
Human Complement Inhibits Myophages against Pseudomonas aeruginosa
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phages and Strains
2.2. Genome Sequencing and Bioinformatics Analysis
2.3. Bacterial Growth
2.4. Serum Preparation and Reagents
2.5. Microplate Reader Assays
2.6. Determination of Bacterial Viability
2.7. Flow Cytometry
2.8. Widefield Fluorescence Microscopy
2.9. C1q ELISA
2.10. Data Analysis and Statistical Testing
3. Results
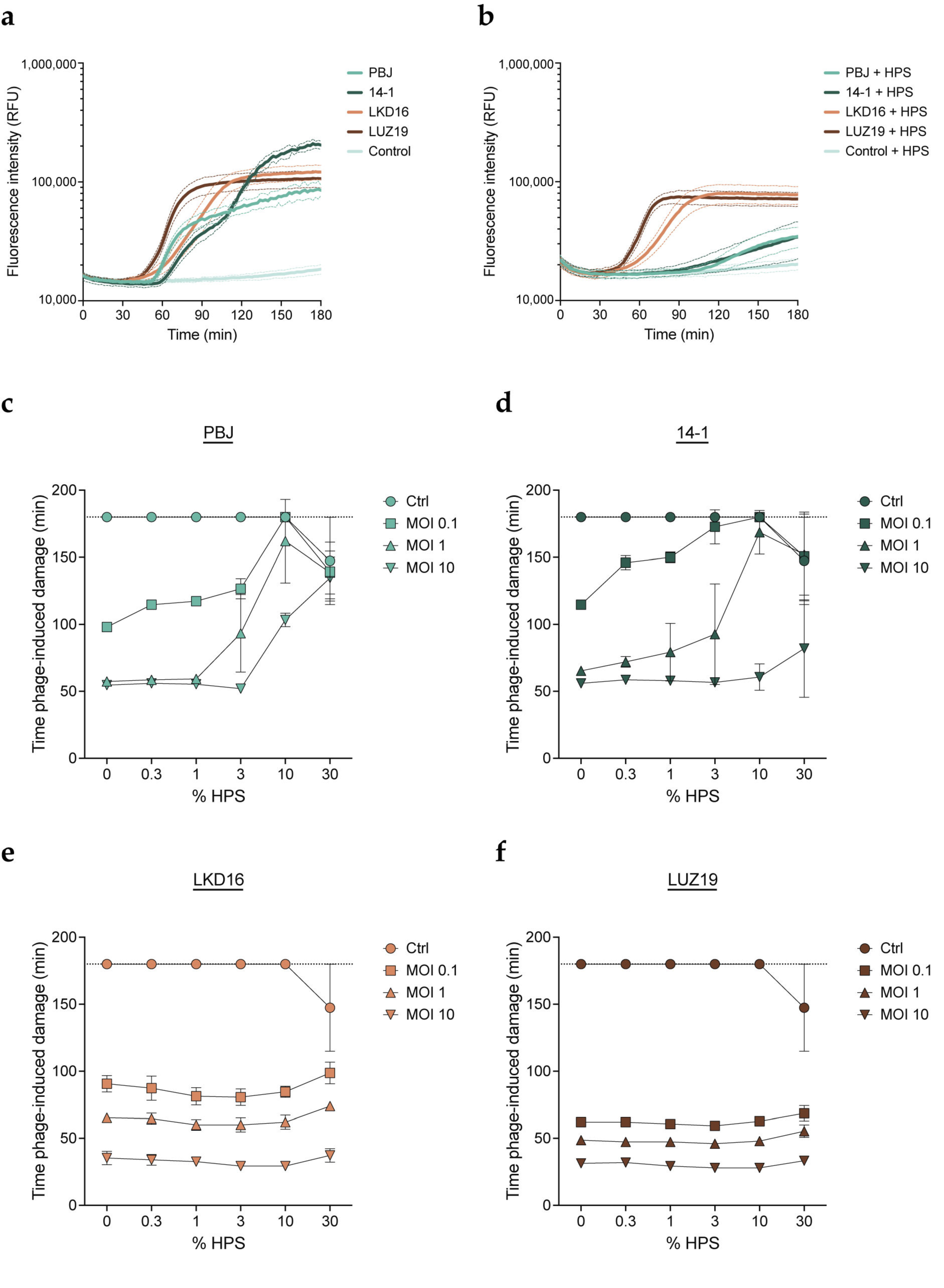
3.1. Human Serum Impairs Activity of Myophages PBJ and 14-1
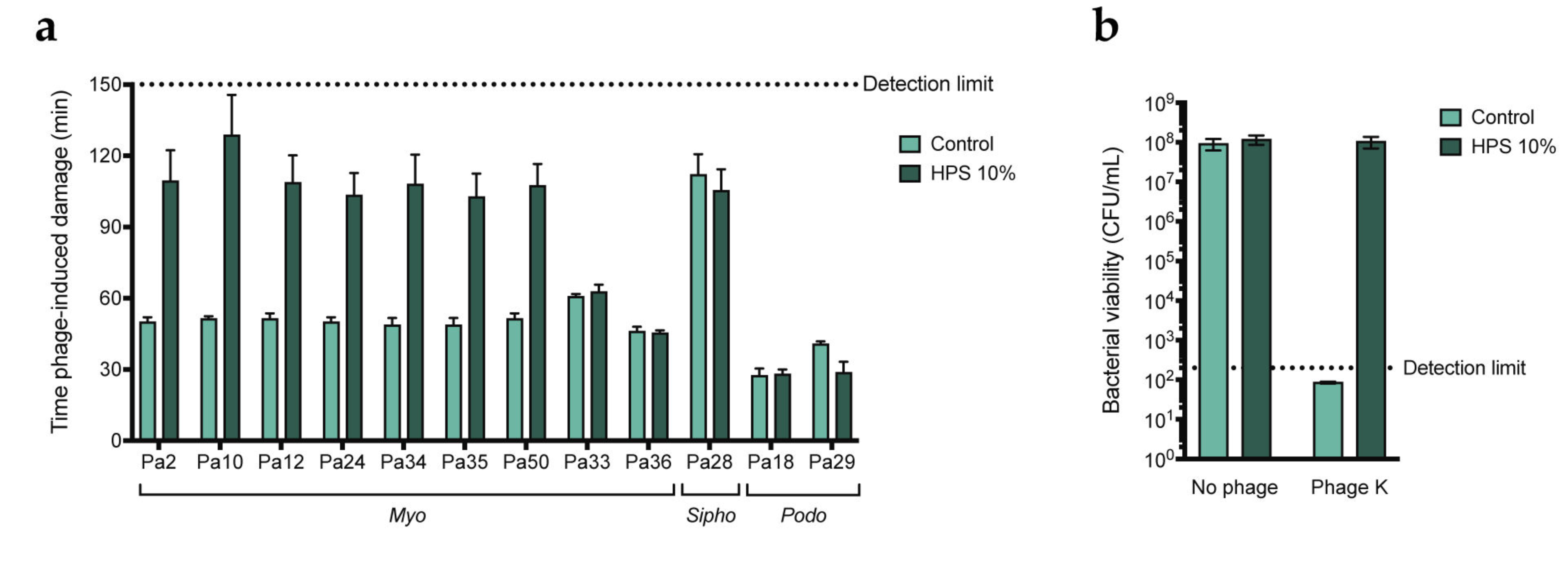
3.2. Serum-Mediated Inhibition Is Found for a Broader Set of Myophages
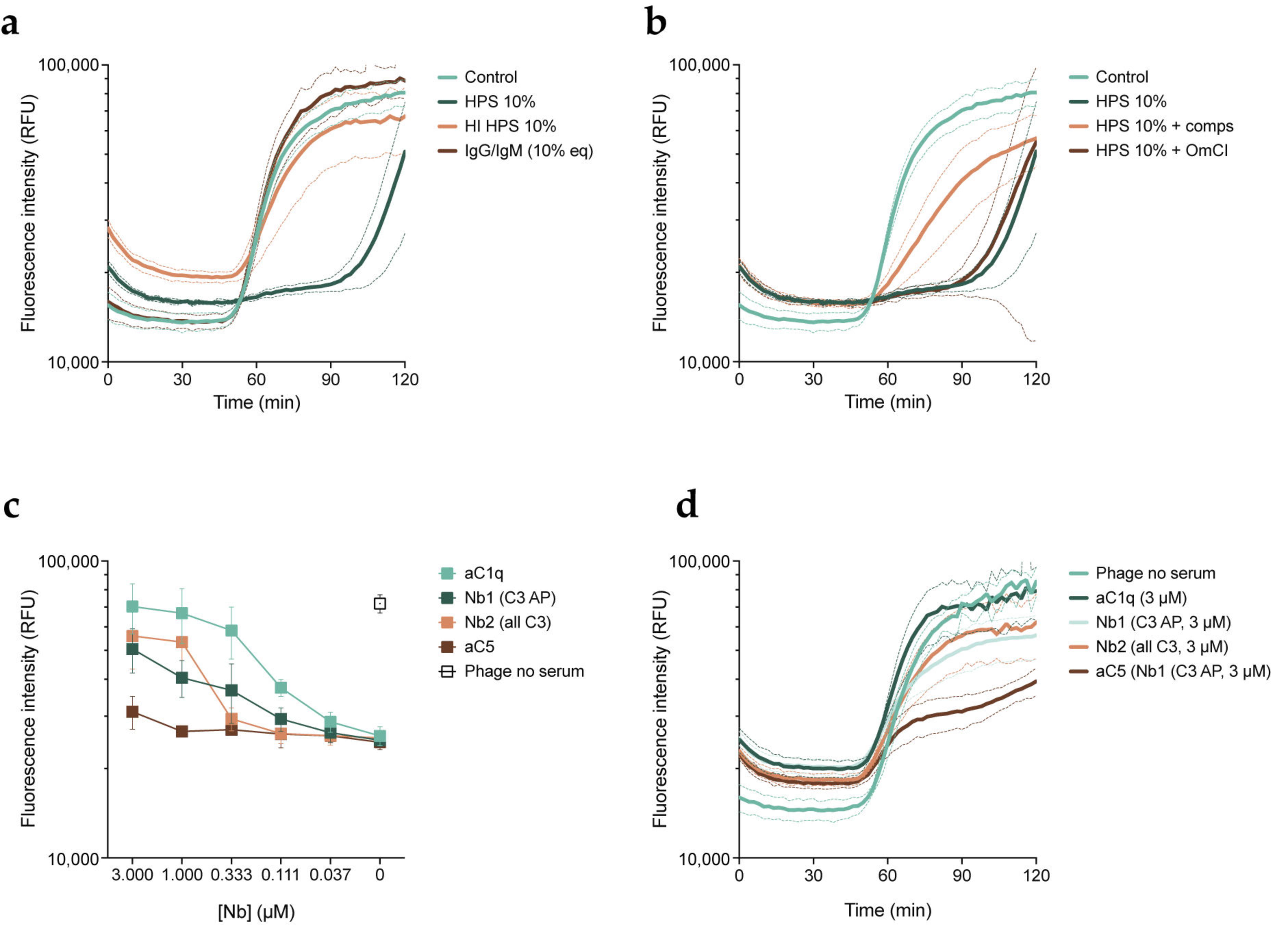
3.3. Phage Inhibition in Serum Is Mediated by the Early Stages of the Complement Cascade
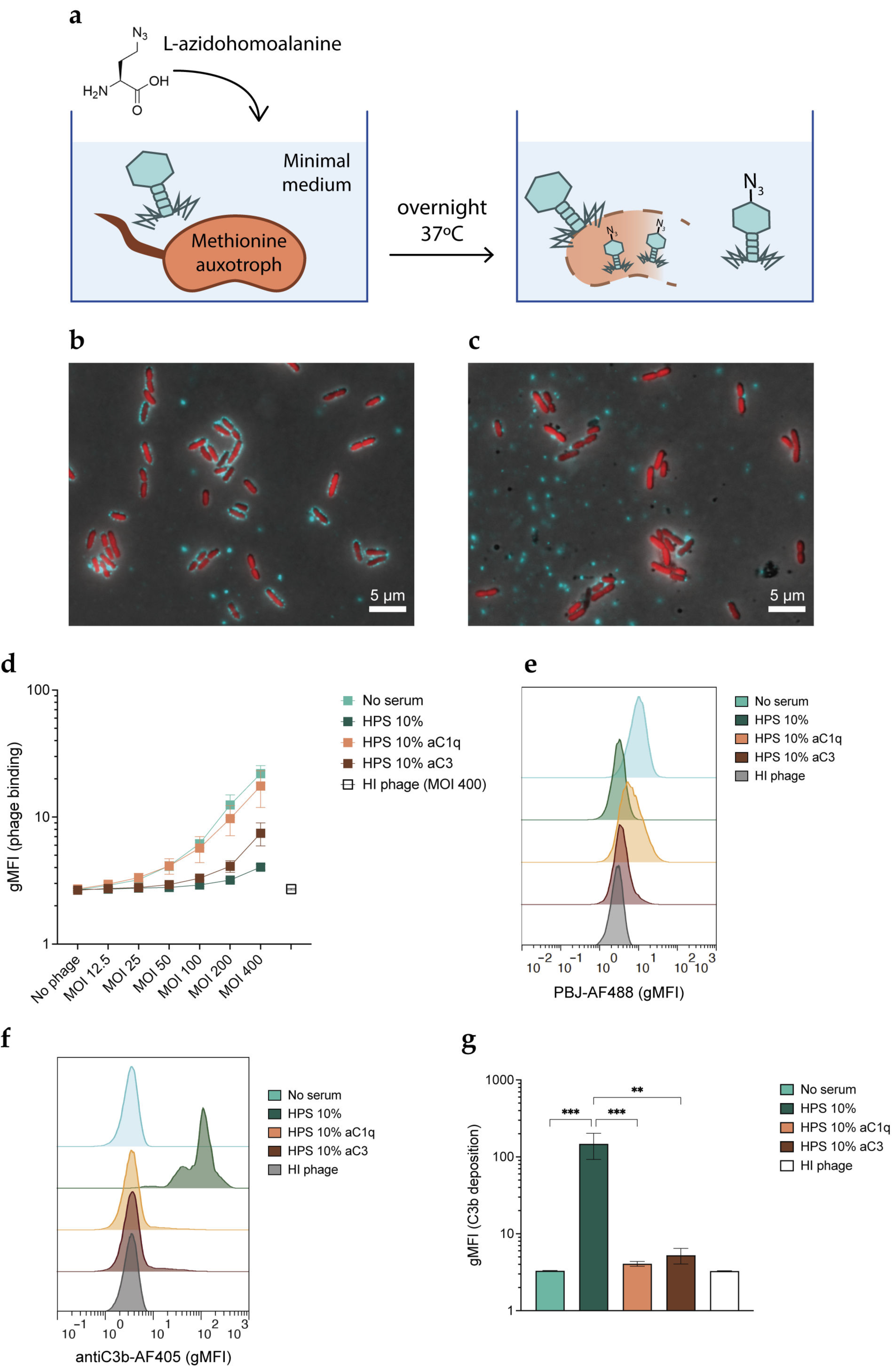
3.4. Inhibition of Phages by Complement Occurs at the Stage of Phage Adsorption
3.5. Complement Inhibitory Effect Is Not Mediated by Receptor Blockage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [PubMed]
- Strathdee, S.A.; Hatfull, G.F.; Mutalik, V.K.; Schooley, R.T. Phage Therapy: From Biological Mechanisms to Future Directions. Cell 2023, 186, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Vilas Boas, D.; Sillankorva, S.; Azeredo, J. Phage Therapy: A Step Forward in the Treatment of Pseudomonas aeruginosa Infections. J. Virol. 2015, 89, 7449–7456. [Google Scholar] [CrossRef] [PubMed]
- Ferry, T.; Kolenda, C.; Laurent, F.; Leboucher, G.; Merabischvilli, M.; Djebara, S.; Gustave, C.-A.; Perpoint, T.; Barrey, C.; Pirnay, J.-P.; et al. Personalized Bacteriophage Therapy to Treat Pandrug-Resistant Spinal Pseudomonas aeruginosa Infection. Nat. Commun. 2022, 13, 4239. [Google Scholar] [CrossRef] [PubMed]
- Simner, P.J.; Cherian, J.; Suh, G.A.; Bergman, Y.; Beisken, S.; Fackler, J.; Lee, M.; Hopkins, R.J.; Tamma, P.D. Combination of Phage Therapy and Cefiderocol to Successfully Treat Pseudomonas aeruginosa Cranial Osteomyelitis. JAC-Antimicrob. Resist. 2022, 4, dlac046. [Google Scholar] [CrossRef] [PubMed]
- Cesta, N.; Pini, M.; Mulas, T.; Materazzi, A.; Ippolito, E.; Wagemans, J.; Kutateladze, M.; Fontana, C.; Sarmati, L.; Tavanti, A.; et al. Application of Phage Therapy in a Case of a Chronic Hip-Prosthetic Joint Infection Due to Pseudomonas aeruginosa: An Italian Real-Life Experience and In Vitro Analysis. Open Forum Infect. Dis. 2023, 10, ofad051. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Floch, R.L.; et al. Efficacy and Tolerability of a Cocktail of Bacteriophages to Treat Burn Wounds Infected by Pseudomonas aeruginosa (PhagoBurn): A Randomised, Controlled, Double-Blind Phase 1/2 Trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Uyttebroek, S.; Chen, B.; Onsea, J.; Ruythooren, F.; Debaveye, Y.; Devolder, D.; Spriet, I.; Depypere, M.; Wagemans, J.; Lavigne, R.; et al. Safety and Efficacy of Phage Therapy in Difficult-to-Treat Infections: A Systematic Review. Lancet Infect. Dis. 2022, 22, e208–e220. [Google Scholar] [CrossRef]
- Stacey, H.J.; De Soir, S.; Jones, J.D. The Safety and Efficacy of Phage Therapy: A Systematic Review of Clinical and Safety Trials. Antibiotics 2022, 11, 1340. [Google Scholar] [CrossRef]
- Pirnay, J.-P.; Djebara, S.; Steurs, G.; Griselain, J.; Cochez, C.; Soir, S.D.; Glonti, T.; Spiessens, A.; Berghe, E.V.; Green, S.; et al. Retrospective, Observational Analysis of the First One Hundred Consecutive Cases of Personalized Bacteriophage Therapy of Difficult-to-Treat Infections Facilitated by a Belgian Consortium. medRxiv 2023. 2023.08.28.23294728. [Google Scholar] [CrossRef]
- Egido, J.E.; Costa, A.R.; Aparicio-Maldonado, C.; Haas, P.-J.; Brouns, S.J.J. Mechanisms and Clinical Importance of Bacteriophage Resistance. FEMS Microbiol. Rev. 2022, 46, fuab048. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Bustamante, C.A.; Dedrick, R.M.; Garlena, R.A.; Russell, D.A.; Hatfull, G.F. Toward a Phage Cocktail for Tuberculosis: Susceptibility and Tuberculocidal Action of Mycobacteriophages against Diverse Mycobacterium tuberculosis Strains. mBio 2021, 12, e00973-21. [Google Scholar] [CrossRef] [PubMed]
- Łusiak-Szelachowska, M.; Żaczek, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Kłak, M.; Fortuna, W.; Letkiewicz, S.; Rogóż, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; et al. Phage Neutralization by Sera of Patients Receiving Phage Therapy. Viral Immunol. 2014, 27, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Hodyra-Stefaniak, K.; Miernikiewicz, P.; Drapała, J.; Drab, M.; Jończyk-Matysiak, E.; Lecion, D.; Kaźmierczak, Z.; Beta, W.; Majewska, J.; Harhala, M.; et al. Mammalian Host-Versus-Phage Immune Response Determines Phage Fate in Vivo. Sci. Rep. 2015, 5, 14802. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.K.; Kulkarni, D.H.; Ozanturk, A.N.; Ma, L.; Kulkarni, H.S. Emerging Roles of the Complement System in Host–Pathogen Interactions. Trends Microbiol. 2022, 30, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Heesterbeek, D.A.; Bardoel, B.W.; Parsons, E.S.; Bennett, I.; Ruyken, M.; Doorduijn, D.J.; Gorham, R.D.; Berends, E.T.; Pyne, A.L.; Hoogenboom, B.W.; et al. Bacterial Killing by Complement Requires Membrane Attack Complex Formation via Surface-Bound C5 Convertases. EMBO J. 2019, 38, e99852. [Google Scholar] [CrossRef]
- Abd El-Aziz, A.M.; Elgaml, A.; Ali, Y.M. Bacteriophage Therapy Increases Complement-Mediated Lysis of Bacteria and Enhances Bacterial Clearance After Acute Lung Infection with Multidrug-Resistant Pseudomonas aeruginosa. J. Infect. Dis. 2019, 219, 1439–1447. [Google Scholar] [CrossRef]
- Ostrycharz, E.; Hukowska-Szematowicz, B. New Insights into the Role of the Complement System in Human Viral Diseases. Biomolecules 2022, 12, 226. [Google Scholar] [CrossRef]
- Hodyra-Stefaniak, K.; Lahutta, K.; Majewska, J.; Kaźmierczak, Z.; Lecion, D.; Harhala, M.; Kęska, W.; Owczarek, B.; Jończyk-Matysiak, E.; Kłopot, A.; et al. Bacteriophages Engineered to Display Foreign Peptides May Become Short-circulating Phages. Microb. Biotechnol. 2019, 12, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.A.; Alwood, A.; Thaipisuttikul, I.; Spencer, D.; Haugen, E.; Ernst, S.; Will, O.; Kaul, R.; Raymond, C.; Levy, R.; et al. Comprehensive Transposon Mutant Library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 14339–14344. [Google Scholar] [CrossRef] [PubMed]
- Barbier, M.; Damron, F.H. Rainbow Vectors for Broad-Range Bacterial Fluorescence Labeling. PLoS ONE 2016, 11, e0146827. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 September 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive Visualization of de Novo Genome Assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Bouras, G.; Nepal, R.; Houtak, G.; Psaltis, A.J.; Wormald, P.-J.; Vreugde, S. Pharokka: A Fast Scalable Bacteriophage Annotation Tool. Bioinformatics 2023, 39, btac776. [Google Scholar] [CrossRef] [PubMed]
- Zwarthoff, S.A.; Magnoni, S.; Aerts, P.C.; van Kessel, K.P.M.; Rooijakkers, S.H.M. Method for Depletion of IgG and IgM from Human Serum as Naive Complement Source. In The Complement System: Innovative Diagnostic and Research Protocols; Roumenina, L.T., Ed.; Methods in Molecular Biology; Springer US: New York, NY, USA, 2021; pp. 21–32. ISBN 978-1-07-161016-9. [Google Scholar]
- Nunn, M.A.; Sharma, A.; Paesen, G.C.; Adamson, S.; Lissina, O.; Willis, A.C.; Nuttall, P.A. Complement Inhibitor of C5 Activation from the Soft Tick Ornithodoros Moubata. J. Immunol. 2005, 174, 2084–2091. [Google Scholar] [CrossRef]
- Laursen, N.S.; Pedersen, D.V.; Gytz, H.; Zarantonello, A.; Bernth Jensen, J.M.; Hansen, A.G.; Thiel, S.; Andersen, G.R. Functional and Structural Characterization of a Potent C1q Inhibitor Targeting the Classical Pathway of the Complement System. Front. Immunol. 2020, 11, 1504. [Google Scholar] [CrossRef]
- Jensen, R.K.; Pihl, R.; Gadeberg, T.A.F.; Jensen, J.K.; Andersen, K.R.; Thiel, S.; Laursen, N.S.; Andersen, G.R. A Potent Complement Factor C3–Specific Nanobody Inhibiting Multiple Functions in the Alternative Pathway of Human and Murine Complement. J. Biol. Chem. 2018, 293, 6269–6281. [Google Scholar] [CrossRef]
- Pedersen, H.; Jensen, R.K.; Hansen, A.G.; Gadeberg, T.A.F.; Thiel, S.; Laursen, N.S.; Andersen, G.R. A C3-Specific Nanobody That Blocks All Three Activation Pathways in the Human and Murine Complement System. J. Biol. Chem. 2020, 295, 8746–8758. [Google Scholar] [CrossRef] [PubMed]
- Yatime, L.; Merle, N.S.; Hansen, A.G.; Friis, N.A.; Østergaard, J.A.; Bjerre, M.; Roumenina, L.T.; Thiel, S.; Kristensen, P.; Andersen, G.R. A Single-Domain Antibody Targeting Complement Component C5 Acts as a Selective Inhibitor of the Terminal Pathway of the Complement System and Thus Functionally Mimicks the C-Terminal Domain of the Staphylococcus aureus SSL7 Protein. Front. Immunol. 2018, 9, 2822. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Miroshnikov, K.; Mattheus, W.; Krylov, V.; Robben, J.; Noben, J.-P.; Vanderschraeghe, S.; Sykilinda, N.; Kropinski, A.M.; Volckaert, G.; et al. Comparative Analysis of the Widespread and Conserved PB1-like Viruses Infecting Pseudomonas aeruginosa. Environ. Microbiol. 2009, 11, 2874–2883. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.C.; Sible, E.; Putonti, C. Pseudomonas PB1-Like Phages: Whole Genomes from Metagenomes Offer Insight into an Abundant Group of Bacteriophages. Viruses 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Lavigne, R.; Mattheus, W.; Chibeu, A.; Hertveldt, K.; Mast, J.; Robben, J.; Volckaert, G. Genomic Analysis of Pseudomonas aeruginosa Phages LKD16 and LKA1: Establishment of the phiKMV Subgroup within the T7 Supergroup. J. Bacteriol. 2006, 188, 6924–6931. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Lecoutere, E.; Wagemans, J.; Cenens, W.; Aertsen, A.; Schoofs, L.; Landuyt, B.; Paeshuyse, J.; Scheer, M.; Schobert, M.; et al. A Multifaceted Study of Pseudomonas aeruginosa Shutdown by Virulent Podovirus LUZ19. mBio 2013, 4, e00061-00013. [Google Scholar] [CrossRef] [PubMed]
- Egido, J.E.; Toner-Bartelds, C.; Costa, A.R.; Brouns, S.J.J.; Rooijakkers, S.H.M.; Bardoel, B.W.; Haas, P.-J. Monitoring Phage-Induced Lysis of Gram-Negatives in Real Time Using a Fluorescent DNA Dye. Sci. Rep. 2023, 13, 856. [Google Scholar] [CrossRef]
- Costa, A.R.; van den Berg, D.F.; Esser, J.Q.; Muralidharan, A.; van den Bossche, H.; Bonilla, B.E.; van der Steen, B.A.; Haagsma, A.C.; Fluit, A.C.; Nobrega, F.L.; et al. Accumulation of Defense Systems in Phage Resistant Strains of Pseudomonas aeruginosa. bioRxiv 2023. 2022.08.12.503731. [Google Scholar] [CrossRef]
- Ricklin, D.; Lambris, J.D. Compstatin: A Complement Inhibitor on Its Way to Clinical Application. Adv. Exp. Med. Biol. 2008, 632, 273–292. [Google Scholar]
- Lamers, C.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Compstatins: The Dawn of Clinical C3-Targeted Complement Inhibition. Trends Pharmacol. Sci. 2022, 43, 629–640. [Google Scholar] [CrossRef]
- Jin, B.-K.; Odongo, S.; Radwanska, M.; Magez, S. Nanobodies: A Review of Generation, Diagnostics and Therapeutics. Int. J. Mol. Sci. 2023, 24, 5994. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, D.R.; Jiang, H.; Kalin, J.H.; Chen, Z.; Cole, P.A. Site-Specific Protein Labeling with NHS-Esters and the Analysis of Ubiquitin Ligase Mechanisms. J. Am. Chem. Soc. 2018, 140, 9374–9378. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.E.; Lemmel, S.A.; Yu, X.; Zhou, Q.A. Bioorthogonal Chemistry and Its Applications. Bioconjug. Chem. 2021, 32, 2457–2479. [Google Scholar] [CrossRef]
- Strable, E.; Prasuhn, D.E., Jr.; Udit, A.K.; Brown, S.; Link, A.J.; Ngo, J.T.; Lander, G.; Quispe, J.; Potter, C.S.; Carragher, B.; et al. Unnatural Amino Acid Incorporation into Virus-Like Particles. Bioconjug. Chem. 2008, 19, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Nawadkar, R.; Ojha, H.; Kumar, J.; Sahu, A. Complement Evasion Strategies of Viruses: An Overview. Front. Microbiol. 2017, 8, 1117. [Google Scholar] [CrossRef] [PubMed]
- Maghsoodi, A.; Chatterjee, A.; Andricioaei, I.; Perkins, N.C. How the Phage T4 Injection Machinery Works Including Energetics, Forces, and Dynamic Pathway. Proc. Natl. Acad. Sci. USA 2019, 116, 25097–25105. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Kaźmierczak, Z.; Letarov, A.; Górski, A. Immunogenicity Studies of Proteins Forming the T4 Phage Head Surface. J. Virol. 2014, 88, 12551–12557. [Google Scholar] [CrossRef]
- Sokoloff, A.V.; Zhang, G.; Sebestyén, M.G.; Wolff, J.A.; Bock, I. The Interactions of Peptides with the Innate Immune System Studied with Use of T7 Phage Peptide Display. Mol. Ther. 2000, 2, 131–139. [Google Scholar] [CrossRef]
- Bíró, A.; Rovó, Z.; Papp, D.; Cervenak, L.; Varga, L.; Füst, G.; Thielens, N.M.; Arlaud, G.J.; Prohászka, Z. Studies on the Interactions between C-Reactive Protein and Complement Proteins. Immunology 2007, 121, 40–50. [Google Scholar] [CrossRef]
- Douradinha, B.; McBurney, S.P.; de Melo, K.M.S.; Smith, A.P.; Krishna, N.K.; Barratt-Boyes, S.M.; Evans, J.D.; Nascimento, E.J.M.; Marques, E.T.A. C1q Binding to Dengue Virus Inhibits Infection of THP-1 and Cellular Inflammatory Responses. Virus Res. 2014, 179, 231–234. [Google Scholar] [CrossRef]
- Varghese, P.M.; Kishore, U.; Rajkumari, R. Human C1q Regulates Influenza A Virus Infection and Inflammatory Response via Its Globular Domain. Int. J. Mol. Sci. 2022, 23, 3045. [Google Scholar] [CrossRef]
- Beirag, N.; Varghese, P.M.; Neto, M.M.; Al Aiyan, A.; Khan, H.A.; Qablan, M.; Shamji, M.H.; Sim, R.B.; Temperton, N.; Kishore, U. Complement Activation-Independent Attenuation of SARS-CoV-2 Infection by C1q and C4b-Binding Protein. Viruses 2023, 15, 1269. [Google Scholar] [CrossRef] [PubMed]
- Gu Liu, C.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.C.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-Antibiotic Synergy Is Driven by a Unique Combination of Antibacterial Mechanism of Action and Stoichiometry. mBio 2020, 11, e01462-20. [Google Scholar] [CrossRef] [PubMed]
- Shinde, P.; Stamatos, N.; Doub, J.B. Human Plasma Significantly Reduces Bacteriophage Infectivity Against Staphylococcus aureus Clinical Isolates. Cureus 2022, 14, e23777. [Google Scholar] [CrossRef] [PubMed]
- Molendijk, M.M.; Phan, M.V.T.; Bode, L.G.M.; Strepis, N.; Prasad, D.K.; Worp, N.; Nieuwenhuijse, D.F.; Schapendonk, C.M.E.; Boekema, B.K.H.L.; Verbon, A.; et al. Microcalorimetry: A Novel Application to Measure In Vitro Phage Susceptibility of Staphylococcus aureus in Human Serum. Viruses 2023, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Pirnay, J.-P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-Controlled Small-Scale Production of a Well-Defined Bacteriophage Cocktail for Use in Human Clinical Trials. PLoS ONE 2009, 4, e4944. [Google Scholar] [CrossRef] [PubMed]
- Duyvejonck, H.; Merabishvili, M.; Vaneechoutte, M.; de Soir, S.; Wright, R.; Friman, V.-P.; Verbeken, G.; De Vos, D.; Pirnay, J.-P.; Van Mechelen, E.; et al. Evaluation of the Stability of Bacteriophages in Different Solutions Suitable for the Production of Magistral Preparations in Belgium. Viruses 2021, 13, 865. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Boeckaerts, D.; Stock, M.; De Baets, B.; Lavigne, R.; van Noort, V.; Briers, Y. Digital Phagograms: Predicting Phage Infectivity through a Multilayer Machine Learning Approach. Curr. Opin. Virol. 2022, 52, 174–181. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egido, J.E.; Dekker, S.O.; Toner-Bartelds, C.; Lood, C.; Rooijakkers, S.H.M.; Bardoel, B.W.; Haas, P.-J. Human Complement Inhibits Myophages against Pseudomonas aeruginosa. Viruses 2023, 15, 2211. https://doi.org/10.3390/v15112211
Egido JE, Dekker SO, Toner-Bartelds C, Lood C, Rooijakkers SHM, Bardoel BW, Haas P-J. Human Complement Inhibits Myophages against Pseudomonas aeruginosa. Viruses. 2023; 15(11):2211. https://doi.org/10.3390/v15112211
Chicago/Turabian StyleEgido, Julia E., Simon O. Dekker, Catherine Toner-Bartelds, Cédric Lood, Suzan H. M. Rooijakkers, Bart W. Bardoel, and Pieter-Jan Haas. 2023. "Human Complement Inhibits Myophages against Pseudomonas aeruginosa" Viruses 15, no. 11: 2211. https://doi.org/10.3390/v15112211
APA StyleEgido, J. E., Dekker, S. O., Toner-Bartelds, C., Lood, C., Rooijakkers, S. H. M., Bardoel, B. W., & Haas, P.-J. (2023). Human Complement Inhibits Myophages against Pseudomonas aeruginosa. Viruses, 15(11), 2211. https://doi.org/10.3390/v15112211