
Web Service for HIV Drug Resistance Prediction Based on Analysis of Amino Acid Substitutions in Main Drug Targets
 , , ,
, , ,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Data Processing
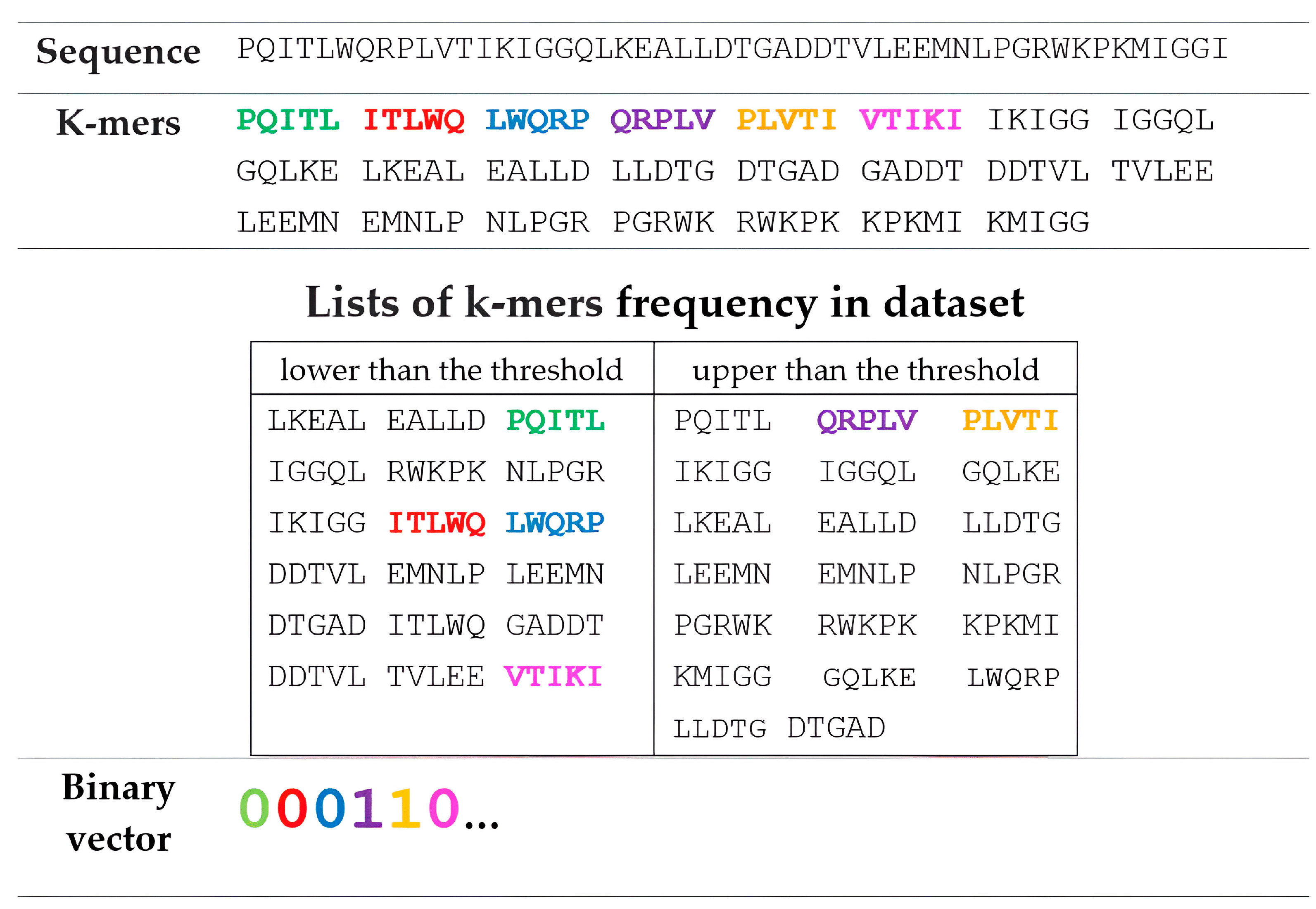
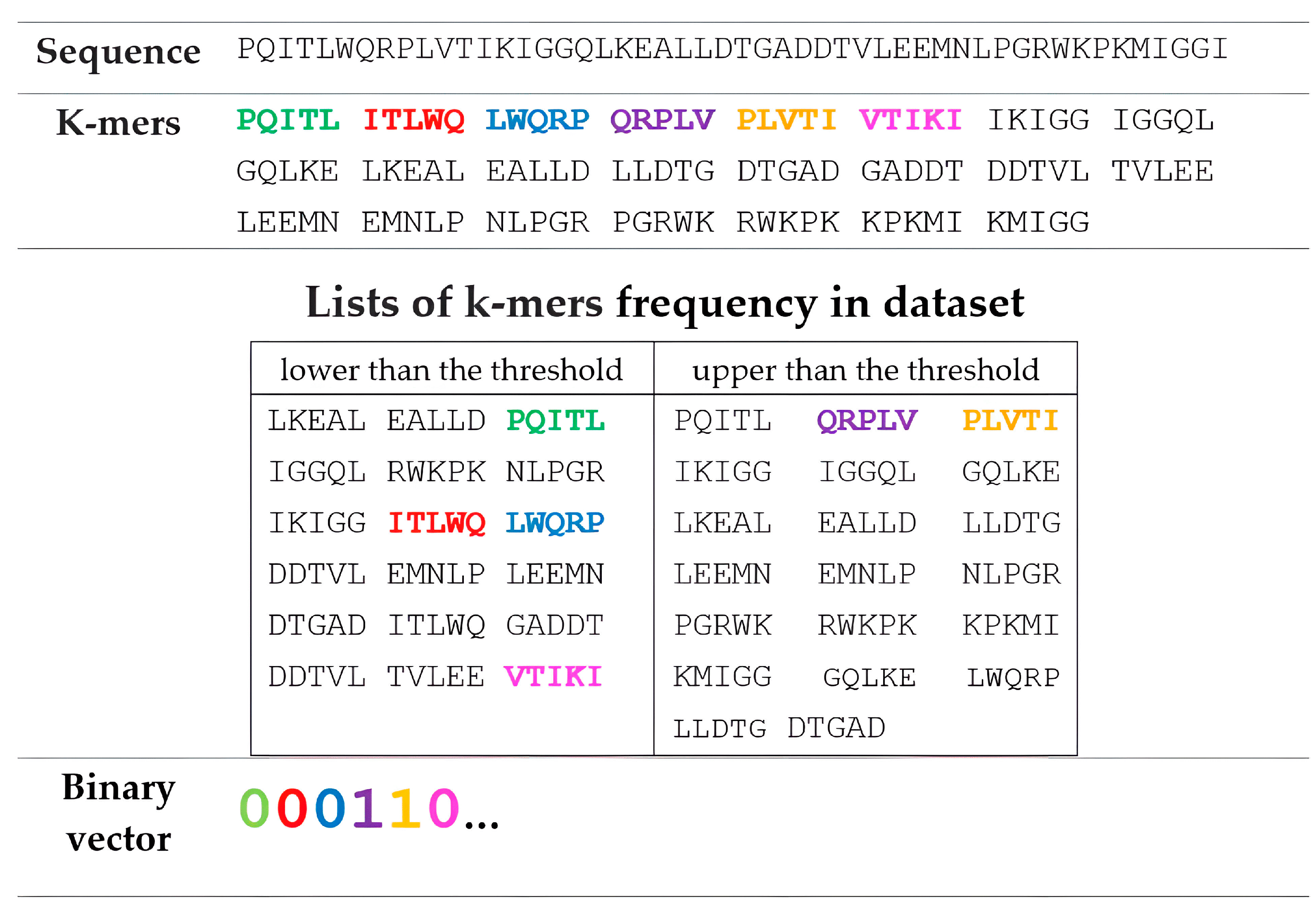
2.2. Representation of Amino Acid Sequences Based on k-mer and String Muti-n-Grams Descriptors
2.3. Model Development and Evaluation
3. Results and Discussion
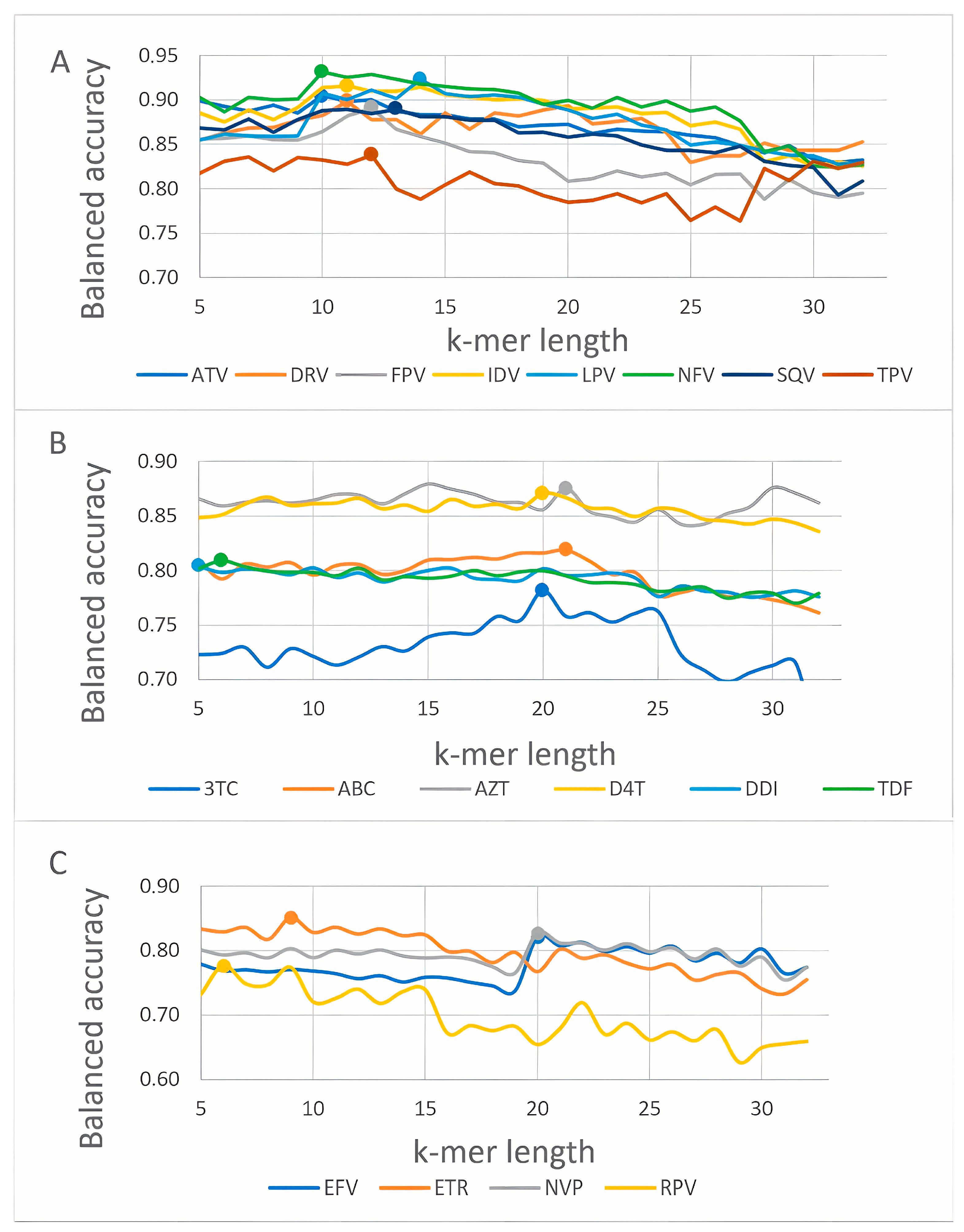
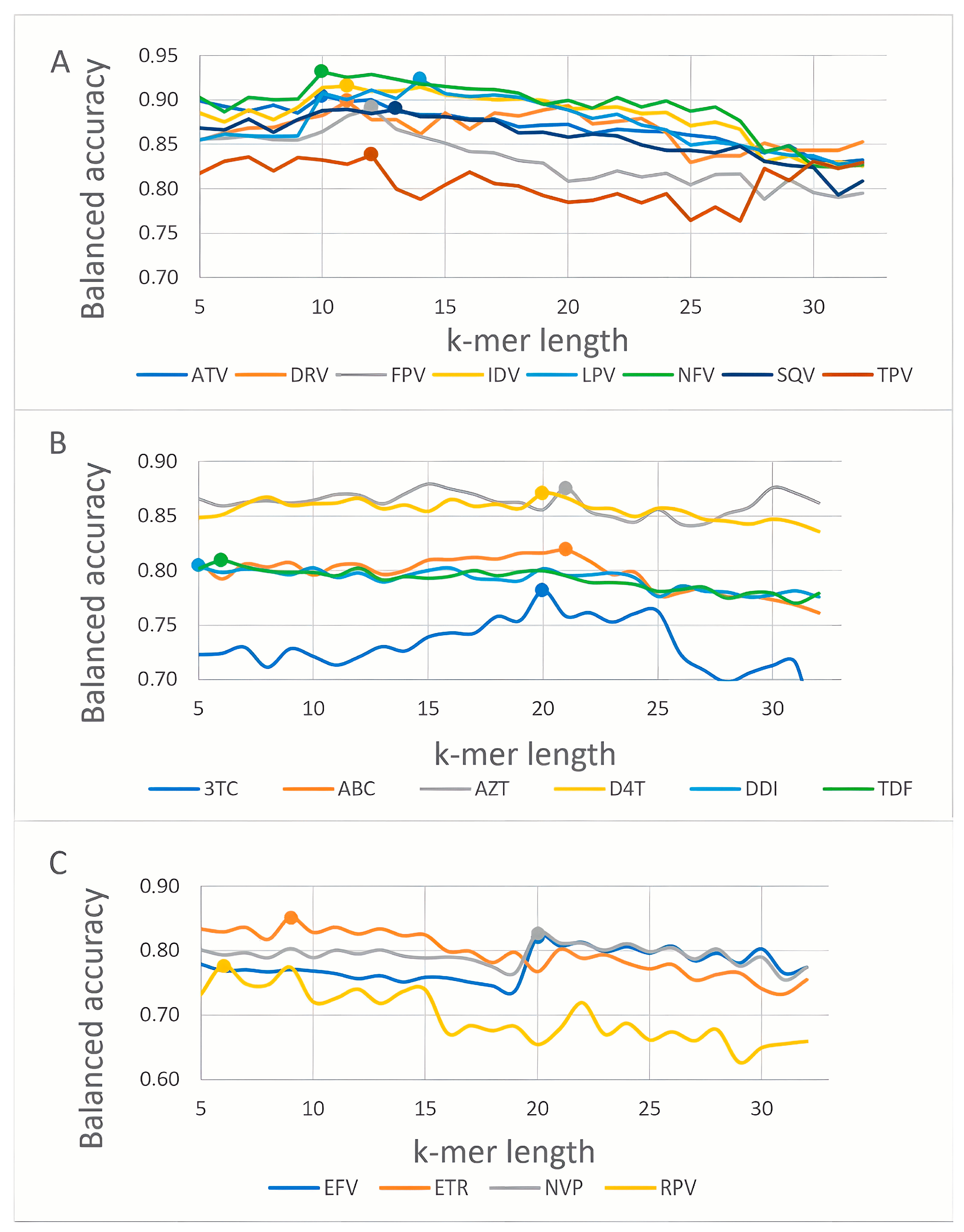
3.1. K-mer Based Model Evaluation
3.2. String Multi-n-Gram Descriptors for HIV-Resistance Prediction
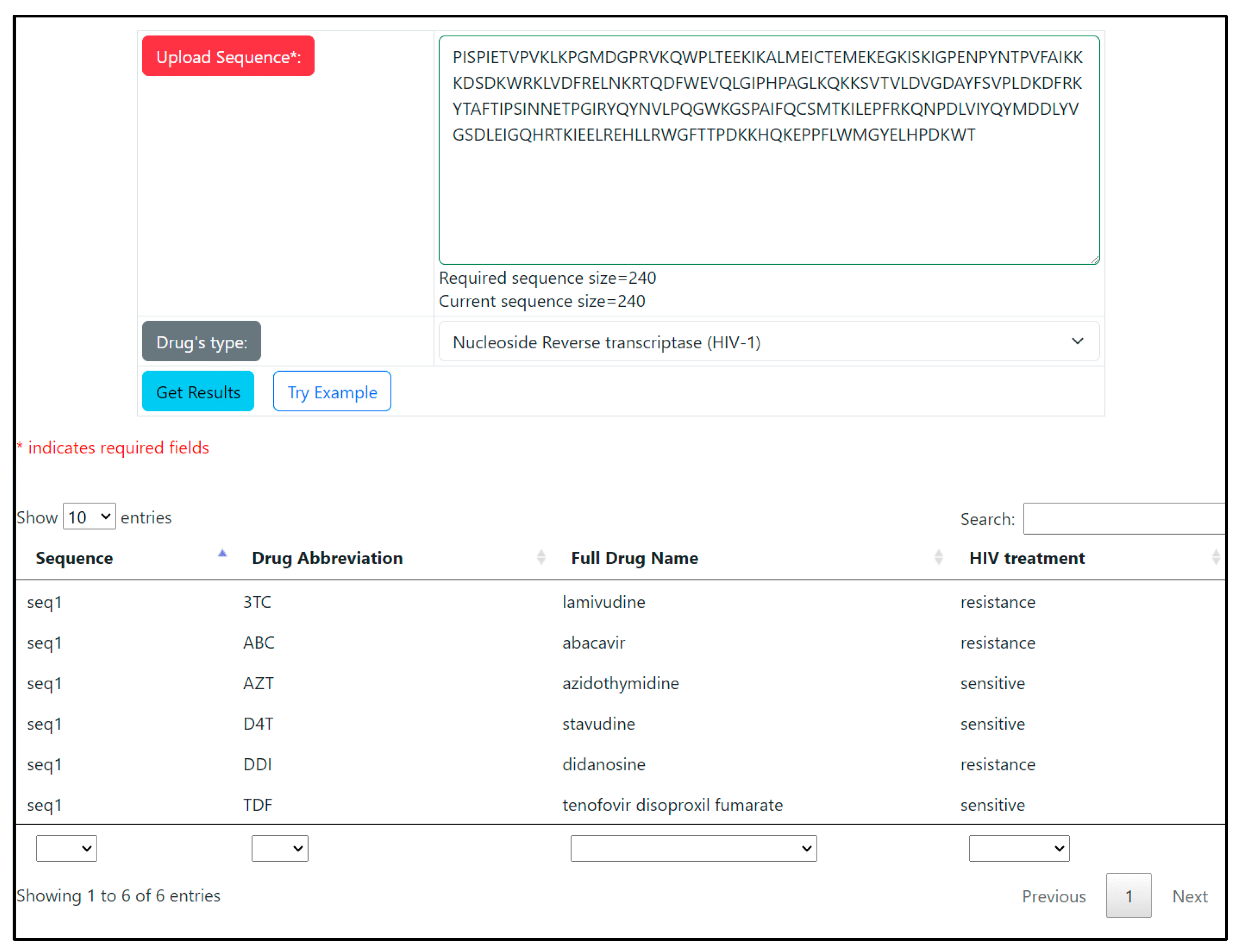
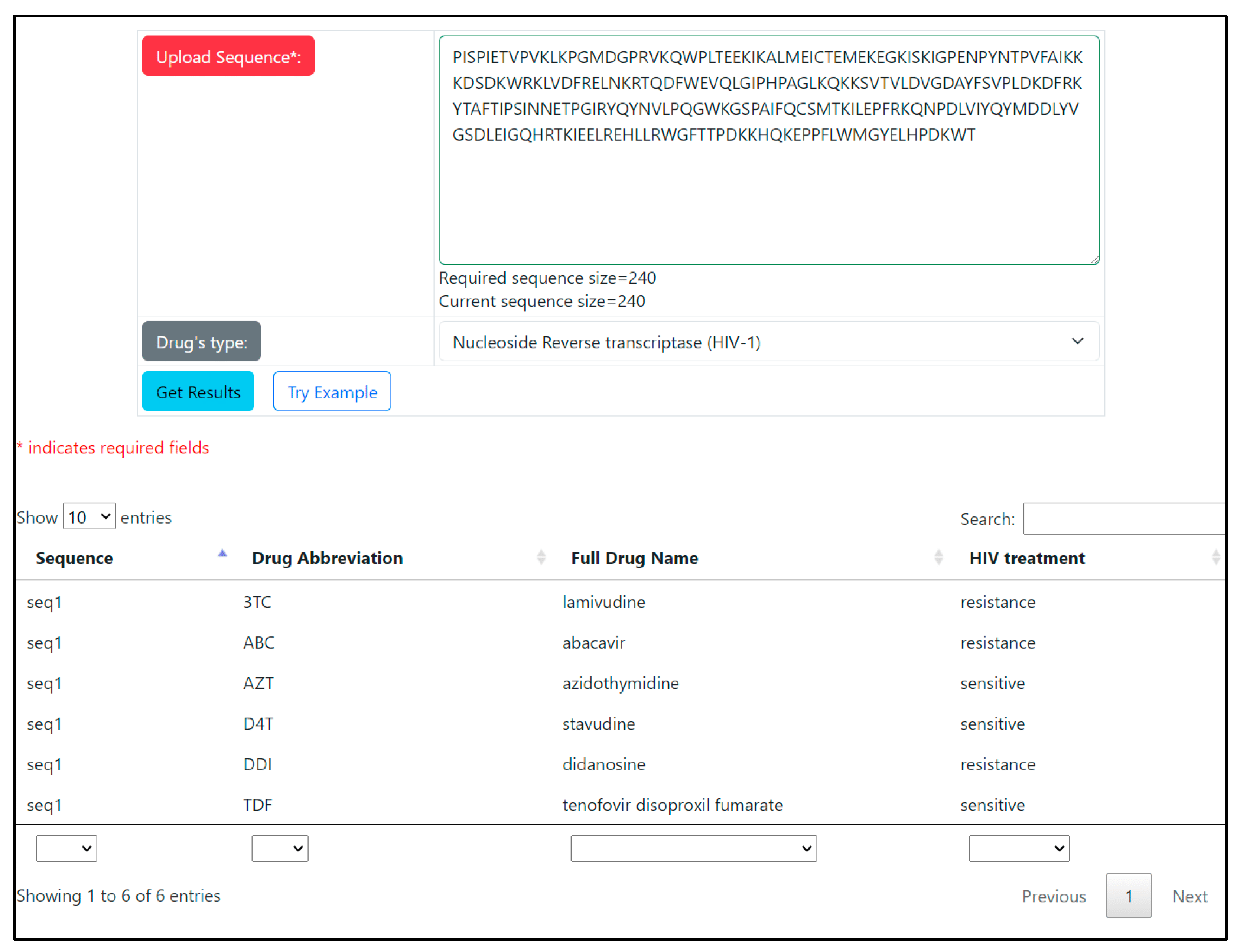
3.3. WEB-Application
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, W.-L.; Kouyos, R.; Scherrer, A.U.; Böni, J.; Shah, C.; Yerly, S.; Klimkait, T.; Aubert, V.; Furrer, H.; Battegay, M.; et al. Assessing the Paradox Between Transmitted and Acquired HIV Type 1 Drug Resistance Mutations in the Swiss HIV Cohort Study From 1998 to 2012. J. Infect. Dis. 2015, 212, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Gregson, J.; Parkin, N.; Haile-Selassie, H.; Tanuri, A.; Andrade Forero, L.; Kaleebu, P.; Watera, C.; Aghokeng, A.; Mutenda, N.; et al. HIV-1 Drug Resistance before Initiation or Re-Initiation of First-Line Antiretroviral Therapy in Low-Income and Middle-Income Countries: A Systematic Review and Meta-Regression Analysis. Lancet Infect. Dis. 2018, 18, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolio, S.; Jordan, M.R.; Giron, A.; Inzaule, S. Epidemiology of HIV Drug Resistance in Low- and Middle-Income Countries and WHO Global Strategy to Monitor Its Emergence. Curr. Opin. HIV AIDS 2022, 17, 229–239. [Google Scholar] [CrossRef]
- Shtrek, S.; Levakhina, L.; Blokh, A.; Pasechnik, O.; Pen’evskaya, N. Prevalence and Spectrum of HIV-1 Resistance Mutations in the Siberian Federal District. Viruses 2022, 14, 2117. [Google Scholar] [CrossRef]
- Khan, F.; Bilal, M.; Khan, M.Y.; Fareezuddin, M. Frequency of Resistance to First-Line Antiretroviral Therapy Observed among Hiv Patients. Pak. J. Med. Sci. 2022, 38, 2011–2015. [Google Scholar] [CrossRef]
- Wei, Q.; Zhao, Y.; Lv, Y.; Kang, X.; Pan, S.; Yao, S.; Wang, L. High Rate of HIV-1 Drug Resistance in Antiretroviral Therapy-Failure Patients in Liaoning Province, China. AIDS Res. Hum. Retrovir. 2022, 38, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.N.; Stover, J.; Cambiano, V.; Nakagawa, F.; Jordan, M.R.; Pillay, D.; Doherty, M.; Revill, P.; Bertagnolio, S. Impact of HIV Drug Resistance on HIV/AIDS-Associated Mortality, New Infections, and Antiretroviral Therapy Program Costs in Sub–Saharan Africa. J. Infect. Dis. 2017, 215, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Hamers, R.L.; Sigaloff, K.C.E.; Kityo, C.; Mugyenyi, P.; De Wit, T.F.R. Emerging HIV-1 Drug Resistance after Roll-out of Antiretroviral Therapy in Sub-Saharan Africa. Curr. Opin. HIV AIDS 2013, 8, 19–26. [Google Scholar] [CrossRef]
- Agwu, A.L.; Fairlie, L. Antiretroviral Treatment, Management Challenges and Outcomes in Perinatally HIV-Infected Adolescents. J. Int. AIDS Soc. 2013, 16, 18579. [Google Scholar] [CrossRef]
- Fitzgerald, F.; Penazzato, M.; Gibb, D. Development of Antiretroviral Resistance in Children with HIV in Low- and Middle-Income Countries. J. Infect. Dis. 2013, 207, S85–S92. [Google Scholar] [CrossRef]
- Mukhatayeva, A.; Mustafa, A.; Dzissyuk, N.; Issanov, A.; Mukhatayev, Z.; Bayserkin, B.; Vermund, S.H.; Ali, S. Antiretroviral Therapy Resistance Mutations among HIV Infected People in Kazakhstan. Sci. Rep. 2022, 12, 17195. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Samudrala, R.; Mittler, J.E. Antivirogram or Phenosense: A Comparison of Their Reproducibility and an Analysis of Their Correlation. Antivir. Ther. 2004, 9, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Geretti, A.M.; Easterbrook, P. Antiretroviral Resistance in Clinical Practice. Int. J. STD AIDS 2001, 12, 145–153. [Google Scholar] [CrossRef]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of Acquired Immunodeficiency Syndrome-Associated Retrovirus in Human and Nonhuman Cells Transfected with an Infectious Molecular Clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Yoon, H.; Abfalterer, W.; Gaschen, B.; Lo, C.; Korber, B. Viral Genome Analysis and Knowledge Management. Methods Mol. Biol. 2013, 939, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, B.; Incardona, F.; Di Teodoro, G.; Mommo, C.; Saladini, F.; Kaiser, R.; Sönnerborg, A.; Lengauer, T.; Zazzi, M.; EuResist Network. Cohort Profile: A European Multidisciplinary Network for the Fight against HIV Drug Resistance (EuResist Network). Trop. Med. Infect. Dis. 2023, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.-Y. Human Immunodeficiency Virus Reverse Transcriptase and Protease Sequence Database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef]
- Gartland, M.; Arnoult, E.; Foley, B.T.; Lataillade, M.; Ackerman, P.; Llamoso, C.; Krystal, M. Prevalence of Gp160 Polymorphisms Known to Be Related to Decreased Susceptibility to Temsavir in Different Subtypes of HIV-1 in the Los Alamos National Laboratory HIV Sequence Database. J. Antimicrob. Chemother. 2021, 76, 2958–2964. [Google Scholar] [CrossRef]
- Valadés-Alcaraz, A.; Reinosa, R.; Holguín, Á. HIV Transmembrane Glycoprotein Conserved Domains and Genetic Markers Across HIV-1 and HIV-2 Variants. Front. Microbiol. 2022, 13, 855232. [Google Scholar] [CrossRef]
- Sutar, J.; Deshpande, S.; Mullick, R.; Hingankar, N.; Patel, V.; Bhattacharya, J. Geospatial HIV-1 Subtype C Gp120 Sequence Diversity and Its Predicted Impact on Broadly Neutralizing Antibody Sensitivity. PLoS ONE 2021, 16, e0251969. [Google Scholar] [CrossRef]
- de Oliveira, T.; Deforche, K.; Cassol, S.; Salminen, M.; Paraskevis, D.; Seebregts, C.; Snoeck, J.; van Rensburg, E.J.; Wensing, A.M.J.; van de Vijver, D.A.; et al. An Automated Genotyping System for Analysis of HIV-1 and Other Microbial Sequences. Bioinformatics 2005, 21, 3797–3800. [Google Scholar] [CrossRef] [PubMed]
- Zazzi, M.; Incardona, F.; Rosen-Zvi, M.; Prosperi, M.; Lengauer, T.; Altmann, A.; Sonnerborg, A.; Lavee, T.; Schülter, E.; Kaiser, R. Predicting Response to Antiretroviral Treatment by Machine Learning: The EuResist Project. Intervirology 2012, 55, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Pironti, A.; Pfeifer, N.; Walter, H.; Jensen, B.-E.O.; Zazzi, M.; Gomes, P.; Kaiser, R.; Lengauer, T. Using Drug Exposure for Predicting Drug Resistance—A Data-Driven Genotypic Interpretation Tool. PLoS ONE 2017, 12, e0174992. [Google Scholar] [CrossRef]
- Meynard, J.-L.; Vray, M.; Morand-Joubert, L.; Race, E.; Descamps, D.; Peytavin, G.; Matheron, S.; Lamotte, C.; Guiramand, S.; Costagliola, D.; et al. Phenotypic or Genotypic Resistance Testing for Choosing Antiretroviral Therapy after Treatment Failure: A Randomized Trial. AIDS 2002, 16, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, O.; Poroikov, V. HIV Resistance Prediction to Reverse Transcriptase Inhibitors: Focus on Open Data. Molecules 2018, 23, 956. [Google Scholar] [CrossRef] [PubMed]
- Ramon, E.; Belanche-Muñoz, L.; Pérez-Enciso, M. HIV Drug Resistance Prediction with Weighted Categorical Kernel Functions. BMC Bioinform. 2019, 20, 410. [Google Scholar] [CrossRef]
- Cai, Q.; Yuan, R.; He, J.; Li, M.; Guo, Y. Predicting HIV Drug Resistance Using Weighted Machine Learning Method at Target Protein Sequence-Level. Mol. Divers. 2021, 25, 1541–1551. [Google Scholar] [CrossRef]
- Ota, R.; So, K.; Tsuda, M.; Higuchi, Y.; Yamashita, F. Prediction of HIV Drug Resistance Based on the 3D Protein Structure: Proposal of Molecular Field Mapping. PLoS ONE 2021, 16, e0255693. [Google Scholar] [CrossRef]
- Tunc, H.; Dogan, B.; Darendeli Kiraz, B.N.; Sari, M.; Durdagi, S.; Kotil, S. Prediction of HIV-1 Protease Resistance Using Genotypic, Phenotypic, and Molecular Information with Artificial Neural Networks. PeerJ 2023, 11, e14987. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Däumer, M.; Oette, M.; Korn, K.; Hoffmann, D.; Kaiser, R.; Lengauer, T.; Selbig, J.; Walter, H. Geno2pheno: Estimating Phenotypic Drug Resistance from HIV-1 Genotypes. Nucleic Acids Res. 2003, 31, 3850–3855. [Google Scholar] [CrossRef]
- Tang, M.W.; Liu, T.F.; Shafer, R.W. The HIVdb System for HIV-1 Genotypic Resistance Interpretation. Intervirology 2012, 55, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.; Hummel, T.; Heider, D. SHIVA—A Web Application for Drug Resistance and Tropism Testing in HIV. BMC Bioinform. 2016, 17, 314. [Google Scholar] [CrossRef] [PubMed]
- Pikalyova, K.; Orlov, A.; Lin, A.; Tarasova, O.; Marcou, M.G.; Horvath, D.; Poroikov, V.; Varnek, A. HIV-1 Drug Resistance Profiling Using Amino Acid Sequence Space Cartography. Bioinformatics 2022, 38, 2307–2314. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, O.; Poroikov, V. Machine Learning in Discovery of New Antivirals and Optimization of Viral Infections Therapy. Curr. Med. Chem. 2021, 28, 7840–7861. [Google Scholar] [CrossRef]
- Steiner, M.C.; Gibson, K.M.; Crandall, K.A. Drug Resistance Prediction Using Deep Learning Techniques on HIV-1 Sequence Data. Viruses 2020, 12, 560. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Yu, X.; Harrison, R.W.; Weber, I.T. Automated Prediction of HIV Drug Resistance from Genotype Data. BMC Bioinform. 2016, 17, 278. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, O.; Biziukova, N.; Filimonov, D.; Poroikov, V. A Computational Approach for the Prediction of HIV Resistance Based on Amino Acid and Nucleotide Descriptors. Molecules 2018, 23, 2751. [Google Scholar] [CrossRef]
- Riemenschneider, M.; Heider, D. Current Approaches in Computational Drug Resistance Prediction in HIV. Curr. HIV Res. 2016, 14, 307–315. [Google Scholar] [CrossRef]
- Lengauer, T.; Sander, O.; Sierra, S.; Thielen, A.; Kaiser, R. Bioinformatics Prediction of HIV Coreceptor Usage. Nat. Biotechnol. 2007, 25, 1407–1410. [Google Scholar] [CrossRef]
- Döring, M.; Büch, J.; Friedrich, G.; Pironti, A.; Kalaghatgi, P.; Knops, E.; Heger, E.; Obermeier, M.; Däumer, M.; Thielen, A.; et al. Geno2pheno[Ngs-Freq]: A Genotypic Interpretation System for Identifying Viral Drug Resistance Using next-Generation Sequencing Data. Nucleic Acids Res. 2018, 46, W271–W277. [Google Scholar] [CrossRef]
- Tarasova, O.A.; Rudik, A.V.; Biziukova, N.Y.; Filimonov, D.A.; Poroikov, V.V. Chemical Named Entity Recognition in the Texts of Scientific Publications Using the Naïve Bayes Classifier Approach. J. Cheminform. 2022, 14, 55. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Guenther, N.; Schonlau, M. Support Vector Machines. Stata J. 2016, 16, 917–937. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of Activity Spectra for Biologically Active Substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Perryman, A.L.; Lin, J.-H.; McCammon, J.A. HIV-1 Protease Molecular Dynamics of a Wild-Type and of the V82F/I84V Mutant: Possible Contributions to Drug Resistance and a Potential New Target Site for Drugs. Protein Sci. 2004, 13, 1108–1123. [Google Scholar] [CrossRef] [PubMed]
- Young, T.P.; Parkin, N.T.; Stawiski, E.; Pilot-Matias, T.; Trinh, R.; Kempf, D.J.; Norton, M. Prevalence, Mutation Patterns, and Effects on Protease Inhibitor Susceptibility of the L76V Mutation in HIV-1 Protease. Antimicrob. Agents Chemother. 2010, 54, 4903–4906. [Google Scholar] [CrossRef]
- Dubois, N.; Khoo, K.K.; Ghossein, S.; Seissler, T.; Wolff, P.; McKinstry, W.J.; Mak, J.; Paillart, J.-C.; Marquet, R.; Bernacchi, S. The C-Terminal P6 Domain of the HIV-1 Pr55 Gag Precursor Is Required for Specific Binding to the Genomic RNA. RNA Biol. 2018, 15, 923–936. [Google Scholar] [CrossRef]
- Yu, F.-H.; Huang, K.-J.; Wang, C.-T. HIV-1 Mutant Assembly, Processing and Infectivity Expresses Pol Independent of Gag. Viruses 2020, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Taylor, J.; Wadhera, G.; Ben-Hur, A.; Brutlag, D.L.; Shafer, R.W. Genotypic Predictors of Human Immunodeficiency Virus Type 1 Drug Resistance. Proc. Natl. Acad. Sci. USA 2006, 103, 17355–17360. [Google Scholar] [CrossRef] [PubMed]
- Heider, D.; Verheyen, J.; Hoffmann, D. Machine Learning on Normalized Protein Sequences. BMC Res. Notes 2011, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, T.; Sing, T. Bioinformatics-Assisted Anti-HIV Therapy. Nat. Rev. Microbiol. 2006, 4, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Druzhilovskiy, D.S.; Rudik, A.V.; Filimonov, D.A.; Gloriozova, T.A.; Lagunin, A.A.; Dmitriev, A.V.; Pogodin, P.V.; Dubovskaya, V.I.; Ivanov, S.M.; Tarasova, O.A.; et al. Computational Platform Way2Drug: From the Prediction of Biological Activity to Drug Repurposing. Russ. Chem. Bull. 2017, 66, 1832–1841. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of Common Molecular Subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Drug | FR Threshold | Susceptible (S) | Resistant (R) | R/S 1 |
|---|---|---|---|---|---|
| PI | Fosamprenavir (FPV) | 4 | 1295 | 891 | 0.69 |
| Atazanavir (ATV) | 3 | 804 | 649 | 0.81 | |
| Indinavir (IDV) | 3 | 1231 | 1003 | 0.81 | |
| Lopinavir (LPV) | 9 | 1069 | 853 | 0.80 | |
| Nelfinavir (NFV) | 3 | 1422 | 874 | 0.62 | |
| Saquinavir (SQV) | 3 | 1297 | 960 | 0.74 | |
| Tipranavir (TPV) | 2 | 731 | 396 | 0.54 | |
| Darunavir (DRV) | 10 | 697 | 130 | 0.19 | |
| NRTI | Lamivudine (3TC) | 3 | 624 | 1417 | 2.27 |
| Abacavir (ABC) | 3 | 806 | 1156 | 1.43 | |
| Zidovudine (AZT) | 3 | 1068 | 1014 | 0.94 | |
| Stavudine (D4T) | 1.5 | 1095 | 1000 | 0.91 | |
| Didanosine (DDI) | 1.5 | 947 | 1148 | 1.21 | |
| Tenofovir (TDF) | 1.5 | 1164 | 544 | 0.46 | |
| NNRTI | Efavirenz (EFV) | 3 | 1072 | 995 | 0.93 |
| Nevirapine (NVP) | 3 | 874 | 1181 | 1.35 | |
| Etravirine (ETR) | 3 | 382 | 172 | 0.45 | |
| Rilpivirine (RPV) | 3 | 121 | 88 | 0.73 |
| Drug | k-mer Length | BA | Se | Sp | Pr | AUC | Method |
|---|---|---|---|---|---|---|---|
| Protease inhibitors | |||||||
| Fosamprenavir (FPV) | 12 | 0.89 | 0.88 | 0.90 | 0.86 | 0.95 | RF |
| Atazanavir (ATV) | 10 | 0.90 | 0.93 | 0.88 | 0.90 | 0.96 | SVM |
| Indinavir (IDV) | 11 | 0.92 | 0.92 | 0.91 | 0.91 | 0.96 | SVM |
| Lopinavir (LPV) | 14 | 0.91 | 0.92 | 0.91 | 0.89 | 0.96 | SVM |
| Nelfinavir (NFV) | 10 | 0.93 | 0.95 | 0.91 | 0.94 | 0.97 | SVM |
| Saquinavir (SQV) | 13 | 0.89 | 0.89 | 0.89 | 0.85 | 0.96 | SVM |
| Tipranavir (TPV) | 12 | 0.84 | 0.78 | 0.89 | 0.81 | 0.92 | RF |
| Darunavir (DRV) | 11 | 0.89 | 0.82 | 0.96 | 0.81 | 0.96 | SVM |
| Nucleoside reverse transcriptase inhibitors | |||||||
| Lamivudine (3TC) | 20 | 0.78 | 0.88 | 0.69 | 0.87 | 0.86 | SVM |
| Abacavir (ABC) | 21 | 0.82 | 0.88 | 0.76 | 0.84 | 0.90 | RF |
| Zidovudine (AZT) | 21 | 0.88 | 0.90 | 0.86 | 0.86 | 0.95 | RF |
| Stavudine (D4T) | 20 | 0.87 | 0.88 | 0.86 | 0.85 | 0.94 | RF |
| Didanosine (DDI) | 5 | 0.80 | 0.83 | 0.77 | 0.81 | 0.88 | RF |
| Tenofovir (TDF) | 6 | 0.81 | 0.70 | 0.92 | 0.80 | 0.90 | RF |
| Non-nucleoside reverse transcriptase inhibitors | |||||||
| Efavirenz (EFV) | 20 | 0.82 | 0.77 | 0.87 | 0.82 | 0.88 | SVM |
| Nevirapine (NVP) | 20 | 0.82 | 0.82 | 0.83 | 0.84 | 0.89 | SVM |
| Etravirine (ETR) | 9 | 0.84 | 0.74 | 0.95 | 0.87 | 0.94 | SVM |
| Rilpivirine (RPV) | 6 | 0.75 | 0.66 | 0.83 | 0.77 | 0.85 | RF |
| Drug | String Length | BA | Se | Sp | Pr | AUC |
|---|---|---|---|---|---|---|
| Protease inhibitors | ||||||
| Fosamprenavir (FPV) | 15 | 0.89 | 0.89 | 0.89 | 0.82 | 0.96 |
| Atazanavir (ATV) | 15 | 0.83 | 0.83 | 0.83 | 0.70 | 0.91 |
| Indinavir (IDV) | 15 | 0.90 | 0.90 | 0.90 | 0.91 | 0.96 |
| Lopinavir (LPV) | 15 | 0.91 | 0.91 | 0.91 | 0.83 | 0.97 |
| Nelfinavir (NFV) | 15 | 0.92 | 0.92 | 0.92 | 0.94 | 0.97 |
| Saquinavir (SQV) | 15 | 0.90 | 0.90 | 0.90 | 0.86 | 0.96 |
| Tipranavir (TPV) | 15 | 0.85 | 0.85 | 0.85 | 0.50 | 0.92 |
| Darunavir (DRV) | 6 | 0.89 | 0.89 | 0.89 | 0.35 | 0.96 |
| Nucleoside reverse transcriptase inhibitors | ||||||
| Lamivudine (3TC) | 15 | 0.82 | 0.82 | 0.82 | 0.86 | 0.90 |
| Abacavir (ABC) | 15 | 0.86 | 0.86 | 0.86 | 0.89 | 0.93 |
| Zidovudine (AZT) | 15 | 0.88 | 0.89 | 0.88 | 0.87 | 0.95 |
| Stavudine (D4T) | 15 | 0.87 | 0.87 | 0.87 | 0.86 | 0.95 |
| Didanosine (DDI) | 15 | 0.82 | 0.82 | 0.82 | 0.81 | 0.90 |
| Tenofovir (TDF) | 15 | 0.80 | 0.80 | 0.80 | 0.53 | 0.89 |
| Non-nucleoside reverse transcriptase inhibitors | ||||||
| Efavirenz (EFV) | 15 | 0.83 | 0.83 | 0.83 | 0.82 | 0.92 |
| Nevirapine (NVP) | 15 | 0.84 | 0.84 | 0.84 | 0.87 | 0.91 |
| Etravirine (ETR) | 6 | 0.84 | 0.84 | 0.84 | 0.34 | 0.90 |
| Rilpivirine (RPV) | 4 | 0.76 | 0.75 | 0.76 | 0.73 | 0.82 |
| Drug | Accuracy Rhee et al. [49] | Accuracy Heider et al. [50]. | Accuracy Beerenwinkel et al. [51] | k-mer Approach | MultiPassR Result |
|---|---|---|---|---|---|
| Protease inhibitors | |||||
| Fosamprenavir (FPV) | -/- | -/- | -/- | 0.87/- | 0.82/- |
| Atazanavir (ATV) | 0.77 */0.76 ** | 0.88 */- ** | - */0.94 ** | 0.90 ***/0.89 ** | 0.80 ***/0.74 ** |
| Indinavir (IDV) | 0.79/- | 0.93/- | -/0.96 | 0.93/0.91 | 0.87/0.85 |
| Lopinavir (LPV) | 0.81/0.79 | 0.92/- | -/0.95 | 0.92/0.87 | 0.87/0.82 |
| Nelfinavir (NFV) | 0.82/- | 0.91/- | -/0.93 | 0.92/0.90 | 0.90/0.87 |
| Saquinavir (SQV) | 0.84/- | 0.89/- | -/0.92 | 0.88/0.81 | 0.87/0.78 |
| Tipranavir (TPV) | -/ | -/- | -/0.86 | 0.85/0.82 | 0.81/0.78 |
| Darunavir (DRV) | -/0.79 | -/- | -/0.90 | 0.92/0.86 | 0.88/0.84 |
| Nucleoside reverse transcriptase inhibitors | |||||
| Lamivudine (3TC) | 0.90/0.97 | 0.90/- | -/0.95 | 0.82/0.79 | 0.81/0.80 |
| Abacavir (ABC) | 0.77/0.90 | 0.88/- | -/0.95 | 0.82/0.76 | 0.84/0.83 |
| Zidovudine (AZT) | 0.76/0.74 | 0.84/- | -/0.87 | 0.87/0.78 | 0.87/0.86 |
| Stavudine (D4T) | 0.78/0.78 | 0.84/- | -/0.89 | 0.87/0.79 | 0.85/0.87 |
| Didanosine (DDI) | 0.75/0.87 | 0.79/- | -/0.77 | 0.81/0.76 | 0.79/0.78 |
| Tenofovir (TDF) | 0.73/0.70 | 0.79/- | -/0.76 | 0.84/0.76 | 0.80/0.76 |
| Non-nucleoside reverse transcriptase inhibitors | |||||
| Efavirenz (EFV) | 0.87/0.85 | 0.88/- | -/0.87 | 0.80/0.75 | 0.81/0.83 |
| Nevirapine (NVP) | 0.91/0.84 | 0.87/- | -/0.89 | 0.79/0.77 | 0.84/0.80 |
| Etravirine (ETR) | -/0.82 | -/- | -/- | 0.84/0.78 | 0.84/0.77 |
| Rilpivirine (RPV) | -/- | -/- | -/- | 0.82/0.75 | 0.74/0.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paremskaia, A.I.; Rudik, A.V.; Filimonov, D.A.; Lagunin, A.A.; Poroikov, V.V.; Tarasova, O.A. Web Service for HIV Drug Resistance Prediction Based on Analysis of Amino Acid Substitutions in Main Drug Targets. Viruses 2023, 15, 2245. https://doi.org/10.3390/v15112245
Paremskaia AI, Rudik AV, Filimonov DA, Lagunin AA, Poroikov VV, Tarasova OA. Web Service for HIV Drug Resistance Prediction Based on Analysis of Amino Acid Substitutions in Main Drug Targets. Viruses. 2023; 15(11):2245. https://doi.org/10.3390/v15112245
Chicago/Turabian StyleParemskaia, Anastasiia Iu., Anastassia V. Rudik, Dmitry A. Filimonov, Alexey A. Lagunin, Vladimir V. Poroikov, and Olga A. Tarasova. 2023. "Web Service for HIV Drug Resistance Prediction Based on Analysis of Amino Acid Substitutions in Main Drug Targets" Viruses 15, no. 11: 2245. https://doi.org/10.3390/v15112245
APA StyleParemskaia, A. I., Rudik, A. V., Filimonov, D. A., Lagunin, A. A., Poroikov, V. V., & Tarasova, O. A. (2023). Web Service for HIV Drug Resistance Prediction Based on Analysis of Amino Acid Substitutions in Main Drug Targets. Viruses, 15(11), 2245. https://doi.org/10.3390/v15112245