 2020. BJDV 2022, 8, 579–592. [Google Scholar] [CrossRef]
- Epstein, J.H.; Anthony, S.J.; Islam, A.; Kilpatrick, A.M.; Ali Khan, S.; Balkey, M.D.; Ross, N.; Smith, I.; Zambrana-Torrelio, C.; Tao, Y.; et al. Nipah Virus Dynamics in Bats and Implications for Spillover to Humans. Proc. Natl. Acad. Sci. USA 2020, 117, 29190–29201. [Google Scholar] [CrossRef]
- IUCN Molossus Molossus; Barquez, R.; Rodriguez, B.; Miller, B.; Diaz, M. The IUCN Red List of Threatened Species 2015: E.T13648A22106602; IUCN: Fontainebleau, France, 2015. [Google Scholar]
- Wilson, D.E.; Reeder, D.M. (Eds.) Mammal Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; ISBN 9780801882210. [Google Scholar]
- Li, Y.; Gordon, E.; Idle, A.; Altan, E.; Seguin, M.A.; Estrada, M.; Deng, X.; Delwart, E. Virome of a Feline Outbreak of Diarrhea and Vomiting Includes Bocaviruses and a Novel Chapparvovirus. Viruses 2020, 12, 506. [Google Scholar] [CrossRef]
- Abreu, W.U.; Rodrigues, L.R.R.; da Costa, A.C.; Morais, V.d.S.; de Paula, A.C.; Barreiros, E.A. Sequências Retrovirais de Interesse Em Medicina Veterinária Identificadas Em Morcegos Urbanos Da Amazônia. RSD 2022, 11, e394111032993. [Google Scholar] [CrossRef]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An Ensemble Strategy That Significantly Improves de Novo Assembly of Microbial Genomes from Metagenomic Next-Generation Sequencing Data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef]
- Katoh, K. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A Multiple Sequence Alignment Method with Reduced Time and Space Complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Strimmer, K.; von Haeseler, A. Likelihood-Mapping: A Simple Method to Visualize Phylogenetic Content of a Sequence Alignment. Proc. Natl. Acad. Sci. USA 1997, 94, 6815–6819. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Pénzes, J.J.; de Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. An Ancient Lineage of Highly Divergent Parvoviruses Infects Both Vertebrate and Invertebrate Hosts. Viruses 2019, 11, 525. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Mietzsch, M.; Pénzes, J.J.; Agbandje-McKenna, M. Twenty-Five Years of Structural Parvovirology. Viruses 2019, 11, 362. [Google Scholar] [CrossRef]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the Family Parvoviridae: A Revised Taxonomy Independent of the Canonical Approach Based on Host Association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; Garcia, M.L.; Curtis Hendrickson, R.; et al. Recent Changes to Virus Taxonomy Ratified by the International Committee on Taxonomy of Viruses. Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Z.; Wang, Y.; Li, W.; Fu, X.; Lin, Y.; Shen, Q.; Wang, X.; Wang, H.; Zhang, W. A Novel Rodent Chapparvovirus in Feces of Wild Rats. Virol. J. 2016, 13, 133. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Klaassen, M.; Bhatta, T.R.; Alexandersen, S. Metagenomic Characterization of Avian Parvoviruses and Picornaviruses from Australian Wild Ducks. Sci. Rep. 2020, 10, 12800. [Google Scholar] [CrossRef]
- Canuti, M.; Verhoeven, J.T.P.; Munro, H.J.; Roul, S.; Ojkic, D.; Robertson, G.J.; Whitney, H.G.; Dufour, S.C.; Lang, A.S. Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis. Viruses 2021, 13, 193. [Google Scholar] [CrossRef]
- Sarker, S. Molecular and Phylogenetic Characterization of a Highly Divergent Novel Parvovirus (Psittaciform Chaphamaparvovirus 2) in Australian Neophema Parrots. Pathogens 2021, 10, 1559. [Google Scholar] [CrossRef]
- Chang, W.-S.; Li, C.-X.; Hall, J.; Eden, J.-S.; Hyndman, T.H.; Holmes, E.C.; Rose, K. Meta-Transcriptomic Discovery of a Divergent Circovirus, and a Chaphamaparvovirus in Captive Reptiles with Proliferative Respiratory Syndrome. Viruses 2020, 12, 1073. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, A.; Delwart, E.; Pankovics, P. Novel Circular Single-Stranded DNA Virus from Turkey Faeces. Arch. Virol. 2014, 159, 2161–2164. [Google Scholar] [CrossRef]
- Palinski, R.M.; Mitra, N.; Hause, B.M. Discovery of a Novel Parvovirinae Virus, Porcine Parvovirus 7, by Metagenomic Sequencing of Porcine Rectal Swabs. Virus Genes 2016, 52, 564–567. [Google Scholar] [CrossRef]
- Du, J.; Wang, W.; Chan, J.F.-W.; Wang, G.; Huang, Y.; Yi, Y.; Zhu, Z.; Peng, R.; Hu, X.; Wu, Y.; et al. Identification of a Novel Ichthyic Parvovirus in Marine Species in Hainan Island, China. Front. Microbiol. 2019, 10, 2815. [Google Scholar] [CrossRef]
- Abayli, H.; Can-Sahna, K. First Detection of Feline Bocaparvovirus 2 and Feline Chaphamaparvovirus in Healthy Cats in Turkey. Vet. Res. Commun. 2022, 46, 127–136. [Google Scholar] [CrossRef]
- Di Profio, F.; Sarchese, V.; Palombieri, A.; Fruci, P.; Massirio, I.; Martella, V.; Fulvio, M.; Di Martino, B. Feline Chaphamaparvovirus in Cats with Enteritis and Upper Respiratory Tract Disease. Transbounding Emerg. Dis. 2022, 69, 660–668. [Google Scholar] [CrossRef]
- Fahsbender, E.; da Costa, A.C.; Gill, D.E.; Milagres, P.F.A.; Brustulin, R.; Monteiro, F.J.C.; Rego, M.O.d.S.; Ribeiro, E.S.D.; Sabino, E.C.; Delwart, E. Plasma Virome of 781 Brazilians with Unexplained Symptoms of Arbovirus Infection Include a Novel Parvovirus and Densovirus. PLoS ONE 2020, 15, e0229993. [Google Scholar] [CrossRef]
- Finoketti, F.; dos Santos, R.N.; Campos, A.A.S.; Zani, A.L.d.S.; Barboza, C.M.; Fernandes, M.E.S.; de Souza, T.d.C.P.; dos Santos, D.D.; Bortolanza, G.W.; Filho, H.O.; et al. Detection of Adenovirus, Papillomavirus and Parvovirus in Brazilian Bats of the Species Artibeus Lituratus and Sturnira Lilium. Arch. Virol. 2019, 164, 1015–1025. [Google Scholar] [CrossRef]
- Van de Vuurst, P.; Diaz, M.M.; Rodríguez-San Pedro, A.; Allendes, J.L.; Brown, N.; Gutierrez, J.D.; Zarza, H.; Vilges de Oliveira, S.; Cárdenas-Canales, E.; Barquez, R.; et al. Desmodus Rotundus Occurrence Record Database. 2022. Available online: https://figshare.com/articles/dataset/Desmodus_rotundus_Occurrence_Record_Database/15025296 (accessed on 9 February 2023).
- IUCN Artibeus Lituratus; Barquez, R.; Perez, S.; Miller, B.; Diaz, M. The IUCN Red List of Threatened Species 2015: E.T2136A21995720; IUCN: Fontainebleau, France, 2015. [Google Scholar]
- Leal, É.; Liang, R.; Liu, Q.; Villanova, F.; Shi, L.; Liang, L.; Li, J.; Witkin, S.S.; Cui, S. Regional Adaptations and Parallel Mutations in Feline Panleukopenia Virus Strains from China Revealed by Nearly-Full Length Genome Analysis. PLoS ONE 2020, 15, e0227705. [Google Scholar] [CrossRef]
- Pereira, C.A.D.; Leal, É.S.; Durigon, É.L. Selective Regimen Shift and Demographic Growth Increase Associated with the Emergence of High-Fitness Variants of Canine Parvovirus. Infect. Genet. Evol. 2007, 7, 399–409. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Size (Base Pairs) | Coverage | Nucleotide Identity | E-Value |
|---|---|---|---|---|
| CHPV_O08 | 4284 | 89% | 78.71% | 0.0 |
| CHPV_B08 | 3797 | 89% | 80.19% | 0.0 |
| CHPV_R04 | 3800 | 91% | 79.61% | 0.0 |
| CHPV_F01 | 4281 | 90% | 78.91% | 0.0 |
| CHPV_R02 | 4001 | 90% | 79.54% | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, E.d.S.F.; Abreu, W.U.; Rodrigues, L.R.R.; Marinho, L.F.; Morais, V.d.S.; Villanova, F.; Pandey, R.P.; Araújo, E.L.L.; Deng, X.; Delwart, E.; et al. Novel Chaphamaparvovirus in Insectivorous Molossus molossus Bats, from the Brazilian Amazon Region. Viruses 2023, 15, 606. https://doi.org/10.3390/v15030606
Ramos EdSF, Abreu WU, Rodrigues LRR, Marinho LF, Morais VdS, Villanova F, Pandey RP, Araújo ELL, Deng X, Delwart E, et al. Novel Chaphamaparvovirus in Insectivorous Molossus molossus Bats, from the Brazilian Amazon Region. Viruses. 2023; 15(3):606. https://doi.org/10.3390/v15030606
Chicago/Turabian StyleRamos, Endrya do Socorro Foro, Wandercleyson Uchôa Abreu, Luis Reginaldo Ribeiro Rodrigues, Luis Fernando Marinho, Vanessa dos Santos Morais, Fabiola Villanova, Ramendra Pati Pandey, Emerson Luiz Lima Araújo, Xutao Deng, Eric Delwart, and et al. 2023. "Novel Chaphamaparvovirus in Insectivorous Molossus molossus Bats, from the Brazilian Amazon Region" Viruses 15, no. 3: 606. https://doi.org/10.3390/v15030606
APA StyleRamos, E. d. S. F., Abreu, W. U., Rodrigues, L. R. R., Marinho, L. F., Morais, V. d. S., Villanova, F., Pandey, R. P., Araújo, E. L. L., Deng, X., Delwart, E., da Costa, A. C., & Leal, E. (2023). Novel Chaphamaparvovirus in Insectivorous Molossus molossus Bats, from the Brazilian Amazon Region. Viruses, 15(3), 606. https://doi.org/10.3390/v15030606