
Genome-Wide Reassortment Analysis of Influenza A H7N9 Viruses Circulating in China during 2013–2019

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequences Collection
2.2. Phylogenetic Analyses and Diversity Estimates
2.3. Phylogenetic Clusters Classification
2.4. Reassortment Detections
2.4.1. Intersubtype Reassortment
2.4.2. Entanglegram and Untanglement
2.4.3. Quantified Investigation Reassortment Events
2.5. Multidimensional Scaling
2.6. Selection Pressure Analysis
3. Results
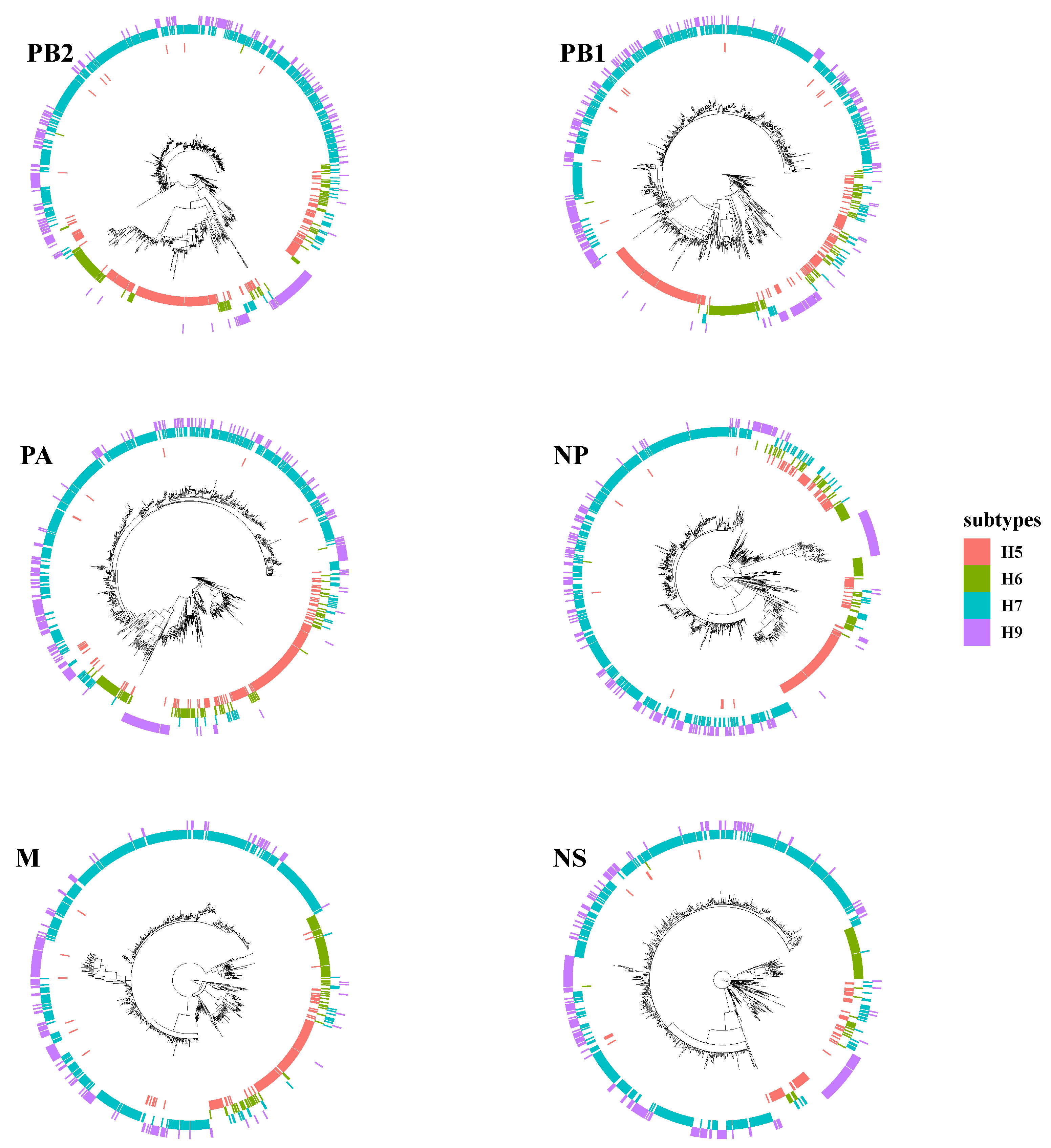
3.1. The Surface Glycoproteins Have Faster Evolutionary Rates but Less Genetic Diversity
3.2. PB2 c05 and M c08 Were the Predominant Clusters in Wave 5
3.3. Intensive Intersubtype Reassortments between H7N9 and H9N2
3.4. Dynamic and Intricate Intrasubtype Reassortment
3.5. HA and NA Have the Lowest Reassortment Rate
3.6. High Reassortment Rate in Waves 3 and 5
3.7. PB2 and PA Shared Coevolutionary History
3.8. High Selection Pressure Acting on HA, NA, NS1, and M2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steel, J.; Lowen, A.C. Influenza A virus reassortment. Curr. Top. Microbiol. Immunol. 2014, 385, 377–401. [Google Scholar] [CrossRef] [PubMed]
- Lowen, A.C. Constraints, Drivers, and Implications of Influenza A Virus Reassortment. Annu. Rev. Virol. 2017, 4, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Maljkovic Berry, I.; Melendrez, M.C.; Li, T.; Hawksworth, A.W.; Brice, G.T.; Blair, P.J.; Halsey, E.S.; Williams, M.; Fernandez, S.; Yoon, I.-K.; et al. Frequency of influenza H3N2 intra-subtype reassortment: Attributes and implications of reassortant spread: Attributes and implications of reassortant spread. BMC Biol. 2016, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Nomikou, K.; Hughes, J.; Wash, R.; Kellam, P.; Breard, E.; Zientara, S.; Palmarini, M.; Biek, R.; Mertens, P. Widespread Reassortment Shapes the Evolution and Epidemiology of Bluetongue Virus following European Invasion. PLoS Pathog. 2015, 11, e1005056. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Lowen, A.C. Implications of segment mismatch for influenza A virus evolution. J. Gen. Virol. 2018, 99, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Simonsen, L.; Viboud, C.; Miller, M.A.; Taylor, J.; George, K.S.; Griesemer, S.B.; Ghedin, E.; Ghedi, E.; Sengamalay, N.A.; et al. Stochastic processes are key determinants of short-term evolution in influenza a virus. PLoS Pathog. 2006, 2, e125. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Viboud, C.; Holmes, E.C.; Koonin, E.V.; Lipman, D.J. Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol. Direct. 2006, 1, 34. [Google Scholar] [CrossRef]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef]
- Lowen, A.C. It’s in the mix: Reassortment of segmented viral genomes. PLoS Pathog. 2018, 14, e1007200. [Google Scholar] [CrossRef]
- de Silva, U.C.; Tanaka, H.; Nakamura, S.; Goto, N.; Yasunaga, T. A comprehensive analysis of reassortment in influenza A virus. Biol. Open 2012, 1, 385–390. [Google Scholar] [CrossRef]
- Wille, M.; Holmes, E.C. The Ecology and Evolution of Influenza Viruses. Cold Spring Harb. Perspect. Med. 2020, 10, a038489. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, H. H7N9 Influenza Virus in China. Cold Spring Harb. Perspect. Med. 2021, 11, a038349. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.-Y.; Wang, J.; Shen, Y.; Zhou, B.; Duan, L.; Cheung, C.-L.; Ma, C.; Lycett, S.J.; Leung, C.Y.-H.; Chen, X.; et al. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 2013, 502, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Wang, S.; Yin, Y.; Zhang, G.; Carter, R.A.; Wang, J.; Xu, G.; Sun, H.; Wang, M.; Wen, C.; et al. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc. Natl. Acad. Sci. USA 2015, 112, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Su, C.; Wang, D.; Peng, Y.; Liu, M.; Hua, S.; Li, T.; Gao, G.F.; Tang, H.; Chen, J.; et al. Sequential reassortments underlie diverse influenza H7N9 genotypes in China. Cell Host Microbe 2013, 14, 446–452. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Zhu, W.; Zhang, Y.; Zou, S.; Bo, H.; Gao, R.; Dong, J.; Huang, W.; Guo, J.; et al. Two Outbreak Sources of Influenza A (H7N9) Viruses Have Been Established in China. J. Virol. 2016, 90, 5561–5573. [Google Scholar] [CrossRef]
- Zhu, Y.; Qi, X.; Cui, L.; Zhou, M.; Wang, H. Human co-infection with novel avian influenza A H7N9 and influenza A H3N2 viruses in Jiangsu province, China. Lancet 2013, 381, 2134. [Google Scholar] [CrossRef]
- Li, J.; Kou, Y.; Yu, X.; Sun, Y.; Zhou, Y.; Pu, X.; Jin, T.; Pan, J.; Gao, G.F. Human co-infection with avian and seasonal influenza viruses, China. Emerg. Infect. Dis. 2014, 20, 1953–1955. [Google Scholar] [CrossRef]
- Pu, Z.; Yang, J.; Shen, X.; Irwin, D.M.; Shen, Y. Human-isolated H7N9 obtained internal genes from duck and human influenza viruses. J. Infect. 2019, 78, 491–503. [Google Scholar] [CrossRef]
- Yang, L.; Xie, J.; Zhang, Y.; Zhu, W.; Li, X.; Wei, H.; Li, Z.; Zhao, L.; Bo, H.; Liu, J.; et al. Emergence of waterfowl-originated gene cassettes in HPAI H7N9 viruses caused severe human infection in Fujian, China. Influenza Other Respir. Viruses 2019, 13, 496–503. [Google Scholar] [CrossRef]
- Jin, Y.; Ren, H.; Teng, Y.; Hu, M.; Peng, X.; Yue, J.; Liang, L. Novel reassortment of avian influenza A(H7N9) virus with subtype H6N6 and H5N6 viruses circulating in Guangdong Province, China. J. Infect. 2017, 75, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Shi, W.; Yang, Y.; Yang, Y.; Liu, X.; Xu, W.; Li, H.; Li, J.; Wang, Q.; Tong, Z.; et al. New Threats from H7N9 Influenza Virus: Spread and Evolution of High- and Low-Pathogenicity Variants with High Genomic Diversity in Wave Five: Spread and Evolution of High- and Low-Pathogenicity Variants with High Genomic Diversity in Wave Five. J. Virol. 2018, 92, e00301-18. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, S.; Bing, G.; Carter, R.A.; Wang, Z.; Wang, J.; Wang, C.; Wang, L.; Wu, G.; Webster, R.G.; et al. Genetic evolution of influenza H9N2 viruses isolated from various hosts in China from 1994 to 2013. Emerg. Microbes Infect. 2017, 6, e106. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Fan, H.; Raghwani, J.; Lam, T.T.-Y.; Li, J.; Pybus, O.G.; Yao, H.-W.; Wo, Y.; Liu, K.; An, X.-P.; et al. Occurrence and reassortment of avian influenza A (H7N9) viruses derived from coinfected birds in China. J. Virol. 2014, 88, 13344–13351. [Google Scholar] [CrossRef]
- Ke, C.; Lu, J.; Wu, J.; Guan, D.; Zou, L.; Song, T.; Yi, L.; Zeng, X.; Liang, L.; Ni, H.; et al. Circulation of reassortant influenza A(H7N9) viruses in poultry and humans, Guangdong Province, China, 2013. Emerg. Infect. Dis. 2014, 20, 2034–2040. [Google Scholar] [CrossRef]
- Qi, W.; Shi, W.; Li, W.; Huang, L.; Li, H.; Wu, Y.; Yan, J.; Jiao, P.; Zhu, B.; Ma, J.; et al. Continuous reassortments with local chicken H9N2 virus underlie the human-infecting influenza A (H7N9) virus in the new influenza season, Guangdong, China. Protein Cell 2014, 5, 878–882. [Google Scholar] [CrossRef]
- Bai, R.; Sikkema, R.S.; Munnink, B.B.O.; Li, C.R.; Wu, J.; Zou, L.; Jing, Y.; Lu, J.; Yuan, R.Y.; Liao, M.; et al. Exploring utility of genomic epidemiology to trace origins of highly pathogenic influenza A/H7N9 in Guangdong. Virus Evol. 2020, 6, veaa097. [Google Scholar] [CrossRef]
- Ma, M.-J.; Yang, Y.; Fang, L.-Q. Highly Pathogenic Avian H7N9 Influenza Viruses: Recent Challenges: Recent Challenges. Trends Microbiol. 2019, 27, 93–95. [Google Scholar] [CrossRef]
- Qiu, Y.; Sun, R.; Hou, G.; Yu, X.; Li, Y.; Li, J.; Zhang, Q.; Zou, F.; Liu, H.; Jiang, W. Novel reassortant H7N2 originating from the H7N9 highly pathogenic avian influenza viruses in China, 2019. J. Infect. 2019, 79, 462–470. [Google Scholar] [CrossRef]
- Nakayama, M.; Uchida, Y.; Shibata, A.; Kobayashi, Y.; Mine, J.; Takemae, N.; Tsunekuni, R.; Tanikawa, T.; Harada, R.; Osaka, H.; et al. A novel H7N3 reassortant originating from the zoonotic H7N9 highly pathogenic avian influenza viruses that has adapted to ducks. Transbound. Emerg. Dis. 2019, 66, 2342–2352. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Li, X.; Cardona, C.J.; Xing, Z. Reassortment and adaptive mutations of an emerging avian influenza virus H7N4 subtype in China. PLoS ONE 2020, 15, e0227597. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, L.; Guo, Z.; Zhang, C.; Wang, Z.; Wen, G.; Zhang, W.; Shang, Y.; Zhang, T.; Jiao, Z.; et al. A Novel Reassortant Avian H7N6 Influenza Virus Is Transmissible in Guinea Pigs via Respiratory Droplets. Front. Microbiol. 2019, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Chen, H.; Li, Q.; Huang, J.; Zhao, M.; Gu, X.; Jiang, K.; Wang, X.; Peng, D.; Liu, X. Enzootic genotype S of H9N2 avian influenza viruses donates internal genes to emerging zoonotic influenza viruses in China. Vet. Microbiol. 2014, 174, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Liu, D.; Shi, W.; Pan, J.; Qi, X.; Li, X.; Guo, X.; Zhou, M.; Li, W.; Li, J.; et al. Dynamic reassortments and genetic heterogeneity of the human-infecting influenza A (H7N9) virus. Nat. Commun. 2014, 5, 3142. [Google Scholar] [CrossRef]
- Pu, J.; Yin, Y.; Liu, J.; Wang, X.; Zhou, Y.; Wang, Z.; Sun, Y.; Sun, H.; Li, F.; Song, J.; et al. Reassortment with dominant chicken H9N2 influenza virus contributed to the fifth H7N9 virus human epidemic. J. Virol. 2021, 95, e01578-20. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- He, D.; Gu, J.; Gu, M.; Wu, H.; Li, J.; Zhan, T.; Chen, Y.; Xu, N.; Ge, Z.; Wang, G.; et al. Genetic and antigenic diversity of H7N9 highly pathogenic avian influenza virus in China. Infect. Genet. Evol. 2021, 93, 104993. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Hill, V.; Baele, G. Bayesian estimation of past population dynamics in BEAST 1.10 using the Skygrid coalescent model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, M.C.F.; Ciccozzi, M.; Fanti, I.; Saladini, F.; Pecorari, M.; Borghi, V.; Di Giambenedetto, S.; Bruzzone, B.; Capetti, A.; Vivarelli, A.; et al. A novel methodology for large-scale phylogeny partition. Nat. Commun. 2011, 2, 321. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Xu, S.; Dai, Z.; Guo, P.; Fu, X.; Liu, S.; Zhou, L.; Tang, W.; Feng, T.; Chen, M.; Zhan, L.; et al. ggtreeExtra: Compact Visualization of Richly Annotated Phylogenetic Data. Mol. Biol. Evol. 2021, 38, 4039–4042. [Google Scholar] [CrossRef]
- Altekar, G.; Dwarkadas, S.; Huelsenbeck, J.P.; Ronquist, F. Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 2004, 20, 407–415. [Google Scholar] [CrossRef]
- Westgeest, K.B.; Russell, C.A.; Lin, X.; Spronken, M.I.J.; Bestebroer, T.M.; Bahl, J.; van Beek, R.; Skepner, E.; Halpin, R.A.; de Jong, J.C.; et al. Genomewide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J. Virol. 2014, 88, 2844–2857. [Google Scholar] [CrossRef]
- Avino, M.; Ng, G.T.; He, Y.; Renaud, M.S.; Jones, B.R.; Poon, A.F.Y. Tree shape-based approaches for the comparative study of cophylogeny. Ecol. Evol. 2019, 9, 6756–6771. [Google Scholar] [CrossRef]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Nagarajan, N.; Kingsford, C. GiRaF: Robust, computational identification of influenza reassortments via graph mining. Nucleic Acids Res. 2011, 39, e34. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Müller, N.F.; Stolz, U.; Dudas, G.; Stadler, T.; Vaughan, T.G. Bayesian inference of reassortment networks reveals fitness benefits of reassortment in human influenza viruses. Proc. Natl. Acad. Sci. USA 2019, 117, 17104–17111. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegrant graphics for data analysis/Hadley Wickham; with contributions by Carson Sievert, 2nd ed.; Springer: Cham, Switzerland, 2016; pp. 9–199. [Google Scholar]
- Jacquot, M.; Rao, P.P.; Yadav, S.; Nomikou, K.; Maan, S.; Jyothi, Y.K.; Reddy, N.; Putty, K.; Hemadri, D.; Singh, K.P.; et al. Contrasting selective patterns across the segmented genome of bluetongue virus in a global reassortment hotspot. Virus Evol. 2019, 5, vez027. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5-A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Chen, R.; Holmes, E.C. Avian influenza virus exhibits rapid evolutionary dynamics. Mol. Biol. Evol. 2006, 23, 2336–2341. [Google Scholar] [CrossRef]
- Cattoli, G.; Fusaro, A.; Monne, I.; Coven, F.; Joannis, T.; El-Hamid, H.S.A.; Hussein, A.A.; Cornelius, C.; Amarin, N.M.; Mancin, M.; et al. Evidence for differing evolutionary dynamics of A/H5N1 viruses among countries applying or not applying avian influenza vaccination in poultry. Vaccine 2011, 29, 9368–9375. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, J.; Jin, X.; Li, X.; Wang, J.; Zhang, M.; Chen, J.; Xie, S.; Qi, W.; Liao, M.; et al. Accelerated Evolution of H7N9 Subtype Influenza Virus under Vaccination Pressure. Virol. Sin. 2021, 36, 1124–1132. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Weng, Z.; Shi, W. Substitution rates of the internal genes in the novel avian H7N9 influenza virus. Clin. Infect. Dis. 2013, 57, 1213–1215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fitch, W.M.; Leiter, J.M.; Li, X.Q.; Palese, P. Positive Darwinian evolution in human influenza A viruses. Proc. Natl. Acad. Sci. USA 1991, 88, 4270–4274. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Natural selection on the influenza virus genome. Mol. Biol. Evol. 2006, 23, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jin, T.; Cui, Y.; Pu, X.; Li, J.; Xu, J.; Liu, G.; Jia, H.; Liu, D.; Song, S.; et al. Influenza H7N9 and H9N2 viruses: Coexistence in poultry linked to human H7N9 infection and genome characteristics. J. Virol. 2014, 88, 3423–3431. [Google Scholar] [CrossRef]
- Marshall, N.; Priyamvada, L.; Ende, Z.; Steel, J.; Lowen, A.C. Influenza virus reassortment occurs with high frequency in the absence of segment mismatch. PLoS Pathog. 2013, 9, e1003421. [Google Scholar] [CrossRef]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef]
- Essere, B.; Yver, M.; Gavazzi, C.; Terrier, O.; Isel, C.; Fournier, E.; Giroux, F.; Textoris, J.; Julien, T.; Socratous, C.; et al. Critical role of segment-specific packaging signals in genetic reassortment of influenza A viruses. Proc. Natl. Acad. Sci. USA 2013, 110, E3840-8. [Google Scholar] [CrossRef]
- Sun, X.; Belser, J.A.; Pappas, C.; Pulit-Penaloza, J.A.; Brock, N.; Zeng, H.; Creager, H.M.; Le, S.; Wilson, M.; Lewis, A.; et al. Risk Assessment of Fifth-Wave H7N9 Influenza A Viruses in Mammalian Models. J. Virol. 2019, 93, e01740-18. [Google Scholar] [CrossRef]
- Hao, X.; Hu, J.; Wang, X.; Gu, M.; Wang, J.; Liu, D.; Gao, Z.; Chen, Y.; Gao, R.; Li, X.; et al. The PB2 and M genes are critical for the superiority of genotype S H9N2 virus to genotype H in optimizing viral fitness of H5Nx and H7N9 avian influenza viruses in mice. Transbound. Emerg. Dis. 2020, 67, 758–768. [Google Scholar] [CrossRef]
- Hao, X.; Wang, X.; Hu, J.; Gu, M.; Wang, J.; Deng, Y.; Jiang, D.; He, D.; Xu, H.; Yang, Y.; et al. The PB2 and M genes of genotype S H9N2 virus contribute to the enhanced fitness of H5Nx and H7N9 avian influenza viruses in chickens. Virology 2019, 535, 218–226. [Google Scholar] [CrossRef]
- Villa, M.; Lässig, M. Fitness cost of reassortment in human influenza. PLoS Pathog. 2017, 13, e1006685. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Lycett, S.J.; Leigh Brown, A.J. Reassortment patterns of avian influenza virus internal segments among different subtypes. BMC Evol. Biol. 2014, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, T.; Yang, W.; Shao, Z.; He, B.; Chen, X.; Wu, L.; Jin, E.; Liu, H.; Chen, J.; et al. Avian Influenza A (H7N9) Virus in a Wild Land Bird in Central China, Late 2015. Virol. Sin. 2018, 33, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Bui, C.; Bethmont, A.; Chughtai, A.A.; Gardner, L.; Sarkar, S.; Hassan, S.; Seale, H.; MacIntyre, C.R. A Systematic Review of the Comparative Epidemiology of Avian and Human Influenza A H5N1 and H7N9 - Lessons and Unanswered Questions. Transbound. Emerg. Dis. 2016, 63, 602–620. [Google Scholar] [CrossRef]
- Bhat, S.; James, J.; Sadeyen, J.-R.; Mahmood, S.; Everest, H.J.; Chang, P.; Walsh, S.; Byrne, A.M.P.; Mollett, B.; Lean, F.; et al. Co-infection of chickens with H9N2 and H7N9 avian influenza viruses leads to emergence of reassortant H9N9 virus with increased fitness for poultry and enhanced zoonotic potential. J. Virol. 2021, 96, e0185621. [Google Scholar] [CrossRef]
- Dugan, V.G.; Chen, R.; Spiro, D.J.; Sengamalay, N.; Zaborsky, J.; Ghedin, E.; Nolting, J.; Swayne, D.E.; Runstadler, J.A.; Happ, G.M.; et al. The evolutionary genetics and emergence of avian influenza viruses in wild birds. PLoS Pathog. 2008, 4, e1000076. [Google Scholar] [CrossRef]
- Dudas, G.; Bedford, T.; Lycett, S.; Rambaut, A. Reassortment between influenza B lineages and the emergence of a coadapted PB1-PB2-HA gene complex. Mol. Biol. Evol. 2015, 32, 162–172. [Google Scholar] [CrossRef]
- Neverov, A.D.; Lezhnina, K.V.; Kondrashov, A.S.; Bazykin, G.A. Intrasubtype reassortments cause adaptive amino acid replacements in H3N2 influenza genes. PLoS Genet. 2014, 10, e1004037. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coding Region | No. of Codons | dN/dS | No. of Selected Sites (% of Codons) | ||||
|---|---|---|---|---|---|---|---|
| Positively Selected | Negatively Selected (SLAC) | ||||||
| SLAC a | FUBAR b | FEL c | MEME d | ||||
| PB2 | 759 | 0.1200 | 4 | 4 | 4 | 10 | 670 (88.16%) |
| PB1 | 757 | 0.0977 | 5 | 6 | 6 | 7 | 657 (75.60%) |
| PA | 716 | 0.1137 | 7 | 4 | 8 | 6 | 573 (79.92%) |
| HA [38] | 564 | 0.2656 | 16 | 13 | 16 | 14 | 374 (66.19%) |
| NP | 498 | 0.1026 | 2 | 1 | 4 | 3 | 413 (82.77%) |
| NA [38] | 465 | 0.2946 | 13 | 8 | 15 | 13 | 280 (60.09%) |
| M1 | 252 | 0.0752 | 0 | 1 | 1 | 1 | 190 (75.10%) |
| M2 | 97 | 0.5398 | 6 | 7 | 8 | 3 | 26 (26.53%) |
| NS1 | 217 | 0.3087 | 13 | 6 | 14 | 10 | 115 (52.75%) |
| NEP | 121 | 0.2766 | 0 | 1 | 1 | 2 | 55 (45.08%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, D.; Wang, X.; Wu, H.; Wang, X.; Yan, Y.; Li, Y.; Zhan, T.; Hao, X.; Hu, J.; Hu, S.; et al. Genome-Wide Reassortment Analysis of Influenza A H7N9 Viruses Circulating in China during 2013–2019. Viruses 2022, 14, 1256. https://doi.org/10.3390/v14061256
He D, Wang X, Wu H, Wang X, Yan Y, Li Y, Zhan T, Hao X, Hu J, Hu S, et al. Genome-Wide Reassortment Analysis of Influenza A H7N9 Viruses Circulating in China during 2013–2019. Viruses. 2022; 14(6):1256. https://doi.org/10.3390/v14061256
Chicago/Turabian StyleHe, Dongchang, Xiyue Wang, Huiguang Wu, Xiaoquan Wang, Yayao Yan, Yang Li, Tiansong Zhan, Xiaoli Hao, Jiao Hu, Shunlin Hu, and et al. 2022. "Genome-Wide Reassortment Analysis of Influenza A H7N9 Viruses Circulating in China during 2013–2019" Viruses 14, no. 6: 1256. https://doi.org/10.3390/v14061256
APA StyleHe, D., Wang, X., Wu, H., Wang, X., Yan, Y., Li, Y., Zhan, T., Hao, X., Hu, J., Hu, S., Liu, X., Ding, C., Su, S., Gu, M., & Liu, X. (2022). Genome-Wide Reassortment Analysis of Influenza A H7N9 Viruses Circulating in China during 2013–2019. Viruses, 14(6), 1256. https://doi.org/10.3390/v14061256