
SARS-CoV-2 Delta Variant N Gene Mutations Reduce Sensitivity to the TaqPath COVID-19 Multiplex Molecular Diagnostic Assay

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Saliva Specimens and Diagnostic Testing
2.2. SARS-CoV-2 Genome Sequencing and Analysis
2.3. Global Data Collection and Analysis
2.4. Plasmids
2.5. Real-Time Quantitative Reverse Transcription PCR (qRT-PCR) Assays
3. Results
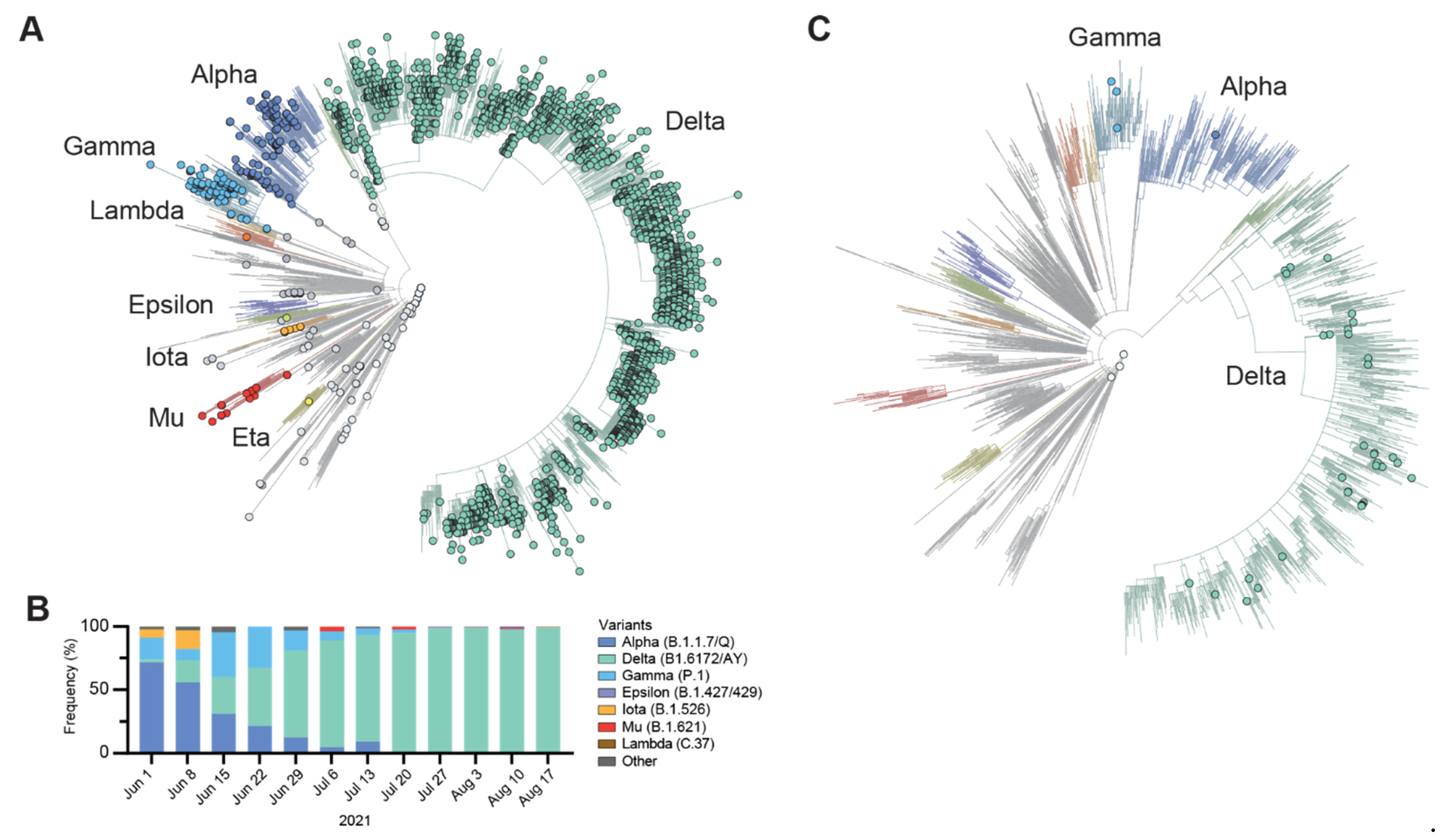
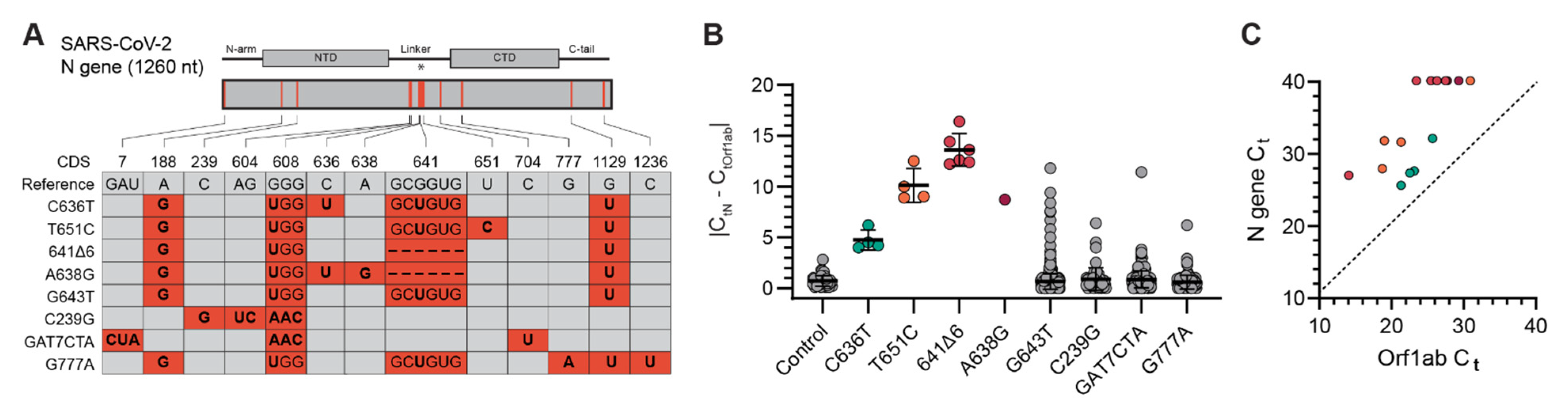
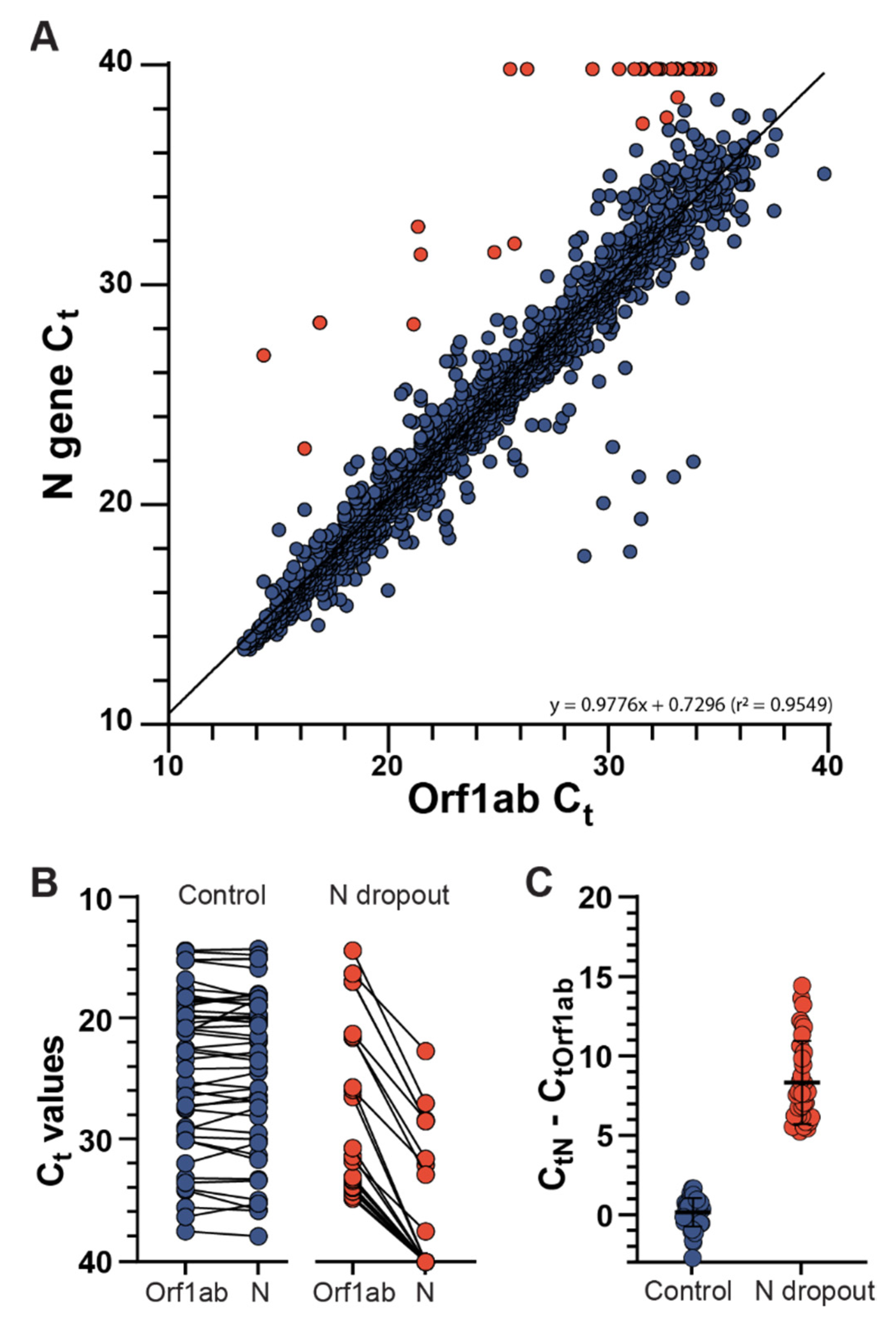
3.1. Identification of SARS-CoV-2 N Gene Target Failures
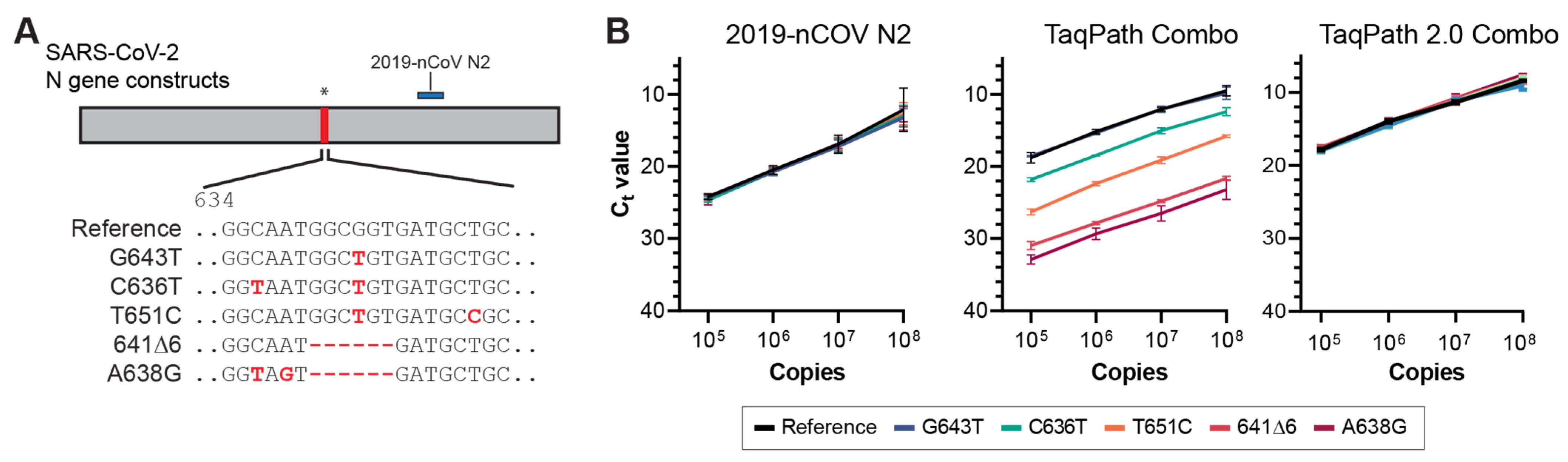
3.2. Next-Generation Sequencing Analysis of Delta Variant N Gene Dropouts
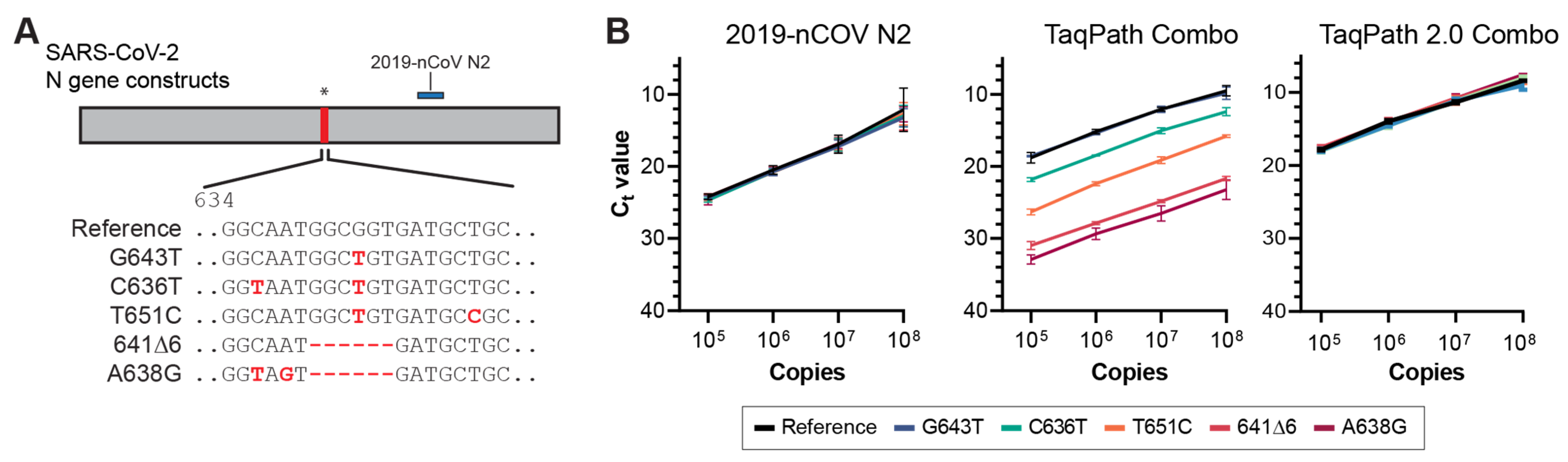
3.3. Molecular Validation of NGTF Mutations
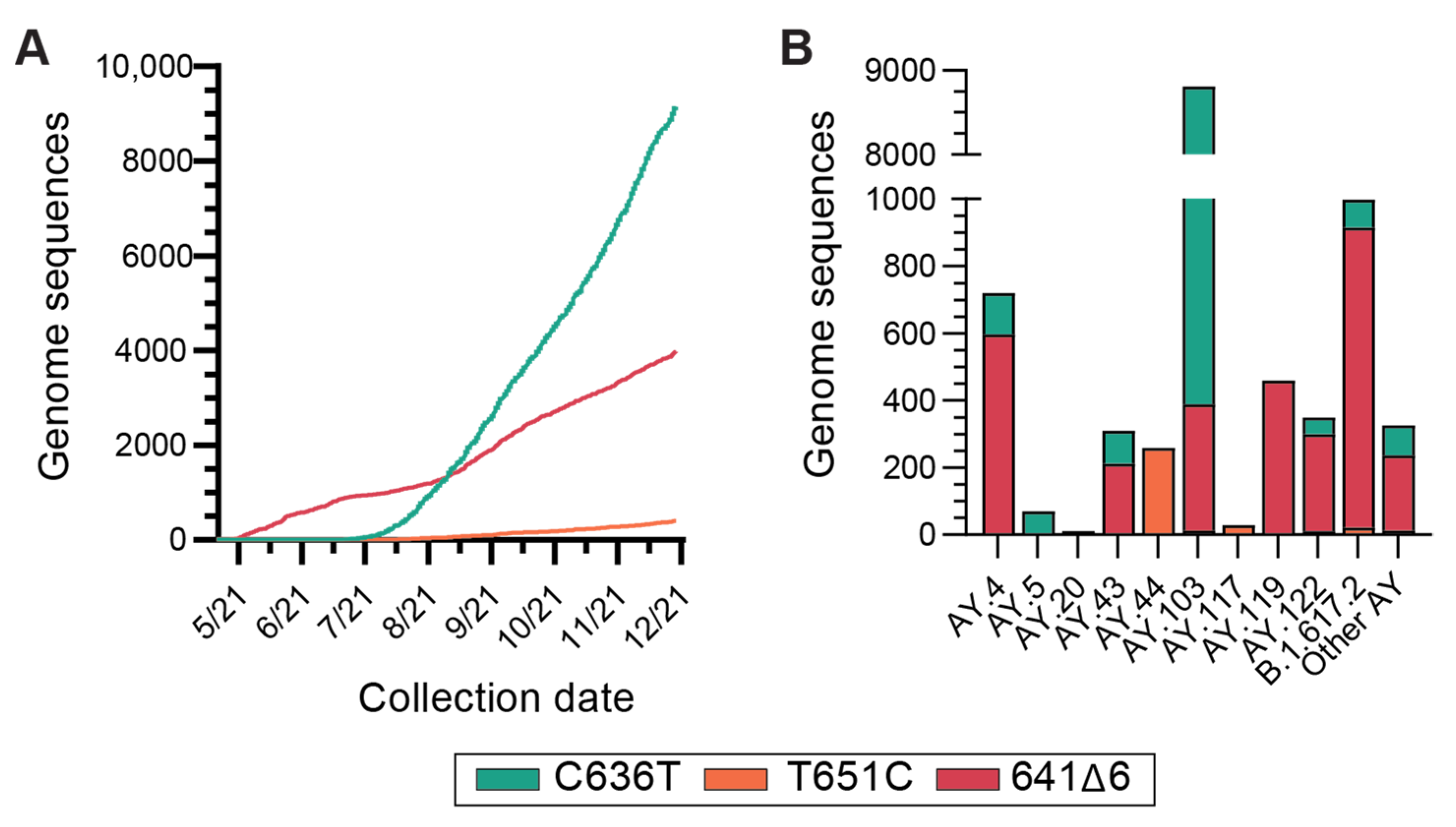
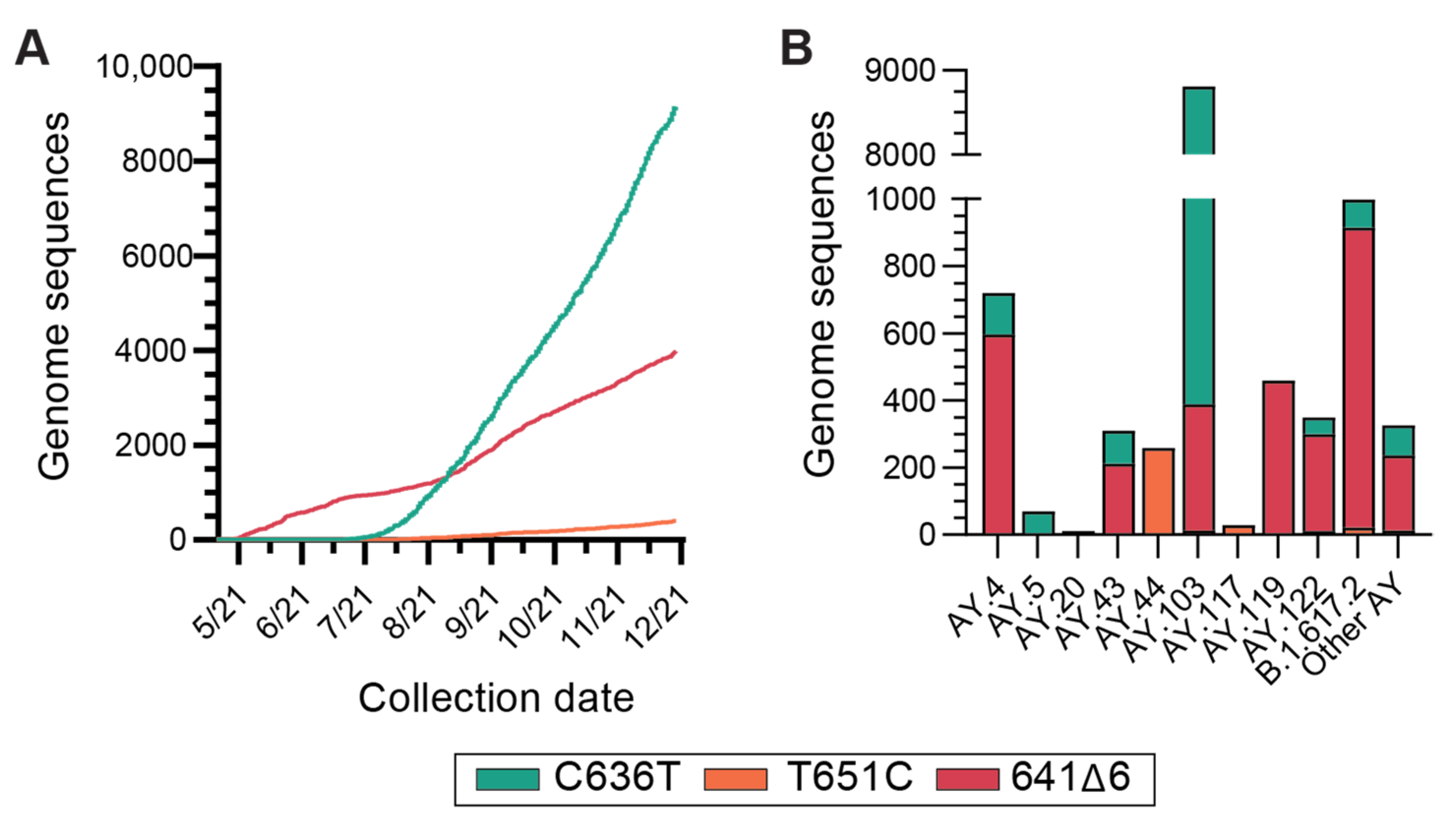
3.4. Global Incidence of Delta Variant N Gene Dropout Mutations
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhatib, M.; Svicher, V.; Salpini, R.; Ambrosio, F.A.; Bellocchi, M.C.; Carioti, L.; Piermatteo, L.; Scutari, R.; Costa, G.; Artese, A.; et al. SARS-CoV-2 Variants and Their Relevant Mutational Profiles: Update Summer 2021. Microbiol. Spectrum. 2021, 9, e0109621. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization HQ. Laboratory Testing for Coronavirus Disease (COVID-19) in Suspected Human Cases: Interim Guidance 19 March 2020. Available online: https://www.who.int/publications/i/item/10665-331501 (accessed on 20 December 2021).
- Centers for Disease Control and Prevention. Research Use Only 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Primers and Probes. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html (accessed on 8 June 2022).
- Artesi, M.; Bontems, S.; Göbbels, P.; Franckh, M.; Maes, P.; Boreux, R.; Meex, C.; Melin, P.; Hayette, M.-P.; Bours, V.; et al. A recurrent mutation at position 26340 of SARS-CoV-2 is associated with failure of the E gene quantitative reverse transcription-PCR utilized in a commercial dual-target diagnostic assay. J. Clin. Microbiol. 2020, 58, e01598-20. [Google Scholar] [CrossRef] [PubMed]
- Tahan, S.; Parikh, B.A.; Droit, L.; Wallace, M.A.; Burnham, C.-A.D.; Wang, D. SARS-CoV-2 E gene variant alters analytical sensitivity characteristics of viral detection using a commercial reverse transcription-PCR assay. J. Clin. Microbiol. 2021, 59, e00075-21. [Google Scholar] [CrossRef] [PubMed]
- Vanaerschot, M.; Mann, S.A.; Webber, J.T.; Kamm, J.; Bell, S.M.; Bell, J.; Hong, S.N.; Nguyen, M.P.; Chan, L.Y.; Bhatt, K.D.; et al. Identification of a polymorphism in the N gene of SARS-CoV-2 that adversely impacts detection by reverse transcription-PCR. J. Clin. Microbiol. 2021, 59, e02369-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Hozumi, Y.; Yin, C.; Wei, G.W. Mutations on COVID-19 diagnostic targets. Genomics 2020, 112, 5204–5213. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization HQ. Enhancing Readiness for Omicron (B.1.1.529): Technical Brief and Priority Actions for Member States; World Health Organization: Geneva, Switzerland, 2021; p. 19.
- Wollschlager, P.; Todt, D.; Gerlitz, N.; Pfaender, S.; Bollinger, T.; Sing, A.; Dangel, A.; Ackermann, N.; Korn, K.; Ensser, A.; et al. SARS-CoV-2 N gene dropout and N gene Ct value shift as indicator for the presence of B.1.1.7 lineage in a commercial multiplex PCR assay. Clin. Microbiol. Infect. 2021, 27, 1353.e1351–1353.e1355. [Google Scholar] [CrossRef] [PubMed]
- Washington, N.L.; White, S.; Barrett, K.M.S.; Cirulli, E.T.; Bolze, A.; Lu, J.T. S gene dropout patterns in SARS-CoV-2 tests suggest spread of the H69del/V70del mutation in the US. medRxiv 2020. [Google Scholar] [CrossRef]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Castellano, S.; Cestari, F.; Faglioni, G.; Tenedini, E.; Marino, M.; Artuso, L.; Manfredini, R.; Luppi, M.; Trenti, T.; Tagliafico, E. iVar, an Interpretation-Oriented Tool to Manage the Update and Revision of Variant Annotation and Classification. Genes 2021, 12, 384. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, A.A.; Hatcher, E.L.; Yankie, L.; Shonkwiler, L.; Brister, J.R.; Karsch-Mizrachi, I.; Nawrocki, E.P. VADR: Validation and annotation of virus sequence submissions to GenBank. BMC Bioinform. 2020, 21, 211. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.Y.; Lee, K.R.; Tarn, W.Y. Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J. 2008, 275, 4152–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moody, R.; Wilson, K.L.; Boer, J.C.; Holien, J.K.; Flanagan, K.L.; Jaworowski, A.; Plebanski, M. Predicted B Cell Epitopes Highlight the Potential for COVID-19 to Drive Self-Reactive Immunity. Front. Bioinform. 2021, 1, 1–21. [Google Scholar] [CrossRef]
- Obermeyer, F.; Schaffner, S.F.; Jankowiak, M.; Barkas, N.; Pyle, J.D.; Park, D.J.; MacInnis, B.L.; Luban, J.; Sabeti, P.C.; Lemieux, J.E. Analysis of 2.1 million SARS-CoV-2 genomes identifies mutations associated with transmissibility. medRxiv 2021. [Google Scholar] [CrossRef]
- Alkhatib, M.; Bellocchi, M.C.; Marchegiani, G.; Grelli, S.; Micheli, V.; Stella, D.; Zerillo, B.; Carioti, L.; Svicher, V.; Rogliani, P.; et al. First Case of a COVID-19 Patient Infected by Delta AY.4 with a Rare Deletion Leading to a N Gene Target Failure by a Specific Real Time PCR Assay: Novel Omicron VOC Might Be Doing Similar Scenario? Microorganisms 2022, 10, 268. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holland, S.C.; Bains, A.; Holland, L.A.; Smith, M.F.; Sullins, R.A.; Mellor, N.J.; Thomas, A.W.; Johnson, N.; Murugan, V.; Lim, E.S. SARS-CoV-2 Delta Variant N Gene Mutations Reduce Sensitivity to the TaqPath COVID-19 Multiplex Molecular Diagnostic Assay. Viruses 2022, 14, 1316. https://doi.org/10.3390/v14061316
Holland SC, Bains A, Holland LA, Smith MF, Sullins RA, Mellor NJ, Thomas AW, Johnson N, Murugan V, Lim ES. SARS-CoV-2 Delta Variant N Gene Mutations Reduce Sensitivity to the TaqPath COVID-19 Multiplex Molecular Diagnostic Assay. Viruses. 2022; 14(6):1316. https://doi.org/10.3390/v14061316
Chicago/Turabian StyleHolland, Steven C., Ajeet Bains, LaRinda A. Holland, Matthew F. Smith, Regan A. Sullins, Nicholas J. Mellor, Alexis W. Thomas, Nathaniel Johnson, Vel Murugan, and Efrem S. Lim. 2022. "SARS-CoV-2 Delta Variant N Gene Mutations Reduce Sensitivity to the TaqPath COVID-19 Multiplex Molecular Diagnostic Assay" Viruses 14, no. 6: 1316. https://doi.org/10.3390/v14061316
APA StyleHolland, S. C., Bains, A., Holland, L. A., Smith, M. F., Sullins, R. A., Mellor, N. J., Thomas, A. W., Johnson, N., Murugan, V., & Lim, E. S. (2022). SARS-CoV-2 Delta Variant N Gene Mutations Reduce Sensitivity to the TaqPath COVID-19 Multiplex Molecular Diagnostic Assay. Viruses, 14(6), 1316. https://doi.org/10.3390/v14061316