

First Genomic Evidence of a Henipa-like Virus in Brazil

 ,
,  , , , ,
, , , ,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
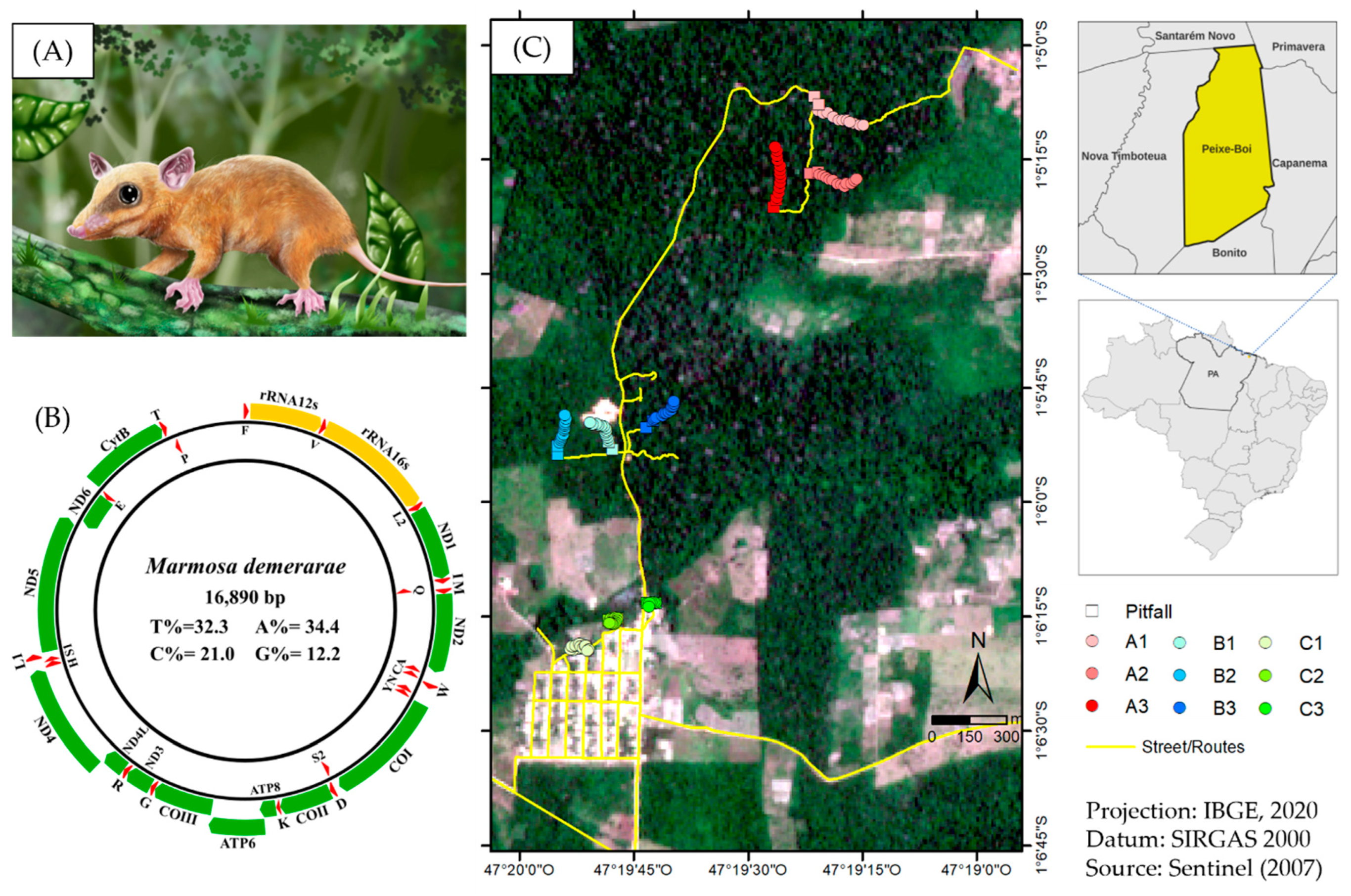
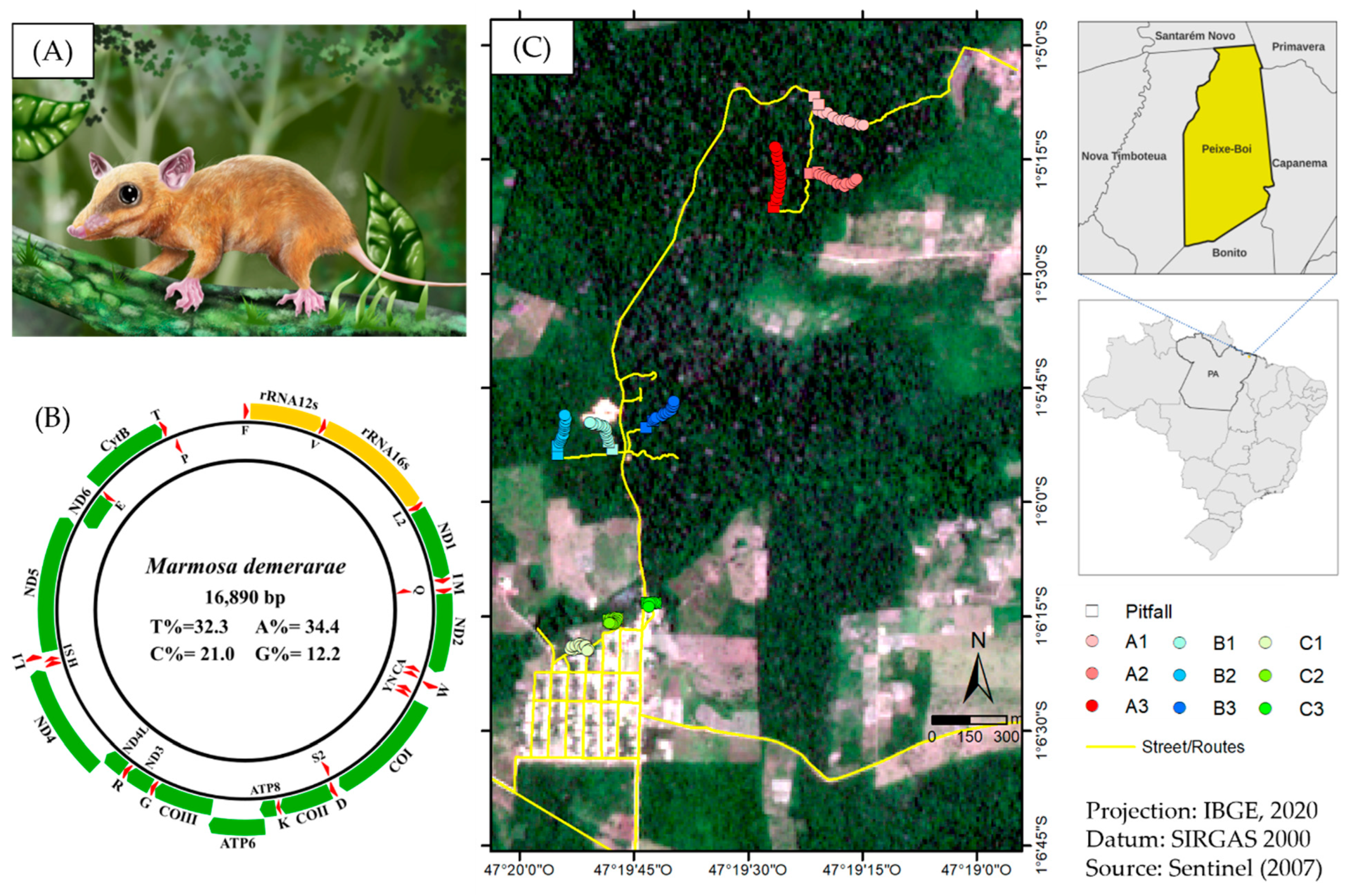
2.1. Samples Collection
2.2. RNA Extraction and cDNA Synthesis
2.3. Sequencing and Sequence Assembly
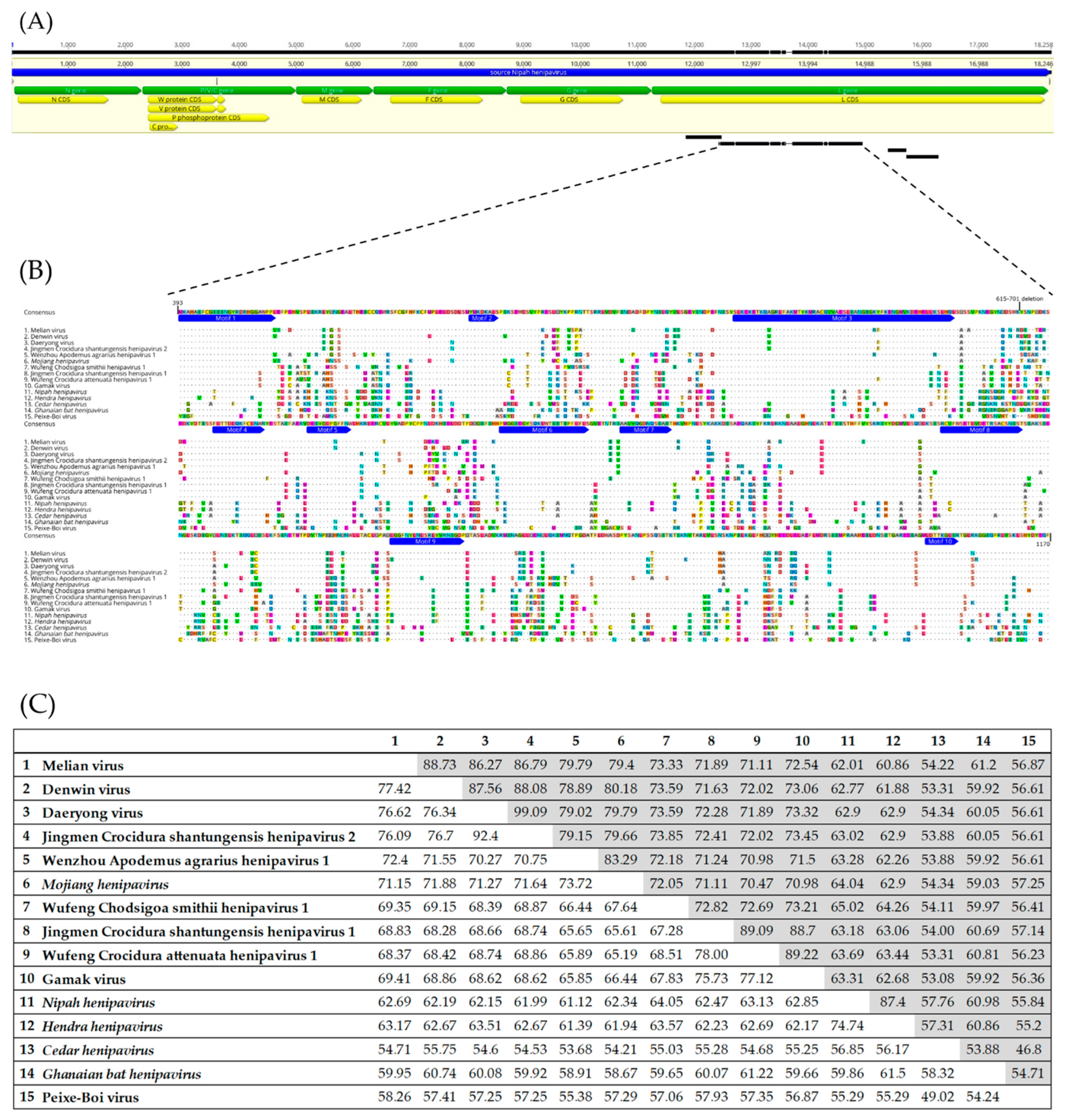
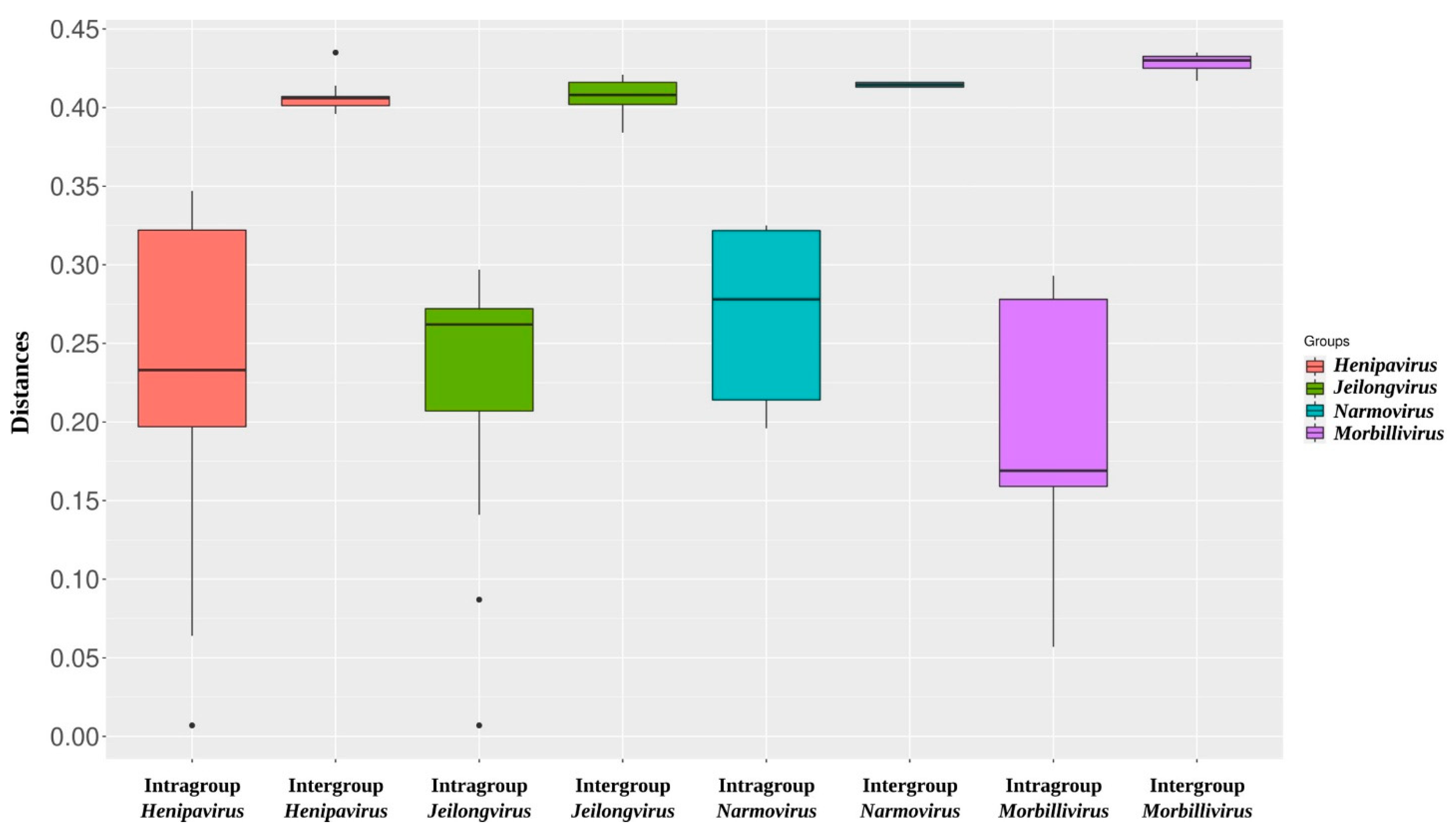
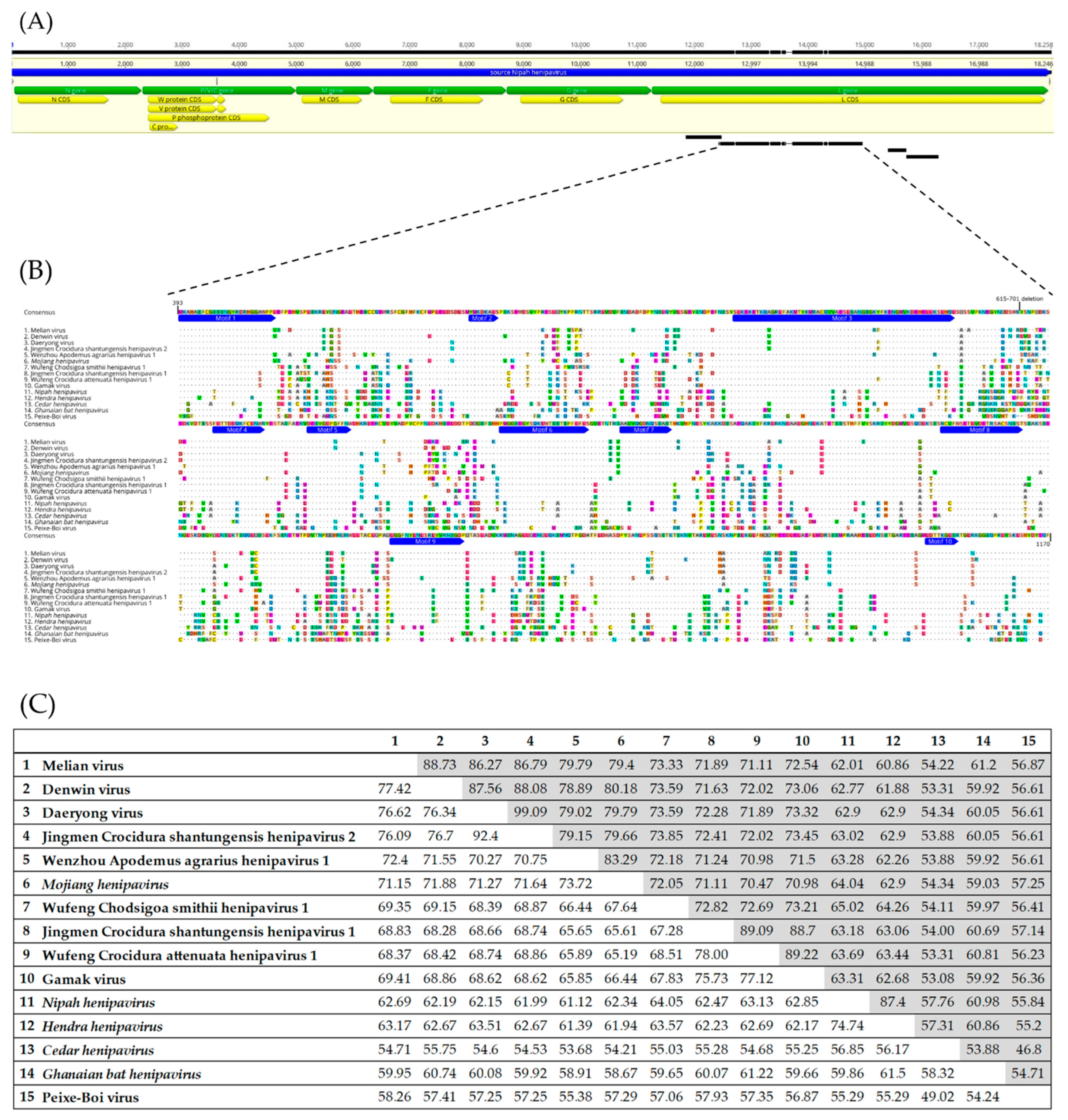
2.4. Alignment and Identity/Divergence Analysis
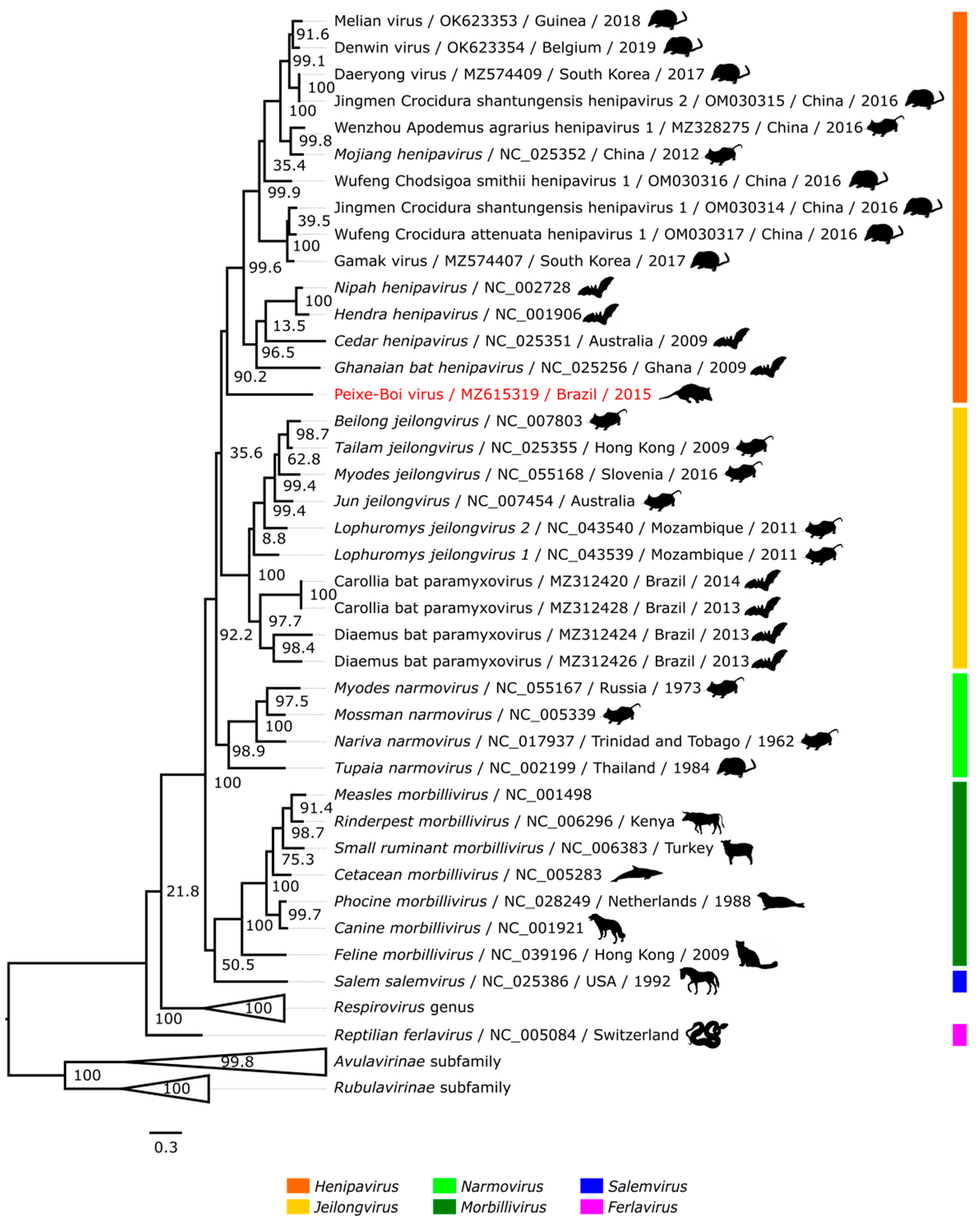
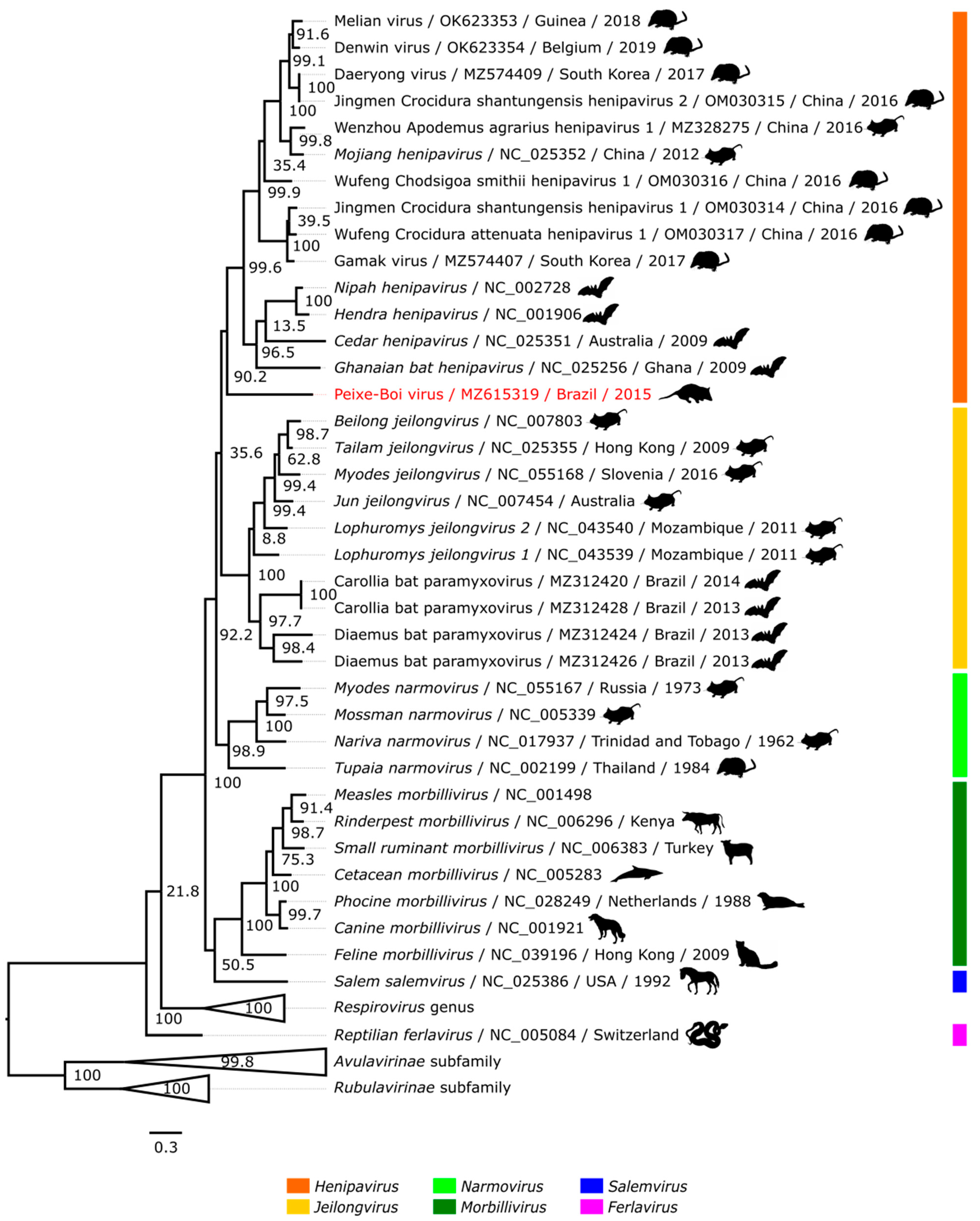
2.5. Phylogenetic Analysis
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.-F.; Crameri, G. Emerging zoonotic viral diseases. Rev. Sci. Tech. 2014, 33, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.K.; Hitchens, P.; Evans, T.S.; Goldstein, T.; Thomas, K.; Clements, A.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; Karesh, W.; et al. Spillover and pandemic properties of zoonotic viruses with high host plasticity. Sci. Rep. 2015, 5, 14830. [Google Scholar] [CrossRef] [PubMed]
- Trovato, M.; Sartorius, R.; D’Apice, L.; Manco, R.; De Berardinis, P. Viral Emerging Diseases: Challenges in Developing Vaccination Strategies. Front. Immunol. 2020, 11, 2130. [Google Scholar] [CrossRef] [PubMed]
- Jornal da USP. Available online: https://jornal.usp.br/ciencias/virus-de-alta-letalidade-ressurgido-no-brasil-apos-20-anos-e-investigado-pela-usp/ (accessed on 23 July 2022).
- Thibault, P.A.; Watkinson, R.E.; Moreira-Soto, A.; Drexler, J.F.; Lee, B. Zoonotic potential of emerging paramyxoviruses: Knows and unknows. Adv. Virus. Res. 2017, 98, 1–55. [Google Scholar] [PubMed]
- International Committee on Taxonomy of Viruses. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 23 July 2022).
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef]
- Epstein, J.H.; Anthony, S.J.; Islam, A.; Kilpatrick, A.M.; Khan, S.A.; Balkey, M.D.; Ross, N.; Smith, I.; Zambrana-Torrelio, C.; Tao, Y.; et al. Nipah virus dynamics in bats and implications for spillover to humans. Proc. Natl. Acad. Sci. USA 2020, 117, 29190–29201. [Google Scholar] [CrossRef]
- Yuen, K.Y.; Fraser, N.S.; Henning, J.; Halpin, K.; Gibson, J.S.; Betzien, L.; Stewart, A.J. Hendra virus: Epidemiology dynamics in relation to climate change, diagnostic tests and control measures. One Health 2021, 12, 100207. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef]
- Pernet, O.; Schneider, B.S.; Beaty, S.M.; LeBreton, M.; Yun, T.E.; Park, A.; Zachariah, T.T.; Bowden, T.A.; Hitchens, P.; Ramirez, C.M.; et al. Evidence for henipavirus spillover into human populations in Africa. Nat. Commun. 2014, 5, 5342. [Google Scholar] [CrossRef]
- Mbu’U, C.M.; Mbacham, W.F.; Gontao, P.; Kamdem, S.L.S.; Nlôga, A.M.N.; Groschup, M.H.; Wade, A.; Fischer, K.; Balkema-Buschmann, A. Henipaviruses at the Interface Between Bats, Livestock and Human Population in Africa. Vector-Borne Zoonotic Dis. 2019, 19, 455–465. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Yang, F.; Ren, X.; Jiang, J.; Dong, J.; Sun, L.; Zhu, Y.; Zhou, H.; Jin, Q. Henipa-like Virus, Mojiang Paramyxovirus, in Rats, China, 2012. Emerg. Infect. Dis. 2014, 20, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- NCBI Virus. Available online: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide&VirusLineage_ss=Henipavirus,%20taxid:260964&utm_source=data-hub&HostLineage_ss=NOT%20Homo%20sapiens%20(human),%20taxid:NOT%209606 (accessed on 23 July 2022).
- Lee, S.-H.; Kim, K.; Kim, J.; No, J.S.; Park, K.; Budhathoki, S.; Lee, S.H.; Lee, J.; Cho, S.H.; Cho, S.; et al. Discovery and Genetic Characterization of Novel Paramyxoviruses Related to the Genus Henipavirus in Crocidura Species in the Republic of Korea. Viruses 2021, 13, 2020. [Google Scholar] [CrossRef] [PubMed]
- Vanmechelen, B.; Meurs, S.; Horemans, M.; Loosen, A.; Maes, T.J.; Laenen, L.; Vergote, V.; Koundouno, F.R.; Magassouba, N.; Konde, M.K.; et al. The characterization of multiple novel paramyxoviruses highlights the diverse nature of the subfamily Orthoparamyxovirinae. Virus Evol. 2022, 8, veac061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-A.; Li, H.; Jiang, F.-C.; Zhu, F.; Zhang, Y.-F.; Chen, J.-J.; Tan, C.-W.; Anderson, D.E.; Fan, H.; Dong, L.-Y.; et al. A Zoonotic Henipavirus in Febrile Patients in China. N. Engl. J. Med. 2022, 387, 470–472. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, J.; Lo, M.K.; Tamin, A.; Ometto, T.L.; Thomazelli, L.M.; Nardi, M.S.; Hurtado, R.F.; Nava, A.; Spiropoulou, C.F.; Rota, P.A.; et al. Antibodies Against Henipa-Like Viruses in Brazilian Bats. Vector-Borne Zoonotic Dis. 2017, 17, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.E.; Seifert, S.N.; Thompson, J.T.; Avanzato, V.; Sterling, S.L.; Yan, L.; Letko, M.C.; Matson, M.J.; Fischer, R.J.; Tremeau-Bravard, A.; et al. Serological Evidence for Henipa-like and Filo-like Viruses in Trinidad Bats. J. Infect. Dis. 2020, 221, 375–382. [Google Scholar] [CrossRef]
- Zhong, S.; Joung, J.-G.; Zheng, Y.; Chen, Y.-R.; Liu, B.; Shao, Y.; Xiang, J.Z.; Fei, Z.; Giovannoni, J.J. High-throughput illumine strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 2011, 940–949. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pharm, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- InterProScan. Available online: https://www.ebi.ac.uk/interpro/search/sequence/ (accessed on 12 September 2022).
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Inkscape. Available online: https://inkscape.org/release/inkscape-1.1/ (accessed on 1 June 2022).
- Wells, H.L.; Loh, E.; Nava, A.; Solorio, M.R.; Lee, M.H.; Sukor, J.R.A.; Navarrete-Macias, I.; Liang, E.; Firth, C.; Epstein, J.H.; et al. Classification of new morbillivirus and jeilongvirus sequences from bats sampled in Brazil and Malaysia. Arch. Virol. 2022, 167, 1977–1987. [Google Scholar] [CrossRef]
- Silva, C.E.F.; de Andrade, R.A.; de Souza, E.M.S.; Eler, E.S.; da Silva, M.N.F.; Feldberg, E. Comparative cytogenetics of some marsupial species (Didelphimorphia, Didelphidae) from the Amazon basin. Comp. Cytogenet. 2017, 11, 703–725. [Google Scholar] [CrossRef]
- Dias, I.M.G.; Almeida, F.C.; Amato, G.; DeSalle, R.; Fonseca, C.G. Delineating geographic boundaries of the woolly mouse opossums, Micoureus demerarae and Micoureus paraguayanus (Didelphimorphia: Didelphidae). Conserv. Genet. 2010, 11, 1579–1585. [Google Scholar] [CrossRef]
- Santori, R.T.; Lessa, L.G.; Astúa, D. Alimentação, nutrição e adaptações alimentares de marsupiais brasileiros. In Os Marsupiais do Brasil: Biologia, Ecologia e Conservação, 2nd ed.; Cáceres, N.C., Ed.; Editora UFMS: Campo Grande, Brazil, 2012; p. 391. ISBN 978-857-613-410-7. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, L.H.A.; da Paz, T.Y.B.; Silva, S.P.d.; Silva, F.S.d.; Barros, B.C.V.d.; Nunes, B.T.D.; Casseb, L.M.N.; Medeiros, D.B.A.; Vasconcelos, P.F.d.C.; Cruz, A.C.R. First Genomic Evidence of a Henipa-like Virus in Brazil. Viruses 2022, 14, 2167. https://doi.org/10.3390/v14102167
Hernández LHA, da Paz TYB, Silva SPd, Silva FSd, Barros BCVd, Nunes BTD, Casseb LMN, Medeiros DBA, Vasconcelos PFdC, Cruz ACR. First Genomic Evidence of a Henipa-like Virus in Brazil. Viruses. 2022; 14(10):2167. https://doi.org/10.3390/v14102167
Chicago/Turabian StyleHernández, Leonardo H. Almeida, Thito Y. Bezerra da Paz, Sandro Patroca da Silva, Fábio S. da Silva, Bruno C. Veloso de Barros, Bruno T. Diniz Nunes, Lívia M. Neves Casseb, Daniele B. Almeida Medeiros, Pedro F. da Costa Vasconcelos, and Ana C. Ribeiro Cruz. 2022. "First Genomic Evidence of a Henipa-like Virus in Brazil" Viruses 14, no. 10: 2167. https://doi.org/10.3390/v14102167
APA StyleHernández, L. H. A., da Paz, T. Y. B., Silva, S. P. d., Silva, F. S. d., Barros, B. C. V. d., Nunes, B. T. D., Casseb, L. M. N., Medeiros, D. B. A., Vasconcelos, P. F. d. C., & Cruz, A. C. R. (2022). First Genomic Evidence of a Henipa-like Virus in Brazil. Viruses, 14(10), 2167. https://doi.org/10.3390/v14102167